

Abstract
A century after Cajal identified a “third element” of the nervous system, many issues have been clarified about the identity and function of one of its major components, the microglia. Here, we review recent findings by microgliologists, highlighting results from imaging studies that are helping provide new views of microglial behavior and function. In vivo imaging in the intact adult rodent CNS has revolutionized our understanding of microglial behaviors in situ and has raised speculation about their function in the uninjured adult brain. Imaging studies in ex vivo mammalian tissue preparations and in intact model organisms including zebrafish are providing insights into microglial behaviors during brain development. These data suggest that microglia play important developmental roles in synapse remodeling, developmental apoptosis, phagocytic clearance, and angiogenesis. Because microglia also contribute to pathology, including neurodevelopmental and neurobehavioral disorders, ischemic injury, and neuropathic pain, promising new results raise the possibility of leveraging microglia for therapeutic roles. Finally, exciting recent work is addressing unanswered questions regarding the nature of microglial-neuronal communication. While it is now apparent that microglia play diverse roles in neural development, behavior, and pathology, future research using neuroimaging techniques will be essential to more fully exploit these intriguing cellular targets for effective therapeutic intervention applied to a variety of conditions.
Keywords: microglia, imaging, motility, migration, development
A Brief History of Microglia
As early as 1913, Spanish neuroanatomist Ramon y Cajal identified a “third element” (non-astrocyte glial cells) of the central nervous system in distinction from the “first element” (neurons) and “second element” (astrocytes). Cells of the “third element” were initially considered mesodermally derived glia or “mesoglia”. Cajal’s student, Pio del Rio Hortega, went on to define microglia and oligodendrocytes as separate components of the “third element” and provided the first comprehensive description of microglia (del Río Hortega, 1919, 1920, 1932). Remarkably, many of Hortega’s descriptions of microglia (which were sometimes referred to as “Hortega cells”) have been validated over the ensuing eight decades since, including the morphological transformation of these cells during injury and disease, a process termed “microglial activation” (Kettenmann et al., 2011).
Interest in microglia has increased considerably in recent years, perhaps in recognition of their potential as cellular targets for therapy during pathology, as discussed below. A PubMed search shows that, of the more than 15,000 research articles on microglia, only 456 were published before 1990. Moreover, thus far in the three years of the current decade, more articles have been published on microglia than all those published in the 1990s combined. While published articles on astrocytes and oligodendrocytes witnessed a 35% and 49% increase, respectively, in the 2000s compared with the 1990s, articles on microglia increased 143% within the same period of time. Moreover, for the first time in history, the Society for Neuroscience had a minisymposium on microglia at its annual meeting in 2011, and the term “microglia” was the sixth most searched term on the meeting’s website (Tremblay et al., 2011; Hughes, 2012).
As alluded to above, microglia have a unique place in the CNS, being derived from the mesoderm as opposed to the neuroectoderm from which other CNS cells are derived (Ransohoff and Cardona, 2010). Since del Rio Hortega’s early descriptions, microglial research was dominated for many decades by intense debate over their origin and nature. There is now general consensus that microglia are derived from two sources: first, from an early embryonic source in the yolk sac; and, secondly, from myeloid precursors that subsequently take up residence in the CNS during embryonic development, forming a stable self-renewing population through adulthood (Ginhoux et al., 2010; Greter and Merad, 2013).
During development, microglial cells in the brain and spinal cord become distributed throughout the gray and white matter, where they develop a highly branched form with intimate relationships with neurons and other glial cells (Fig. 1). Historically, descriptions of microglia centered on the remarkable morphological transformations they undergo in response to tissue injury or disease. Based largely on static microscopic images of fixed tissues, microglia were ‘observed’ to transform or “activate” in response to virtually all CNS pathological conditions, however minor the perturbation to homeostasis (Kreutzberg, 1996). With the focus on microglial morphological responses to pathology, the concept of microglia as a “resting” or “quiescent” cell in the healthy CNS was developed in contradistinction to an “activated” cell in the pathological CNS. Since microglia were thought to be quiescent (i.e., inactive) in the uninjured state, microglial studies were largely focused on the injured and diseased CNS to determine microglial behavior and functions, making microglial behavior during normal physiology a relatively neglected research endeavor. Thus, it is only recently that novel behaviors and functions of microglia in the uninjured brain are beginning to be unraveled, overturning the idea that microglia are “quiescent” under normal conditions in the adult CNS.
Figure 1. Morphology of ‘resting/surveillant’ microglia in developing mouse neocortex.

Confocal image shows GFP-expressing microglia (green) and YFP-expressing pyramidal neurons (yellow) in cortical tissue from an early postnatal double-transgenic reporter mouse (CX3CR1GFP/+:Thy1-YFP). Note the fine microglia branches intercalated among the dendritic arbors of the pyramidal neurons.
Microglia as Surveying Cells in the CNS
Microglial Surveillance in the Adult CNS
The first time-lapse imaging studies of microglia in living mammalian brain tissues (ex vivo preparations) were performed in the 1990s. These imaging studies directly demonstrated that microglia are morphologically dynamic and capable of active phagocytosis (Smith et al., 1990; Brockhaus et al., 1996; Haas et al., 1996; Dailey and Waite, 1999). This led to the suggestion that microglia are equipped with receptors to detect pathological aberrations in situ (Kreutzberg, 1996; Brockhaus et al., 1996). Even though some of these early studies utilized widefield or confocal imaging to view cells deep within ex vivo tissue preparations, the motile activity of microglia was largely presumed to be a by-product of inevitable tissue injury that occurred during excision of brain tissues. It was commonly believed that “resting” microglia in uninjured brain tissues were structurally virtually inactive. This notion was overturned in 2005 with the publication of the first in vivo imaging studies in the uninjured brain (Davalos et al., 2005; Nimmerjahn et al., 2005). These two seminal studies, along with more recent in vivo imaging in the mouse cortex (Kim and Dustin, 2006; Wake et al., 2009) and spinal cord (Davalos et al., 2008), showed that microglia in the uninjured adult CNS are far from being morphologically static.
