

Abstract
The advent of high-throughput biology has catalyzed a remarkable improvement in our ability to identify new genes. A large fraction of newly discovered genes have an unknown functional role, particularly when they are specific to a particular lineage or organism. These genes, currently labeled “hypothetical,” might support important biological cell functions and could potentially serve as targets for medical, diagnostic, or pharmacogenomic studies. An important challenge to the scientific community is to associate these newly predicted genes with a biological function that can be validated by experimental screens. In the absence of sequence or structural homology to known genes, we must rely on advanced biotechnological methods, such as DNA chips and protein–protein interaction screens as well as computational techniques to assign putative functions to these genes. In this article, we propose an effective methodology for combining biological evidence obtained in several high-throughput experimental screens and integrating this evidence in a way that provides consistent functional assignments to hypothetical genes. We use the visualization method of propagation diagrams to illustrate the flow of functional evidence that supports the functional assignments produced by the algorithm. Our results contain a number of predictions and furnish strong evidence that integration of functional information is indeed a promising direction for improving the accuracy and robustness of functional genomics.
Recent advances in genomic sequencing have generated an astounding number of new genes whose biological functions remain a mystery. Although sequence homology (1) provides clues that suggest a functional assignment for many newly sequenced genes, >35% of genes in prokaryotic organisms are annotated as “function unknown.” In eukaryotes, functional annotation is an even more daunting challenge, especially as we expand sequencing beyond model organisms and their close relatives. For example, >60% of the genes in Plasmodium falciparum are “hypothetical” proteins (2).
Several research groups (3, 4) have popularized the framework of a “functional-linkage graph” as a promising step toward obtaining a detailed understanding of the functional relationships between proteins. In a typical functional-linkage graph, each node corresponds to a protein, and an edge connects two proteins if some experimental or computational procedure suggests that these proteins might share the same function. For instance, two proteins might be linked if they test positive in a yeast two-hybrid screen (5) or if their gene-expression patterns are correlated in several experimental conditions. Such a link usually does not provide information on which specific functional annotation the proteins share.
Many authors have explored the idea of “integrative functional genomics,” which combines information from multiple sources to facilitate functional annotation of newly discovered genes. In the context of functional-linkage graphs, the working hypothesis underlying integrative functional genomics is that if we can establish a putative functional linkage between two proteins in two different and independently conducted experiments, then the probability of genuine functional linkage between these proteins increases. Indeed, this article and prior work provide a confirmation of this intuitive hypothesis (4, 6).
For instance, Marcotte et al. (4) describe a purely local integration of functional links. This simple “conjunctive integration” method generates a new functional-linkage graph by including exactly the edges that can be confirmed in each source graph. This conservative approach is likely to generate a high false-negative rate. An alternative “disjunctive integration” approach inserts an edge in the integrated graph if it is supported by an edge in any source graph. This overly permissive approach tends to increase the false-positive rate. A compromise can be achieved by regarding each source of evidence as an expert and by combining these experts by using probabilistic evidence-integration schemes that take into account (6, 7) the correlations between the predictions of different experts.
Integrated databases, such as bind (9), predictome (10), and string (11), have assembled a large collection of putative functional links between proteins by including information provided by diverse computational and experimental screens. Although these functional-linkage databases are a valuable source of information, they do not provide a comprehensive mechanism for making accurate functional predictions by fully exploiting the evidence encoded in the graph. Many edges in these databases are based on large-scale experimental screens, such as the yeast two-hybrid method (12, 13) and mass spectrometry-based techniques (14, 15), for determining protein–protein interactions (PPI). However, these experimental screens have inherently high false-positive rates and significant false-negative rates (16). It is tempting to use functional links to transfer a functional annotation from a labeled to a newly discovered protein. However, transferring a functional annotation from an annotated protein to its hypothetical neighbor across every link in a graph is likely to result in a very high error rate.
To increase the robustness of transferring annotation to neighboring nodes in a functional-linkage graph, researchers have proposed a simple local-threshold rule (17, 18) for functional assignment (often referred to as “guilt by association”). This rule is based on the hypothesis that if some fraction of the neighbors of a given protein p are annotated with function f, this functional annotation can be transferred to p. Fig. 1 illustrates this idea. The gray nodes in Fig. 1 represent hypothetical proteins. Red nodes correspond to proteins annotated with the function “ribosome biogenesis,” whereas the blue nodes correspond to proteins with a different functional role. In Fig. 1, we can use the guilt-by-association rule to label node YMR049C with the function “ribosome biogenesis” because 6 of its 11 neighbors have that function. Although this threshold rule is easy to apply, it has fundamental limitations. For instance, node YER006W has two blue neighbors and two red neighbors. What should its functional assignment be? This issue is just one of many subtle problems that arise in automated annotation when functional-linkage graphs are used. In many currently available functional-linkage graphs based on PPI screens, a large fraction of proteins do not have any annotated neighbors. Some researchers have suggested generalizing the neighborhood rule to include nodes at a distance that is greater than one link (19). However, it is not clear what the appropriate distance is and whether neighborhoods of different sizes should be selected for different nodes or for different functions. A more fundamental question is whether functional assignments that are based on local information should be trusted, given the high degree of noise in PPI data. Zhou et al. (8) describe a nonlocal approach that propagates of functional labels to nonneighboring nodes. Their method determines function based on the shortest path in the functional-linkage graph defined by correlation between gene-expression profiles. Their work provides additional motivation for obtaining functional evidence from nonneighboring nodes in functional-linkage graphs. It would be advantageous to develop a method that provides the capability to propagate evidence systematically across the entire graph by taking into account the global constraints and structure of the graph and to integrate diverse sources of information, such as microarray data and interactions reported in the literature.
Fig. 1.

A subgraph of the functional-linkage graph in S. cerevisiae showing proteins annotated with the function “ribosome biogenesis” (GO:0007046). To improve readability, we display only interactions in which hypothetical proteins are involved.
The main contribution of this article is a framework for achieving both objectives: the integration and propagation of evidence. We apply this framework to propagate evidence in functional-linkage graphs formed by integrating information from PPI and gene-expression data. Our method can be understood as repeated application of the local-threshold rule until the network reaches a state that is ideally maximally consistent with the integrated evidence.
In addition to providing an effective methodology for evidence integration and propagation in functional-linkage networks, we also propose a method for visualizing the flow of information in the functional-linkage graph. We present a visualization paradigm called a “propagation diagram” that allows us to track the sequence of interactions by which a particular protein receives an annotation. We believe these diagrams may provide useful insights to biologists who are interested in assessing the reasoning behind the putative functional predictions produced by the algorithm.
We have applied our method to a collection of PPI data and gene-expression data for S. cerevisiae and produced new functional predictions for >200 proteins of unknown function. Functional categories were based on Gene Ontology (GO). We restricted our predictions to GO functions that we have cross-validated as having at least 75% precision and recall on average (see Performance Evaluation for definitions). These predictions span functions from all three GO hierarchies and include functions related to DNA repair, cell cycle, rRNA processing, and RNA transport. We perform leave-one-out cross-validation and demonstrate that integrating gene expression and PPI yields both improved precision and recall. Finally, we provide a list of hypothetical genes whose predicted annotation appears to be consistent with other evidence (e.g., with information in published literature) but have been difficult to annotate unambiguously by using the simple local neighborhood rule.
Methods
We map the functional-linkage graph into a variant of a discrete-state Hopfield network. Hopfield networks are a class of neural architectures, inspired originally by statistical physics and used subsequently in computational neuroscience (20). There is a one-to-one correspondence between the nodes and edges of the functional-linkage graph and the network we use.