These in vivo imaging studies showed that microglial movements in the normal adult brain consisted of a stationary cell body (soma) with extensively motile branches (projections). Only 5% of microglial somata travelled 1–2 μm/hr while microglial processes were remodeled continuously at a velocity of about 88 μm/hr (Nimmerjahn et al., 2005). The concept of the microglia as surveying cells of the normal adult CNS was then introduced and microgliologists recommended replacing the misnomer of “resting” microglia with the idea of microglia as “surveying” cells of the CNS (Raivich, 2005; Hanisch and Kettenmann, 2007).
The observed differences between the behavior of microglial somata and processes is important for proper descriptions of microglial morphological activity. For consistency, we recommend that a distinction be made in the microglia literature between microglial process remodeling and microglial soma movement. We suggest that microglial process dynamism be referred to as “microglial motility,” while microglial soma movement or translocation be referred to as “microglial migration;” together microglial motility and migration can be referred to as “microglial mobility.” In this light, it may be said that microglia in the uninjured adult CNS are motile, non-migratory cells functioning to persistently survey the CNS to detect aberrations from homeostasis and respond accordingly.
Microglial Surveillance in the Developing CNS
As noted above, in vivo imaging of microglia in the uninjured adult mouse CNS revealed that microglia are stationary (non-migratory), albeit motile cells. However, whether microglia in the developing rodent CNS are also stationary, motile cells has yet to be determined because there are no published studies in the developing mammalian CNS in vivo.
Thus far, microglial imaging in the developing mammalian CNS have only been performed in excised neonatal or embryonic tissues rather than in the intact CNS. Here, unlike the behavior in adults where only about 5% of microglial somata migrated at 1–2 μm/hr, microglia in the developing brain have been shown to be highly migratory. Amoeboid microglia in acutely excised slices of corpus callosum from postnatal day (P) 5–9 mice were observed to migrate at rates up to 30 μm/hr (Brockhaus et al., 1996). Subsequently, microglia at P4 in the rat hippocampus were reported to migrate at up to 118 μm/hr (Stence et al., 2001). Furthermore, embryonic cortical mouse microglia were reported to migrate at up to 31 μm/hr on embryonic day (E)14.5 (Swinnen et al., 2013). These results suggest that microglia in developing rodent brain are not stationary but migratory. However, because microglia are exquisitely sensitive to injury, it is possible that tissue injury, induced during excision, may account in large measure for this migration impetus. Nevertheless, several observations suggest that this may not be the case.
First, even after local injury in the adult cortex in vivo, microglial somata remain stationary for up to 5 hours, suggesting that acute tissue injury may not be sufficient to induce microglial migration for several hours in adults (Nimmerjahn et al., 2005) despite the fact that within two hours of tissue excision, many microglia in neonatal tissue slices show some degree of migration (Stence et al., 2001; Kurpius et al., 2007).
Secondly, as with in vivo observations, microglia in cortical brain slices from previously uninjured adult mice were non-migratory (Carbonell et al., 2005). However, when a forebrain stab lesion was performed unilaterally in vivo, migratory microglia were found only in the injured hemisphere beginning 1 day and peaking 3 days after the lesion. These observations suggest that brain tissue excision itself may not be sufficient to induce microglial migration, at least in the short term.
Finally, although there are currently no published studies on microglial mobility in the developing rodent brain in vivo, recent imaging in the intact zebrafish brain suggest that microglia cell bodies are not stationary. During two hours of imaging of fluorescently labeled microglia in transgenic zebrafish at 3–4 days post-fertilization (dpf), Sieger et al. (2012) showed that even without neuronal injury, microglia are migratory. Additionally, Svahn et al. (2013) quantified microglial migration 3–10 dpf in zebrafish and reported that migration was highest at about 4 dpf, when microglia migrate at an average of ~20 μm/hr, and dropped significantly a day later when they migrated on average at ~8 μm/hr. A similar developmental decrease in migration was reported in embryonic mouse cortical slices between E14.5 and E17.5 (Swinnen et al., 2013). These results suggest that the rate and frequency of microglial migration may decrease with increasing developmental age. We may thus think of microglia as sentinels of the CNS: patrolling the nervous tissue as mobile phagocytes during early development and surveying it chiefly as stationary cells with motile branches in adulthood.
Whether developmental differences in microglial migration are cell autonomous or dependent on the tissue milieu is presently unknown. Microglial migration necessarily depends on interactions with neighboring cells and the extracellular matrix (ECM), and expression and activation of cell surface integrins in cultured microglia can be altered by changes in ECM components and cytokines (Milner and Campbell, 2002, 2003). However, little is known about how microglial interactions with the ECM may change during development.
Surveying microglia in the adult appear to have their cell body anchored in place, implying a strong interaction with the ECM. The transition from a stationary to a migratory amoeboid phenotype likely involves cell detachment from the ECM. Recent work is providing insights into microglial degradation of ECM, which may facilitate migration. Here, cultured rat microglia reminiscent of activated microglia during development were shown to possess podosomes, structures similar to those expressed on invading cancer cells, by which they could degrade components of the ECM (Vincent et al., 2012; Siddiqui et al., 2012). Although this has not been tested in vivo, an intriguing possibility is that during development microglia possess such structures to facilitate their migration through brain tissues, and that this machinery is down-regulated during maturation, resulting in the anchoring of microglial cell bodies in adults. Mobilization of microglia after tissue injury may necessitate upregulation of ECM degradation machinery. Future work would be necessary to determine whether there are indeed developmental differences in microglial migration in vivo and, if so, whether this may relate to differences in the composition of brain ECM or interaction of microglia with the ECM.
Microglial Functions in the Developing Brain
Although imaging studies of microglia in living tissues have been largely performed in the adult brain, awareness of neuropathological conditions during human development has led to increased exploration of the developing brain. During neural development, microglia increase in density and undergo a progressive ramification, or increase in number and complexity of branches. In the rodent brain, microglia start in the embryonic stage as roundish, amoeboid cells with a few short projections; progress during the perinatal period into more complex cells with a few branchy extensions (so-called “primitive ramified microglia”); and in the late postnatal period they mature into highly ramified cells (Fig. 2; Dalmau et al., 1997; Dalmau et al., 1998). There is considerable heterogeneity in the morphological appearance of microglia in developing tissues, and this seems to vary with local levels of developmental apoptosis. The later stages of morphological maturation correlate with other developmental changes in CNS tissues, including a decline in developmental apoptosis, formation of neuronal arbors, and synaptogenesis.
Figure 2. Microglial Ramification from Development to Adulthood.

A, Microglia from a neonatal mouse hippocampus showing a central soma and several projections. B, Schematic representation of the progressive ramification of microglia through development into adulthood. Brain-resident microglia are first observed during embryonic development as amoeboid cells with few projections. Beginning prenatally and extending into the early postnatal period, microglia begin to transform into cells with more defined primary projections with little secondary branching. By the second postnatal week onwards, microglial morphology consists of more elaborate primary, secondary and even tertiary branching in their projections.
Recent research is beginning to elucidate previously unrecognized functions of microglia during development. We will briefly consider studies over the past decade indicating that microglia function, by analogy, as: (i) “plumbers” in angiogenesis; (ii) “gardeners” in the induction of neuronal apoptosis; (iii) “morticians” in phagocytic clearance of dead cells; and (iv) “electricians” in synapse remodeling.
Microglia as “Plumbers”: Roles in Formation and Maintenance of Blood Vessels
It has long been evident that developing microglia localize in close proximity to blood vessels (Ashwell et al., 1989; Thomas, 1999). It was initially assumed that this may simply reflect entry of microglia into the brain through these vessels. However, some microglia are present in the brain parenchyma prior to its vascularization, suggesting that at least some level of microglial dispersion in the brain occurs independent of the cerebral vasculature (Ginhoux et al., 2010). Recent data, discussed below, now provide evidence that microglia facilitate angiogenic sprouting during development.
Targeted deletion of PU.1, a transcription factor required for the development of certain hematopoietic cell lineages, results in mice lacking brain-resident microglia (Scott et al., 1994; McKercher et al., 1996; Beers et al., 2006). In PU.1-deficient mice, where even yolk sac-derived microglia are absent, vascular branching and complexity is significantly reduced. Similar results are observed in mice deficient in colony stimulating factor (CSF1), which is also important for microglial development (Kubota et al., 2009). Vascularization defects were reported in both the mouse hindbrain and retina. Moreover, time-lapse imaging in the developing zebrafish brain provided direct visual evidence for microglia promoting the fusion of nearby blood vessels (Fantin et al., 2010).
A subsequent study confirmed the spatio-temporal association of microglia with developing vessels during the first week of postnatal development in the mouse retina (Rymo et al., 2011). These researchers used the CSF1-deficient mice, which lack microglia in the retina during development and show a corresponding lower level of vessel branching complexity. The sufficiency of microglia to promote angiogenesis in vitro was tested using an aortic ring culture system. Aortic rings cultured without microglia, or with embryonic fibroblasts, failed to elaborate complex branching vessels. However, in the presence of microglia, complex vessels were formed, indicating that microglia can induce sprouting. Furthermore, conditioned media from microglia was able to induce sprouting, suggesting that diffusible signals from microglia contribute to angiogenesis. The specific factor(s) that mediate angiogenesis remain to be identified. Together, these studies provide evidence that microglia, like plumbers that assemble the pipes, are important in regulating developmental angiogenesis.
Interestingly, microglia also respond to damaged or leaky pipes. Using transcranial imaging in adult mice, Nimmerjahn et al. (2005) showed that laser-induced focal blood vessel damage rapidly activates nearby microglia, which extend processes to surround injury sites within minutes. This is thought to be a protective mechanism, shielding the parenchymal tissue from vessel leakage. Finally, time-lapse imaging in developing rat hippocampal slices showed that migrating microglia can contact and track along branching capillaries, even when they lack blood flow (Grossmann et al., 2002). Together, these studies indicate intimate relationships between microglia and blood vessels that may contribute to the construction and maintenance of the vascular system in the CNS.
Microglia as “Gardeners”: Induction of Developmental Neuronal Apoptosis
During development, more neurons are generated than will survive to adulthood, and many die as a result of a developmental pruning program known as programmed cell death (PCD) or developmental apoptosis (Burek and Oppenheim, 1996). Microglia are now thought to play active roles in the induction of neuronal cell death. The first suggestion of this was based on the observation that microglia often colocalize with dying neurons (Ashwell et al., 1989; Dalmau et al., 1998; Marin-Teva et al., 2004; Peri and Nusslein-Volhard, 2008; Svahn et al., 2013). This evidence, in and of itself, could not distinguish between an active, causal role for microglia from a merely responsive role during PCD. However, more recent data suggest a direct, causal role for microglia.
During embryonic development, the vertebrate retina originates as a projection of the developing brain, making the retina an extension of the CNS. When the developing chick retina was excised before microglial entry and colonization, normal developmental cell death was substantially reduced (Frade and Barde, 1998). Nerve growth factor (NGF) was also absent from the developing chick retina when microglia were absent. When cultured microglia were added to the developing eye cups, NGF was detected and neuronal cell death was restored. These results suggest that microglial NGF release provides instructive cues for programmed neuronal cell death in developing tissues.
Similar results were obtained in the developing mouse cerebellum. Here, a significant number of Purkinje cells undergo PCD during the first postnatal week. However, when endogenous microglia were depleted by clodronate-containing liposomes in freshly prepared cerebellar slices, the number of surviving Purkinje cells after three days in culture was dramatically increased (Marin-Teva et al., 2004). In this case, NGF was not the apoptosis-inducing factor; rather superoxide production by microglia was found to be responsible. One potential caveat with these experimental approaches is that microglia in excised tissue slices and cell culture are at least partially activated, and the contributions of microglia to developmental cell death may differ depending on the activation state of microglia.
As in the cerebellum, developmental apoptosis occurs in the rodent hippocampus during the first postnatal week (Marin-Teva et al., 2004; Knuesel et al., 2005; Murase et al., 2011). A peak in neuronal cell death occurs around birth in the developing mouse subicular complex adjacent to the hippocampus (Knuesel et al., 2005; Wakselman et al., 2008). During this period, microglia are often found in close proximity to cleaved caspase-3 positive neurons. Interestingly, loss of CD11b and DAP12, which in the brain are exclusively expressed by microglia, resulted in a significant reduction in cleaved caspase-3 positive neuronal staining, suggesting a reduction in PCD under these conditions. As in the cerebellum, neuronal apoptosis in the hippocampus was driven by microglial superoxide production (Wakselman et al, 2008). These studies in the cerebellum and hippocampus provide evidence that microglial superoxide production controls PCD, and suggests a potential role for NADPH oxidase (NOX). Although NOX does not seem to play a critical role during perinatal stroke (Doverhag et al., 2008), its role in microglia-mediated developmental apoptosis has not been definitively tested.