In this study, we constructed a distinct network for each function in GO. Each node in the network can be in one of three discrete states. Given a particular GO function f, the state of a node is +1 if the protein is annotated with f and -1 if the protein is annotated with a different function (in the same GO hierarchy). The state of annotated nodes does not change during the execution of the algorithm. We set the initial state of all hypothetical proteins at 0, corresponding to an uncertain state. (An alternate and more typical approach sets the initial state of a hypothetical protein randomly to -1 or +1. Our approach avoids creating a bias in the initial state of a hypothetical protein.) Each edge in the network has a real-valued weight. The weighted edge between two nodes represents a noisy putative functional relationship between the corresponding proteins. The larger the weight, the more evidence we have that the two proteins share the same function. If there is no evidence that two proteins share the same function, we set the weight of the edge connecting them at 0. Our approach readily supports negative edge weights, although we do not use them in this article.
The goal of our procedure is to assign a state of -1 or +1 to the nodes whose initial state is equal to 0. We assign the putative functional annotation f to all hypothetical proteins to which the procedure assigns a state of +1. Intuitively, we would like to assign states to these nodes in such a way that the two nodes connected by an edge receive the same functional assignment; we say that such an edge is “consistent.” Because it is not always possible to ensure that every edge is consistent, it is desirable to compute a maximally consistent state assignment (one in which the weighted sum of consistent edges is as large as possible). This approach formalizes and generalizes the simpler local neighborhood rule used in prior studies. We can achieve such a maximally consistent assignment by minimizing the following “energy” function:
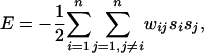 |
where n is the number of nodes in the network; wij is the weight of the edge connecting proteins i and j; and si is the state assigned to protein i. In this equation, a consistent edge makes a positive contribution to E, and an inconsistent edge makes a negative contribution to E. Therefore, minimizing E maximizes the weighted sum of consistent edges. In the case where all of the edges have unit weight, we maximize the number of consistent edges. The problem of computing maximally consistent assignments is computationally intractable by reduction from computing maximal cuts in graphs (21).
Given the computational intractability of identifying maximally consistent assignments, we must rely on heuristics. We employ a local search procedure, which is an instance of iterative gradient descent. Our algorithm applies the following activation rule, which defines the dynamic behavior of the network, iteratively to each node of the network until convergence (i.e., when further application of this rule does not change the state of any node).
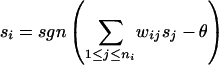 |
Here, ni is the number of neighbors of protein i and θ is an “activation threshold”. The right side of this equation computes the weighted sum of the states of the neighbors of node i and compares this sum with θ: if the sum is >θ, then the state of node i is set to +1, otherwise it is set to -1. This rule is a variant of the local guilt-by-association rule used in earlier studies. Iterative application of this rule achieves a more globally consistent functional annotation to all of the proteins in the network than a single application of this rule.
Our algorithm applies the rule serially to each node in the network. A single iteration of the algorithm updates (if necessary) all of the nodes in the network. The algorithm repeats these iterations until convergence. It can be shown easily that the update rule changes the value of the energy function monotonically and is, therefore, guaranteed to reach a local minimum (21, 22). Moreover, this solution is guaranteed to be a half-approximation to the best solution (21). For networks with unit cost edge weights, the maximum number of updates is bounded by 2n2. When the edge weights are real-valued, in the worst case, the network might need an exponential number of applications of the local update rule to reach convergence. In practice, we have noted that our networks converge within two or three iterations over all of the nodes.
As an illustration of the algorithm, consider the network shown in Fig. 1. We set the activation threshold to 0. Initially, ORF YER006W has an ambiguous vote from its neighbors (two in state +1, two in state -1, and one in state 0). When the algorithm first processes ORF YMR049C, it uses the update rule to assign a state of +1 to YMR049C (because YMR049C has six neighbors in state +1 and at most five neighbors in state -1). This change modifies the neighborhood of YER006W. The next time the algorithm processes YER006W, it can assign a state of +1 to YER006W. We see a similar effect for ORF YMR290C.