In addition, recent evidence in the spinal cord shows a correlation between microglial arrival during embryonic development and motor neuron cell death (Rigato et al., 2011). Similarly, microglia accumulate in regions of developmental cell death in the embryonic cortex (Swinnen et al., 2013). However, whether microglial arrival in these CNS regions causally instructs neuronal cell death has not yet been determined.
Together, these studies argue for novel roles by microglia in regulating neural development by controlling neuronal numbers during early postnatal (and possibly embryonic) development. Since this has been observed in several regions of the CNS including the retina (Frade and Barde, 1998), cerebellum (Marin-Teva et al., 2004), hippocampus (Wakselman et al., 2008), and possibly the embyonic cortex and spinal cord (Rigato et al., 2011; Swinnen et al., 2013; Rigato et al., 2012), it suggests that microglial regulation of PCD may be widespread in the developing CNS. Thus, the ability of microglia to induce the demise of exuberant neurons, as well as their ability to eliminate inappropriate neuronal connections during early development, has earned microglia the title of developmental “gardeners” that prune cellular elements to enhance neurological function (Hughes, 2012).
Microglia as “Morticians”: Phagocytic Clearance of Dead Cells
Perhaps the most widely recognized function of microglial cells is their role in phagocytic clearance of cellular debris (for reviews, see Napoli and Neumann, 2009; Neumann et al., 2009). This role has long been recognized during both disease pathology (Penfield, 1925) and normal development (Ferrer et al., 1990). When cells die, they may release cytotoxic substances into the extracellular milieu, thereby injuring otherwise healthy cells nearby. Rapid and efficient phagocytosis, then, is thought to be essential for proper nervous system development as well as tissue restoration following injury.
Microglia have been recognized as CNS phagocytes in many species, including leech (Masuda-Nakagawa et al., 1990; Ngu et al., 2007), goldfish (Battisti et al., 1995), chick (Caldero et al., 2009), rat (Petersen and Dailey, 2004), mouse (Sierra et al., 2010), and human (Penfield, 1925). In mammalian brain tissues, ex vivo tissue preparations have been especially useful for studying the dynamic process of microglial engagement and clearance of dead cells (See Fig. 3 and Movie 1; Brockhaus et al., 1996; Petersen and Dailey, 2004). In recent years, some researchers have turned to the zebrafish to study microglial behavior. This is a highly accessible in vivo model system, given its optical transparency and well characterized developmental program. Two studies in zebrafish have demonstrated the efficiency of microglia phagocytosis during vertebrate brain development (Peri and Nusslein-Volhard, 2008; Svahn et al, 2013). In these studies, apoptotic cell debris was never found outside of microglia. Additionally, when foreign bacteria (E. coli) were introduced to the zebrafish brain, microglia rapidly engulfed them soon after injection (Peri and Nusslein-Volhard, 2008). Thus, zebrafish microglia are very effective phagocytes in the developing brain, and this model system holds great promise for combining optical and genetic approaches to elucidate microglial function in a vertebrate system in vivo.
Figure 3. Microglial motility facilitates rapid mobilization to injured neurons.
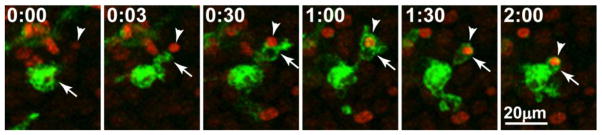
In this case, injury was experimentally induced in a mouse hippocampal tissue slice by focal laser burn. Within minutes after the injury, an activated amoeboid microglial cell (arrow) extends a branch toward a Sytox-labeled nucleus of an injured neuron (arrowhead), contacts it, and engulfs it. Time is shown in hr:min.
Similar to the developing zebrafish brain, microglia in the young adult mouse hippocampus rapidly phagocytize newborn apoptotic neurons (Sierra et al., 2010). Engulfment of an apoptotic cell was estimated to take 60–90 minutes indicating a high phagocytic efficiency. A previous confocal time-lapse study in excised neonatal rat brain tissue slices demonstrated that microglia engulf newly dying (likely necrotic) cells on average within ~33 minutes (Peterson and Dailey, 2004). Likewise, even during peak periods of developmental apoptosis, microglia efficiently phagocytize nearby apoptotic bodies in the neonatal mouse hippocampus in vivo (See Fig. 4 and Movie 2). Together, these results show that, like morticians who undertake to remove dead corpses, microglia are effective at clearing both naturally occurring apoptotic cell debris and foreign pathogens.
Figure 4. Microglial engagement of early and late stage apoptotic cells in the developing mouse hippocampus.