In this article, we compare two types of schemes for assigning edge weights. The first variant attempts to capture only qualitative functional links between proteins. In our case, these are PPI links. Therefore, we assign a weight of 1 to each edge of the network. In the second scheme, we integrate gene-expression measurements from a set of 300 yeast knockout experiments performed by Hughes et al. (23). The weight of an edge in the integrated network is the absolute value of the correlation coefficient of the gene-expression profiles of the pair of interacting proteins. Weighting edges in this manner improves the probability that the network will assign consistent functions to pairs of proteins for which both the PPI and the gene-expression data sets contain evidence of shared function; in the rest of this article, we refer to these variants as the “PPI-only” and “integrated” networks, respectively.
We use leave-one-out cross-validation to evaluate our methodology. For each function f and for each protein annotated with this function, we change the initial state of the protein to 0 and apply the procedure to check whether the network assigns a final state of +1 to that protein. To measure the false-positive rate, we perform a similar operation for an equal number of proteins with the initial state -1. We iterate this procedure over all functions. If TP is the total number of proteins for which we correctly predict a final state of +1, FP is the total number of proteins for which we incorrectly predict a final state of +1, and FN is the total number of proteins for which we incorrectly predict a final state of -1 (we compute these numbers by summing over all of the functions), then the “precision” of our procedure is TP/(TP + FP), and the “recall,” or sensitivity, of our approach is TP/(TP + FN). Note that we do not report the specificity of the procedure. The majority of functions induce a network with a large number of proteins in initial state -1. Thus, even a trivial algorithm that assigns all proteins to state -1 will achieve high specificity. To compare different versions of our algorithm, we use the “F-measure,” which is the harmonic mean of the precision and the recall; the harmonic mean of two numbers x and y is 2xy/(x+ y). This measure is commonly used in information retrieval (24). Typically, improving the precision of an algorithm decreases its recall and vice versa. The F-measure aims to combine both criteria, so that a higher F-measure corresponds to a better performance of the algorithm.
Software Availability. The software implementing our technique is available at http://genomics10.bu.edu/gain and http://bioinformatics.cs.vt.edu/gain.
Results and Discussion
In this study, we used a PPI network derived from the interactions in the S. cerevisiae grid data set. We used only those interactions that were confirmed by at least two publications. The resulting network contains 997 distinct interactions among 1,004 proteins. We used GO for functional annotations. The hierarchical structure of GO enabled us to modify the functional annotations as follows: if a protein p was annotated with a function f, then we annotated p with every function f′ that was an ancestor of f in GO. This process resulted in a total of 1,395 functions annotating the 1,004 proteins.
We found the following interesting results. (i) Leave-one-out cross-validation demonstrates that the integrated network created from the PPI graph augmented with gene-expression correlations has superior predictive ability in comparison with the PPI-only network. (ii) A detailed examination of the improved predictions provides numerous examples of proteins that are annotated correctly (in cross-validation trials) only in the integrated network. (iii) Many of the putative functional annotations made by our technique have support in the literature. (iv) By deploying the methodology of propagation diagrams introduced in this article, we demonstrate that our technique is indeed capable of annotating hypothetical proteins whose only neighbors are hypothetical proteins. These proteins cannot be annotated by the purely local guilt-by-association rules. All results in this section use an activation threshold of 0.
Results of Cross-Validation. The PPI-only network achieves 93.6% precision and 63.7% recall over the 828 GO functions that have an F-measure >0. The functions with the highest F-measure include those related to DNA-dependent transcription and regulation of transcription from the biological-process hierarchy. To compare the two versions of our algorithm, we restricted our attention to the 440 functions for which our method makes at least one prediction for a hypothetical protein. For many of these 440 functions, we observe that the F-measure of the integrated network is significantly higher than the F-measure of the PPI-only network. More specifically, the F-measure for 168 functions increased in the integrated network. The F-measure did not change for 227 functions and decreased only for 45 functions. Fig. 3, which is published as supporting information on the PNAS web site, demonstrates this improvement visually by showing a plot the F-measures of these functions for the PPI-only and integrated networks.