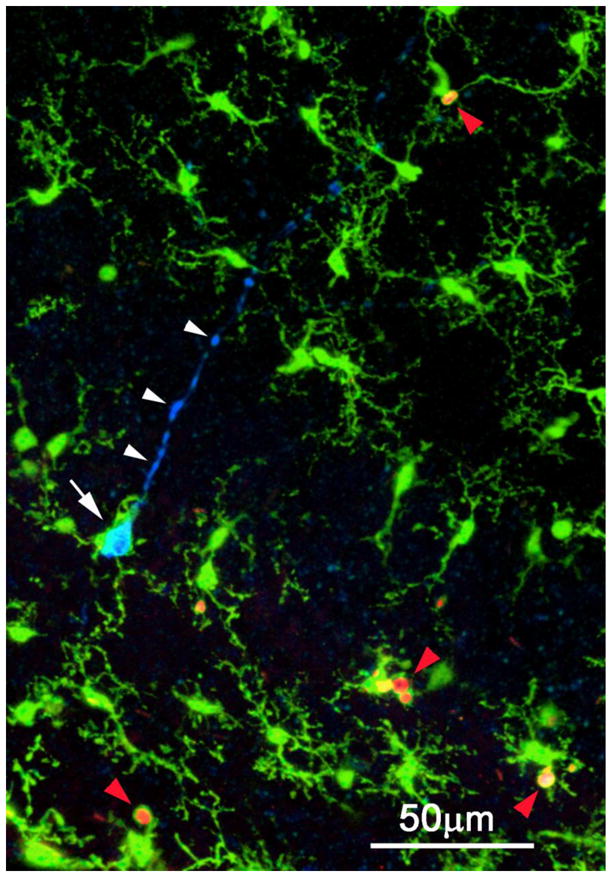
Three-channel confocal image showing primitive ramified microglia (green), cleaved caspase-3 immunostained early apoptotic pyramidal neuron (blue), and PSVue-stained late stage apoptotic bodies (red). A microglial cell (white arrow) enwraps the soma of an apoptotic pyramidal neuron labeled by CC3 antibodies. Note labeling of the primary apical dendrite (white arrowheads), which has a blebby, degenerating appearance. Other microglia nearby are in the process of phagocytosing late stage apoptotic bodies labeled with PSVue+ (red arrowheads). The cells are dying by natural, developmental apoptosis.
Microglia as “Electricians”: Evidence for Synapse Remodeling
There is growing evidence that microglia monitor the functional state of synapses and respond to changes in synaptic activity during development. Three studies in the past few years have been especially instrumental in promoting this concept. First, using two-photon in vivo imaging and electron microscopy in the visual cortex of 3–5 week old mice, Tremblay et al. (2010) reported differences in microglial behavior during visual deprivation. During normal visual experience, microglial processes preferentially associated with a subset of dendritic spines that were small and transiently growing, and these were frequently eliminated. Conversely, when mice were deprived of sensory input, microglia preferentially associated with a subset of large dendritic spines that were shrinking. This phenotype could be rescued by a return to sensory visual exposure. This study suggests that microglia are important for synapse remodeling. However, whether microglia are necessary for such remodeling will have to be determined by microglial depletion studies.
Another study performed in second and third postnatal week mice provided evidence that cytokine signaling regulates microglial elimination of neuronal synapses (Paolicelli et al., 2011). Dendritic spine density was significantly increased in hippocampal neurons in mice lacking the fractalkine receptor (CX3CR1), which in the brain is expressed almost exclusively in microglia. Moreover, neurons from mice lacking the fractalkine receptor displayed physiological properties of synaptic strength, plasticity, and excitability that are characteristic of immature circuitry. These results suggest that microglia may modulate the maturation of neuronal structure and physiology via fractalkine receptor signaling.
Finally, Schafer et al. (2012) provided evidence for microglial engulfment of presynaptic material in an activity dependent manner during the first postnatal week in the developing mouse retinogeniculate system. Greater engulfment of synaptic material was observed when neuronal activity was inhibited. Additionally, complement signaling was identified as a putative signaling pathway by which microglia identify and eliminate synaptic elements (Schafer et al., 2012). Linnartz et al. (2012) suggested that this complement-dependent synapse elimination is regulated by sialic acid, which forms the cap of the neuronal glycocalyx. The presence of sialic acid prevented complement binding and thus microglial clearance of synapses. However, when neurons were de-sialylated, microglial-dependent clearance was enhanced, suggesting that neuronal cell surface glycosylation may function upstream of complement tagging and subsequent elimination by microglia. Nevertheless, since the sialic acid study was performed in dissociated cell cultures, the significance of this mechanism during in vivo development remains to be determined. Moreover, for all these studies, the extent to which engulfment of presynaptic material arises from pruning at the single synapse level, or removal of whole dying cells, remains unclear without direct visualization of the processes in situ.
In addition to these results during normal physiology, microglial interactions with neurons can be altered in experimental pathology. For example, during ischemia, microglial processes reportedly make much more prolonged contacts with synapses, some of which are subsequently eliminated, suggesting that microglia monitor the functional state of synapses and potentially contribute to the elimination of non-functional ones (Wake et al., 2009).
Together, these results raise the intriguing possibility that microglia participate in the establishment or selection of appropriate neuronal connections during development and into adulthood. These observations have led to the suggestion that, while themselves not electrically excitable, microglia modulate the structure and function of electrically excitable neurons, thereby playing roles during development as “brain electricians”: appropriately wiring neuronal brain circuits (Graeber, 2010; Graeber and Streit, 2010). It is envisioned that microglia “listen in” on neuronal communication at synapses and respond to strengthen certain connections or eliminate others. On account of this involvement in regulating synaptic connectivity, it has been proposed that microglia are bone fide synaptic partners (Paolicelli and Gross, 2011), thereby extending the three-way (tri-partite) synapse model which included astrocytes (or presynaptic Schwann cells in the PNS) in pre- and post-synaptic communication (Araque et al., 1999) to a four-way (quad-partite) synapse model that includes microglia (Schafer et al., 2013). However, a greater mechanistic understanding of microglia function at the synapse will be needed to firmly establish this concept.
Microglia as Cellular Targets during Pathology: Disease, Injury, and Behavior
Recent interest in microglia has centered not only on understanding their basic biological functions but also on their therapeutic potential during pathological conditions. Recent evidence indicates the potential promise of microglia as targets for therapeutic intervention during pathology. Here we focus on studies showing the importance of microglia for (1) pathological grooming, as an example of microglia modulation of a behavior disorder; (2) Rett syndrome, as an example of a genetic neurodevelopmental brain disorder; (3) ischemic stroke, as an example of acute brain injury, and (4) neuropathic pain as an example of a chronic pathophysiological condition.
Pathological Grooming
For decades, microglia have been well known to play important roles during disease and injury. However, until recently, it was not evident that they are important for regulating normal behavior. It was first observed that Hoxb8 mutant mice displayed excessive grooming of both themselves and wild type littermates, leading to hair loss and sometimes skin lesions. This pathological phenotype has been suggested as a mouse model for studying obsessive compulsive behavior in humans. In early studies, the authors concluded that, although Hoxb8 is expressed in both the central and peripheral nervous system, the basis of the excessive grooming phenotype was more than likely aberrations in the central rather than peripheral nervous system (Greer and Capecchi, 2002).