The functions with the highest improved F-measure include “cytoskeleton regulatory protein-binding activity” from the molecular function hierarchy and “mitotic checkpoint,” “ribosome assembly,” and “actin filament organization” from the biological-process hierarchy. The proteins annotated with these functions are parts of protein complexes in S. cerevisiae (9). Although complexes are relatively easy to annotate by using only PPI data, it is possible that interactions in some complexes (highly connected subgraphs in the PPI-only network) correspond to false-positives in yeast 2-hybrid screens. In these cases, the integrated network weeds out false interactions, enabling us to achieve a higher F-measure.
Improved Functional Predictions in Cross-Validation in the Integrated Network. To illustrate the improved cross-validation performance of the integrated network, we tracked the predictions for individual genes. Considering only GO functions that have good performance in the cross-validation study (F-measure ≥0.75), we searched for genes that changed from false negatives in the PPI-only network to true positives in the integrated network. These genes confirm the usefulness of integrating multiple information sources.
Examples of such genes include SEC31, a gene whose product in known to be involved in protein transport from the endoplasmic reticulum (ER) to the Golgi body. When we performed the cross-validation study, the PPI-only network did not predict the following functions for gene SEC31: “ER to Golgi transport” (GO:0006888), “protein secretion” (GO:0009306), “protein transport” (GO:0015031), and “protein metabolism” (GO:0019538). The integrated network made all of these predictions correctly.
Another example is the CDC42 gene, whose protein product is known to be a member of the Rho subfamily of Ras-like proteins. Some annotations of CDC42 that are “corrected” by the integrated network include “establishment and/or maintenance of cell polarity” (GO:0030012), “budding” (GO:0007114), and “shmoo tip” (GO:0005937). A list of genes whose predictions change from “wrong” in the PPI-only network to “correct” in the integrated network is available in Data Set 1, which is published as supporting information on the PNAS web site.
Evaluating the Plausibility of New Functional Annotations. We applied the technique to the grid interactions network, limiting ourselves to interactions that were confirmed by at least two publications. Recall that we built a separate network for each function in GO and assigned the function as a putative annotation to a hypothetical protein if the network computed a final state of +1 for that protein. We assessed the plausibility of our predictions by using various means. Many proteins are annotated in at most two GO hierarchies. In such cases, the plausibility of a new functional prediction in one GO hierarchy can be assessed by looking at the already existing annotations for those proteins. We also looked for related information in a search of the PubMed database. The Saccharomyces Genome Database descriptions for the protein of interest were sometimes useful. A list of all putative functional predictions made by our algorithm using the integrated network is provided in Table 1, which is published as supporting information on the PNAS web site.
Our technique assigns the protein PKC1 to the GO cellular component “1,3-beta-glucan synthase complex” (GO:0000148), a multienzyme complex that catalyzes the synthesis of glucan, a major structural component of the yeast cell wall. PKC1 is known to be involved in the monitoring of the state of the cell wall during growth and morphogenesis; it has been assigned the GO molecular function “protein kinase C activity” (GO:0004697), the GO biological processes “cell wall organization and biogenesis” (GO:0007047) and “protein amino acid phosphorylation” (GO:0006468), and the cellular component category “intracellular” (GO:0005622). Our technique refines the cellular-component assignment from “intracellular” to “1,3-beta-glucan synthase complex,” which is consistent with the annotations of PKC1 in the other GO hierarchies.
The NHP10 gene is an “HMG1-box containing protein.” It has been hypothesized that HMG1 proteins are potent architectural elements of chromatin that are able to induce strong bends and untwist DNA structure (25). Assigning NHP10 the biological process “chromatin modeling” (GO:0006338) and the cellular component “chromatin remodeling complex” (GO:0016585) is in accordance with this hypothesis. Ref. 26 obtains the same prediction by using a probabilistic annotation technique.
The UFO1 protein is described as an F-box protein in the Saccharomyces Genome Database. The cellular component “nuclear ubiquitin ligase complex,” assigned to UFO1, is consistent with its GO molecular function “ubiquitin–protein ligase activity” (GO:0004842), as well as the biological processes “ubiquitin-dependent protein catabolism” (GO:0006511) and “response to DNA damage” (GO:0006974).