More recently, a set of elegant experiments has confirmed that defects in the CNS are responsible for the anomalous grooming behavior and provided a direct link to microglia as the cellular culprits (Chen et al., 2010). First, cell lineage tracing experiments showed that both prenatal and postnatal Hoxb8 expressing cells are of hematopoietic origin and reside in the brain as microglia. This was confirmed both morphologically and molecularly (CD11b and Iba1 were used as microglial markers). However, only about 40% of microglia express Hoxb8. Additionally, total microglial numbers are diminished in Hoxb8 mutants by about 15%.
Next, transplantation of wild type bone marrow cells into irradiated Hoxb8 mutant mice rescued both hair loss and grooming behavior in 60% of recipient animals, with 40% becoming indistinguishable from wild type animals 5 months after treatment (Chen et al., 2010). Conversely, transfer of Hoxb8 deficient bone marrow cells into wild type mice resulted in abnormal behaviors similar to that observed in mutant mice in 20% of the mouse recipients. Moreover, conditional deletion of Hoxb8 in the hematopoietic system (but not the spinal cord) was able to reproduce the grooming phenotype, while transplantation rescue was still recapitulated with bone marrow from mice that do not produce B and T cells, suggesting that microglia may be the primary Hoxb8-expressing cells responsible for the behavior.
Rett Syndrome
Rett Syndrome is a debilitating X-linked neurological disorder that predominantly affects young girls after birth. Symptoms manifest as a progressive loss of neurological ability (Chahrour and Zoghbi, 2007). Since the gene involved in Rett syndrome, Mecp2, has also been shown to be aberrant in autism spectrum disorders, developing potential therapies against the syndrome could also provide opportunities for treatment of autism spectrum disorders (Derecki et al., 2010).
Glial cells have been suggested to induce neuronal abnormalities in Rett syndrome. Application of conditioned media from MECP2-deficient mixed glial cultures to wildtype neurons induced dendritic abnormalities. However, these abnormalities were absent when conditioned media from pure astrocyte cultures were used, suggesting involvement of non-astrocytic glial cells (Maezawa et al., 2009). Interestingly, conditioned media from dissociated microglial cultures deficient for MECP2 caused dendritic and synaptic abnormalities resulting from increased glutamate release by MECP2-deficient microglia (Maezawa and Jin, 2010). These in vitro observations were subsequently translated to an in vivo mouse model of Rett syndrome where bone marrow from normal donor animals was transplanted into MECP2-deficient animals after irradiation. Transplanted cells took up residence in the brain and displayed characteristics of resident microglia. Remarkably, successful transplantation attenuated disease symptoms, improving lifespan, breathing ability, and motor function. Moreover, microglial phagocytic activity was implicated in expression of the disease because inhibition of phagocytosis after successful microglial transplantation prevented a reduction of disease symptoms (Derecki et al., 2012). These results suggest that: (1) Rett syndrome is a disease of aberrant microglia and, more importantly, (2) therapeutic strategies involving microglial transplantation to diseased individuals hold remarkable promise.
Ischemic Stroke
Stroke is a leading cause of death and disability worldwide affecting especially the aged and the very young (Nelson and Lynch, 2004). The most common type of stroke is ischemic stroke, occurring in about 80% of stroke victims, and is usually caused by occlusion of a cerebral blood vessel (Guo et al., 2010; Weinstein et al., 2010). Although research on ischemia has traditionally focused on delineating pathophysiological mechanisms of neuronal death, and such studies yielded promising candidates for therapeutic approaches, thus far they have not yet translated into successful clinical trials. In this light, novel approaches involving the elucidation of pathophysiological mechanisms employed by glia during ischemia have been proposed to complement neuronal based studies (Nedergaard and Dirnagl, 2005; Barretto et al., 2011).
Although it is clear that ischemic injury activates microglia, there is intense debate regarding the role of microglia in stroke (Lai and Todd, 2006; Yenari et al., 2010). On the one hand, several studies have highlighted detrimental activities of microglia during stroke progression. For example, earlier studies reported that inhibition of microglial activation by minocycline improved stroke outcome (Yrjanheikki et al., 1998, 1999). It has been suggested that production of inflammatory factors by activated microglia is detrimental to the survival of other cells of the CNS during ischemia (Deng et al., 2011), perhaps due to production of reactive oxygen species (ROS). Indeed, evidence has been presented that microglial generation of ROS is regulated by the voltage-gated proton channel Hv1, and that Hv1-dependent ROS production is responsible for a substantial fraction of brain damage at early time points after ischemic stroke (Wu et al., 2012). On the other hand, other studies indicate that elimination of endogenous microglia during stroke worsened the ischemic injury (Lalancette-Hebert et al., 2007; Faustino et al., 2011). Thus, microglia may play different roles at different times (early versus late stages) following stroke. Nevertheless, considerable evidence implicates microglia as potential targets in ameliorating the effects of ischemia, which we now consider.
First, using ex vivo organotypic hippocampal slice cultures, Neumann et al. (2006) showed that introduction of naïve BV2 microglial cells provided significant neuroprotection when applied as late as 4 hours after the onset of simulated ischemia. These ex vivo results have also been confirmed by in vivo studies. For example, using a 2 hour transient occlusion model, injection of microglia within an hour after onset of experimental ischemia improved neuronal survival (Kitamura et al., 2004) and behavioral function (Kitamura et al., 2005).
Similarly, when microglia were injected into brains 4–7 days prior to experimental global ischemia, functional deficits in neurons were prevented (Hayashi et al., 2006). Moreover, an increased number of surviving neurons was observed following transient global ischemia in gerbils when microglia were introduced as late as 24 hours after ischemic onset (Imai et al., 2007). Furthermore, when human microglia were transplanted into rats subjected to transient focal ischemia as late as 48 hours after the insult, protection was still observed, including enhanced secretion of neurotrophic factors, reduction of apoptotic cells, and functional recovery in motor, sensory, reflex, and balance tests (Narantuya et al., 2010).
Currently, the most recognized treatment for stroke involves thrombolytic therapy, which is efficient if administered within a narrow window of 3 – 6 hours after the onset of ischemia (Wardlaw et al., 2009). The results from animal models summarized above suggest that microglia can be used for ischemic therapy even during delayed post-ischemic periods, potentially extending the window for effective treatment.