ORF YKL067W has known “nucleoside-diphosphate kinase (NDK) activity” (GO:0004550). Experimental studies have suggested that nucleoside-diphosphate kinase functions not only as a simple enzyme but is also likely to interfere with the mating pheromone signal transduction in Schizosaccharomyces pombe (27). Our prediction of “signal transduction” (GO:0007165) from the biological-process hierarchy confirms this suggestion, whereas the second prediction “spindle pole body” (GO:0005816) from the cellular-component hierarchy suggests its involvement in a specific component in the yeast mating cycle.
Our method also makes consistent predictions in different hierarchies for proteins that have no known functions in any of the three hierarchies. One example of such a prediction is ORF YML053C, for which we predict the molecular function “glyceraldehyde 3-phosphate dehydrogenase (phosphorylating) activity,” the biological process “glycolysis,” and the cellular component “lipid particle.” Two other examples are the ORFs YCR099C and YBL059W, which are predicted to have the biological process ER to Golgi transport and the cellular component “COPII vesicle coat”; vesicles with COPII coats are associated with ER membranes at steady state (28).
Propagation Diagrams. We monitor and trace the flow of functional information in the network by using a graphical representation that we call “propagation diagram.” In Fig. 2, we display an example of such a diagram for the function “ribosome biogenesis.” In this diagram, red nodes correspond to proteins annotated with “ribosome biogenesis,” blue nodes to proteins annotated with some other function, and gray nodes to hypothetical proteins. Our technique annotates hypothetical proteins with the function either as a result of direct evidence from its neighbors (black arrows) or by means of propagation (green arrows). These arrows mark edges along which functional evidence travels in the network. A black arrow signifies propagation of evidence from an already-annotated protein. A green arrow denotes the propagation of information from one hypothetical node to another. In Fig. 4, which is published as supporting information on the PNAS web site, YER006W has two blue and two red neighbors causing an ambiguity in its functional annotation. The six red neighbors of YMR049C let us annotate YMR049C with the function. This annotation then propagates to YER006W, resolving the ambiguity in its neighborhood. YMR290C becomes annotated in a similar way. This example demonstrates that our approach provides new putative annotations for hypothetical proteins that cannot be annotated by simple local calculations. Fig. 4 displays other propagation diagrams.
Fig. 2.

Example of a propagation diagram, demonstrating the flow of functional evidence in the network.
Conclusion
In this article, we have demonstrated an effective method to interpret functional-linkage networks as a medium for inferring gene function by integrating the evidence captured by protein–protein interactions and gene-expression data. This framework provides two important capabilities. It provides (i) a promising methodology for propagating functional information across functional-linkage graphs to genes that cannot be annotated with certainty solely by examining their neighbors in the graph and (ii) the integration of diverse types of experimental evidence about functional similarity with the propagation procedures.
The approach described in this article suggests possible avenues for research. In many cases, a confidence level can be associated with a given link. In such cases, statistically robust schemes are needed for incorporating this measure of certainty into the network. Our current networks support one functional assignment at a time. It is relatively easy to generalize this approach to a more general constraint network in which nodes can receive multiple functional labels. The more general framework will provide a general language for expressing probabilistic dependencies, integrity constraints, and inconsistencies that we would like to impose on the process of assigning functional labels to a node and its neighbors. Our current implementation is guaranteed only to attain a local minimum, although it usually converges almost instantly to such a solution. This problem can be addressed by developing a stochastic variant of the algorithm. However, these variants do not allow us to use propagation diagrams to illustrate the transfer of functional assignments and help us obtain an intuitive interpretation of the result.
An important aspect of functional annotation is the assessment of the statistical significance of putative functional assignments. Currently, the precision and/or recall values obtained from cross-validation provide an indirect measure of confidence in our predictions. These values are implicitly Bonferroni-corrected for the testing of multiple hypotheses. In the future, we plan to compute a P value for each putative functional annotation by using the following procedure. For each function f, we create multiple randomized instances of the Hopfield network by shuffling the initial states (+1 or -1) of the annotated proteins and apply our technique to each such network. The P value for assigning f to a hypothetical protein h is calculated as the fraction of networks in which h is annotated with f. This confidence is then normalized by using a Bonferroni correction. Because this procedure is computationally intensive, we have chosen to rely on the cross-validation results as an estimate of the statistical significance of our predictions.
In this article, we have focused on the integration and propagation of experimental data. In the context of annotation of microbial genomes, additional evidence for functional linkage may be obtained by using computational methods, such as gene fusion, chromosomal proximity or phylogenetic profiles (29–33). Naturally, information-extraction methods, such as the cooccurrence of protein names in scientific abstracts (34, 35) may be applicable also as a source of evidence to be integrated by using our methodology.
Note. We have learned recently that Vazquez et al. (36) have independently developed an approach similar to ours. However, by integrating various sources of data such as protein–protein interaction and gene-expression correlation, our analysis improves the coverage and the accuracy of functional annotation. Furthermore, the optimization scheme presented in this article is computationally efficient and facilitates the visualization of the flow of evidence in the network by using propagation diagrams, as illustrated in Fig. 2.
Supplementary Material
Acknowledgments
We thank John Rachlin, Naren Ramakrishnan, and one reviewer for numerous suggestions on the manuscript; and Charles DeLisi, Itai Yanai, and Joe Mellor for prior discussion related to predictome. This work was supported in part by National Science Foundation Grants 0239435 and 9980088.
Abbreviations: PPI, protein–protein interaction; GO, Gene Ontology; ER, endoplasmic reticulum.
References
- 1.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gardner, M. J., Shallom, S. J., Carlton, J. M., Salzberg, S. L., Nene, V., Shoaibi, A., Ciecko, A., Lynn, J., Rizzo, M., Weaver, B., et al. (2002) Nature 419, 531-534. [DOI] [PubMed] [Google Scholar]
- 3.Yanai, I., Mellor, J. C. & DeLisi, C. (2002) Trends Genet. 18, 176-179. [DOI] [PubMed] [Google Scholar]
- 4.Marcotte, E. M., Pellegrini, M., Thompson, M. J., Yeates, T. O. & Eisenberg, D. (1999) Nature 402, 83-86. [DOI] [PubMed] [Google Scholar]
- 5.Fields, S. & Song, O. (1989) Nature 340, 245-246. [DOI] [PubMed] [Google Scholar]
- 6.Troyanskaya, O. G., Dolinski, K., Owen, A. B., Altman, R. B. & Botstein, D. (2003) Proc. Natl. Acad. Sci. USA 100, 8348-8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pavlovic, V., Garg, A. & Kasif, S. (2002) Bioinformatics 18, 19-27. [DOI] [PubMed] [Google Scholar]