In summary, microglial functions have been shown to be important during neuropathological behaviors such as excessive grooming, neurological disease such as Rett syndrome, and acute brain injury such as ischemia. Moreover, as has been demonstrated for all three conditions, microglial transplantation therapies have been used to successfully ameliorate associated defects. The implications of these results are two-fold: (1) although the mechanistic details are not clear, microglia perform important physiological roles in the normally functioning CNS, and (2) microglial replacement or augmentation holds promise for improvement of these human-relevant conditions.
Neuropathic Pain
Pain serves an important function in organisms as a warning signal against potentially harmful stimuli. However, pain can become pathological, in which case the organism becomes especially sensitive to normally harmless stimuli, a phenomenon known as allodynia. While normal pain lasts only for the duration of the stimulus or shortly thereafter, pathological or neuropathic pain is persistent, being present even in the absence of an immediate stimulus (Inoue and Tsuda, 2012). As in the conditions discussed above, microglial dysfunction has been implicated in the maintenance of neuropathic pain. Here we briefly consider the evidence for microglial purinergic receptors in the progression of neuropathic pain.
Adenosine triphosphate (ATP) has long been recognized as an essential intracellular molecule for cellular energy. About 40 years ago, evidence for an extracellular function of ATP and its metabolites began to emerge (Burnstock, 1972). It is now well recognized that virtually all cells express ATP receptors, including G-protein coupled metabotropic (P2Y) receptors and ATP-gated ionotropic (P2X) receptors (Burnstock et al., 2011). Although ATP is normally maintained at high intracellular levels (1–5 mM), extracellular levels of ATP are very low (<1 μM). During certain pathological conditions, extracellular ATP levels may rise and activate additional purinergic receptors.
Microglia express several purinergic receptors (Inoue, 2008), three of which have been implicated in microglial dysfunction during neuropathic pain after peripheral nerve injury (Tsuda et al., 2013). In the CNS, P2Y12 receptors are uniquely expressed in microglia and have been shown to be critical for directed branch extension and chemotaxis of microglia toward sources of exogenous ATP/ADP and traumatically injured cells (Honda et al., 2001; Haynes et al., 2006). Microglial P2Y12 receptors were implicated in neuropathic pain when it was observed: (1) that they were selectively upregulated in microglia at both mRNA and protein levels in the ipsilateral spinal cord after nerve injury (Kobayashi et al., 2008; Tozaki-Saito et al., 2008); (2) that pharmacologic inhibition (Kobayashi et al., 2008; Tozaki-Saito et al., 2008), antisense knockdown (Kobayashi et al., 2008), and genetic depletion (Tozaki-Saito et al., 2008) of these receptors attenuated allodynia and other pathological pain behaviors in response to nerve injury; and (3) application of a P2Y12 agonist reconstituted pain behaviors (Kobayashi et al., 2008).
In addition to metabotrophic P2Y12 receptors, both P2X4 and P2X7 ionotropic receptors have been implicated in neuropathic pain. First, both P2X4 (Tsuda et al., 2003; Ulmann et al., 2008) and P2X7 (Kobayashi et al., 2011; He et al., 2012) receptor expression is upregulated in microglia after nerve injury, and pharmacological inhibition of P2X4 (Tsuda et al., 2003) and P2X7 (Honore et al., 2006; McGaraughty et al., 2007; Kobayashi et al., 2011) receptors have been shown to reduce pathological pain symptoms. Similar improvements were reported in response to application of antisense oligonucleotides for P2X4 receptors (Tsuda et al., 2003). Additionally, genetic ablation of either the P2X4 (Tsuda et al., 2009) or P2X7 (Chessell et al., 2005) receptors attenuated pathological pain responses. Moreover, injection of P2X4-activated microglia into naïve rats was sufficient to elicit neuropathic pain responses (Tsuda et al., 2003).
Given that microglia play a central role in neuropathic pain, the downstream processes by which microglia contribute to pain mechanisms in neurons have been of considerable interest (Zhuo et al., 2011). Coull et al. (2005) demonstrated that brain-derived neurotrophic factor (BDNF) produced by ATP-stimulated microglia reverses the anionic potential of GABA-activated currents to make GABA signaling excitatory rather than inhibitory during peripheral nerve neurotransmission. BDNF signaling through the Trk-B receptor was shown to be both necessary and sufficient for mediating the change in GABA-ergic transmission during neuropathic pain (Coull et al., 2005). As expected, BDNF release was markedly reduced in P2X4 null tissues (Ulmann et al., 2008), suggesting that at least P2X4 activation of microglia results in BDNF release, which subsequently increases neuronal hypersensitivity (Trang et al., 2011).
Together, these studies and others argue that microglial dysfunction is paramount in the initiation and propagation of at least some forms of neuropathic pain. Could microglia be viable therapeutic targets to treat chronic pains? It is interesting that microglia-depleted brains were recently shown to be rapidly repopulated by monocyte derived cells within two weeks (Varvel et al., 2012). Moreover, these cells developed similar functional properties to naturally resident microglia. Given that repopulation of the CNS with “naive” microglia can have favorable outcomes, coupled with the observation that blood derived macrophages have beneficial roles during spinal cord injury (Shechter et al., 2009), it is tempting to speculate that microglial replacement may be a viable therapeutic option for ameliorating certain forms of chronic pain.
Outstanding Questions on Microglial-Neuronal Communication
The studies considered above indicate that microglia play important roles during brain development and maintenance. However, many questions regarding the nature and extent of microglial-neuronal communication remain unanswered. It is now well established that neurons can modulate microglial activation states. Indeed, it has been suggested that since microglia express cognate receptors for neuron-specific ligands, including CD200L and CX3CL1, healthy neurons may keep microglia in an “off” or “silent” (low activation) state (Biber et al., 2007). On the other hand, injured neurons and glia may turn microglia “on” by increasing the release of purines, thereby activating both metabotropic (P2Y12) and ionotropic (P2X4 and P2X7) purinergic receptors in microglia. As noted above, these purinergic receptors have been implicated in the pathogenesis of neuropathic pain.