- 8.Zhou, X., Kao, M. C. & Wong, W. H. (2002) Proc. Natl. Acad. Sci. USA 99, 12783-12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bader, G. D., Betel, D. & Hogue, C. W. (2003) Nucleic Acids Res. 31, 248-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mellor, J. C., Yanai, I., Clodfelter, K. H., Mintseris, J. & DeLisi, C. (2002) Nucleic Acids Res. 30, 306-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Mering, C., Huynen, M., Jaeggi, D., Schmidt, S., Bork, P. & Snel, B. (2003) Nucleic Acids Res. 31, 258-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito, T., Chiba, T., Ozawa, R., Yoshida, M., Hattori, M. & Sakaki, Y. (2001) Proc. Natl. Acad. Sci. USA 98, 4569-4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uetz, P., Giot, L., Cagney, G., Mansfield, T. A., Judson, R. S., Knight, J. R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., et al. (2000) Nature 403, 623-627. [DOI] [PubMed] [Google Scholar]
- 14.Ho, Y., Gruhler, A., Heilbut, A., Bader, G. D., Moore, L., Adams, S. L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., et al. (2002) Nature 415, 180-183. [DOI] [PubMed] [Google Scholar]
- 15.Gavin, A. C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J. M., Michon, A. M., Cruciat, C. M., et al. (2002) Nature 415, 141-147. [DOI] [PubMed] [Google Scholar]
- 16.von Mering, C., Krause, R., Snel, B., Cornell, M., Oliver, S. G., Fields, S. & Bork, P. (2002) Nature 417, 399-403. [DOI] [PubMed] [Google Scholar]
- 17.Tong, A. H., Drees, B., Nardelli, G., Bader, G. D., Brannetti, B., Castagnoli, L., Evangelista, M., Ferracuti, S., Nelson, B., Paoluzi, S., et al. (2002) Science 295, 321-324. [DOI] [PubMed] [Google Scholar]
- 18.Schwikowski, B., Uetz, P. & Fields, S. (2000) Nat. Biotechnol. 18, 1257-1261. [DOI] [PubMed] [Google Scholar]
- 19.Hishigaki, H., Nakai, K., Ono, T., Tanigami, A. & Takagi, T. (2001) Yeast 18, 523-531. [DOI] [PubMed] [Google Scholar]
- 20.Hopfield, J. J. & Tank, D. W. (1986) Science 233, 625-633. [DOI] [PubMed] [Google Scholar]
- 21.Kasif, S., Banerjee, S., Delcher, A. & Sullivan, G. (1993) Ann. Math. Artif. Intell. 9, 327-344. [Google Scholar]
- 22.Hopfield, J. J. (1982) Proc. Natl. Acad. Sci. USA 79, 2554-2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes, T. R., Marton, M. J., Jones, A. R., Roberts, C. J., Stoughton, R., Armour, C. D., Bennett, H. A., Coffey, E., Dai, H., He, Y. D., et al. (2000) Cell 102, 109-126. [DOI] [PubMed] [Google Scholar]
- 24.Salton, G. & McGill, M. J. (1983) Introduction to Modern Information Retrieval (McGraw–Hill, New York).
- 25.Wisniewski, J. R., Krohn, N. M., Heyduk, E., Grasser, K. D. & Heyduk, T. (1999) Biochim. Biophys. Acta 1447, 25-34. [DOI] [PubMed] [Google Scholar]
- 26.Letovsky, S. & Kasif, S. (2003) Bioinformatics 19, Suppl. 1, I197-I204. [DOI] [PubMed] [Google Scholar]
- 27.Izumiya, H. & Yamamoto, M. (1995) J. Biol. Chem. 270, 27859-27864. [DOI] [PubMed] [Google Scholar]
- 28.Kirchhausen, T. (2000) Nat. Rev. Mol. Cell Biol. 1, 187-198. [DOI] [PubMed] [Google Scholar]
- 29.Zheng, Y., Szustakowski, J. D., Fortnow, L., Roberts, R. J. & Kasif, S. (2002) Genome Res. 12, 1221-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu, J., Kasif, S. & DeLisi, C. (2003) Bioinformatics 19, 1524-1530. [DOI] [PubMed] [Google Scholar]
- 31.Zheng, Y., Roberts, R. J. & Kasif, S. (2002) Genome Biol. 3, RESEARCH0060. [DOI] [PMC free article] [PubMed]
- 32.Yanai, I., Derti, A. & DeLisi, C. (2001) Proc. Natl. Acad. Sci. USA 98, 7940-7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pellegrini, M., Marcotte, E. M., Thompson, M. J., Eisenberg, D. & Yeates, T. O. (1999) Proc. Natl. Acad. Sci. USA 96, 4285-4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcotte, E. M., Xenarios, I. & Eisenberg, D. (2001) Bioinformatics 17, 359-363. [DOI] [PubMed] [Google Scholar]
- 35.Raychaudhuri, S., Schutze, H. & Altman, R. B. (2002) Genome Res. 12, 1582-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vazquez, A., Flammini, A., Maritan, A. & Vespigniani, A. (2003) Nat. Biotechnol. 21, 697-700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.