Imaging studies have clearly demonstrated that activation of purinergic receptors strongly affects microglia motility and migration. As discussed above, pharmacological studies have implicated both P2X4 and P2Y12 receptors in microglial chemotaxis (Honda et al., 2001; Oshawa et al., 2007; Wu et al., 2007), and previous studies using knockout mice have shown that P2Y12 receptors are required for directed microglial branch extension and migration during purine-based chemotaxis (Haynes et al., 2006). These mechanisms are likely relevant for microglial mobilization following traumatic injury and other conditions involving necrotic cell death. Despite these studies implicating P2X4 and P2Y12 receptors in purine-based chemotaxis, the specific receptor(s) controlling basal microglial motility are unknown. It seems likely that basal motility is driven by ongoing release of purines because apyrase, an enzyme that degrades ATP and ADP, rapidly and reversibly abolishes microglial motility in neonatal hippocampal slices (Kurpius et al., 2007) and in the neocortex in vivo (Davalos et al., 2005). There is evidence that activation of P2Y receptors is required for the basal motility of microglia (Wu et al., 2007). It has been argued that connexin- or pannexin-like hemi-channels may be involved in tonic release of purines that drive basal microglial motility because flufenamic acid (FFA), a pharmacological blocker of these channels, reversibly inhibits baseline microglial motility in vivo (Davalos et al., 2005) and in tissue slices (See Fig. 5 and Movie 3). FFA was also shown to inhibit ATP-induced microglial chemotaxis in vivo (Davalos et al., 2005) and in tissue slices (Wu et al., 2007). Because pannexins are expressed in hippocampal neurons (Vogt et al., 2005), astrocytes (Santiago et al., 2011), and microglia (Rigato et al., 2012), ATP may be released via pannexins from any of these cells by an ATP-induced ATP release mechanism (Davalos et al., 2005; Fontainhas et al., 2011; Dou et al., 2012). However, studies implicating pannexin and connexin channels must be interpreted cautiously because FFA may have multiple targets, and other non-selective cation channels may be involved. It appears that ATP-gated P2X7 receptors, which can form large pores that may facilitate ATP release, are not involved because there is no obvious deficiency in basal microglial motility or chemotaxis to injured cells in hippocampal tissue slices from P2X7–knockout mice (See Fig. 6 and Movie 4). Thus, the identity of the purinergic receptor(s) driving basal microglial motility has not been firmly established.
Figure 5. Pharmacological modulation of microglial motility by a non-steroidal anti-inflammatory drug (NSAID), flufenamic acid (FFA).

(A) Time-lapse imaging of microglia in a live brain tissue slice from a GFP-reporter mouse shows that FFA (100 μM) reversibly inhibits microglial motility. Top row shows raw fluorescence images. Bottom row shows ‘difference images,’ which indicate motile changes between sequential time-points in an image sequence. Note the lack of motility during FFA. (B) Quantification of the motility index (MI), based on the difference images, shows slow decline during FFA and recovery during washout. For details of methods, see Eyo & Dailey (2012). FFA has been shown to block extension of processes from microglia (Davalos et al., 2005; Wu et al., 2007) and astrocytes (Kim and Dustin, 2006) after laser injury. FFA may inhibit microglial motility by blocking connexin hemichannels. FFA is an anthranilic acid derivative with analgesic, anti-inflammatory, and antipyretic properties.
Figure 6. Time-lapse multiphoton imaging sequences showing rapid mobilization of microglia to injured neurons in a live brain tissue slice from a neonatal mouse lacking the P2X7 receptor (P2X7−/−).

Microglia cell bodies and branches (green) are visible due to expression of green fluorescent protein (GFP) in this mouse line (P2X7−/−:CX3CR1GFP/+). The many healthy cells in the tissue are unlabeled. (A) Time-lapse movie under baseline conditions (00:00–00:17) show continual remodeling of microglial branches. Focal tissue injury (00:17) was induced along a line by brief exposure to high intensity laser light (white line between white arrowheads). Within minutes, injured cells begin to take up a membrane-impermeable red fluorescent DNA-binding dye, ToPro3, and nearby microglia extend branches (yellow arrowheads) toward the laser-damaged cells (00:28). Within a couple of hours, stationary microglia have transformed to activated fusiform or amoeboid microglia that migrate and accumulate near the injured cells (01:47). (B) Higher-magnification view of the same time-lapse sequence shows migration of a microglia cell (yellow arrowheads) toward the injured cells. Microglia respond to tissue injury even though they lack the P2X7 purinoceptor in these knockout mice. Time is shown in hr:min. See supplementary Movie 4.
Whether other classic neurotransmitters regulate microglial motility is currently hotly debated. The first study investigating the possibility of neurotransmitter release modulating microglial motility in situ was performed by Nimmerjahn et al. (2005) during in vivo two-photon imaging of cortical adult microglia. Topical application of bicuculline, an inhibitor of GABA-ergic neurotransmission, increased both excitatory synaptic activity and microglial sampling volume. Assuming that this drug does not modulate microglial activity directly, this result suggests that neuronal activity may determine the rate of microglial tissue sampling (Nimmerjahn et al., 2005).
Combining electrophysiological and confocal imaging approaches, Wu and Zhou (2008) investigated the effects of local neurotransmitter release (glutamate and GABA) and synaptic plasticity on microglia in adult mouse brain slices and failed to detect changes in microglial motility or electrophysiological properties. Furthermore, these electrophysiological results suggest that surveillant (unactivated) microglia are not equipped to directly respond to these neurotransmitters. However, microglia in culture and spinal cord slices from 10–15 day old mice were shown to migrate toward a source of glutamate in an ATP-independent manner, although prolonged glutamate exposure was required (Liu et al., 2009).
Together, these studies raise the possibility of developmental and/or regional differences in microglial responsiveness to glutamate. To address the possibility that spinal microglia may behave differently than brain microglia, Chen et al. (2010) investigated the ability of microglia to respond to a diverse set of neurotransmitters in the adult spinal cord. Microglia motility was altered upon application of ATP but not in response to several different neurotransmitters or cytokines, suggesting that microglia from brain and spinal cord are equally unresponsive to exogenous neurotransmitters. Studies in retina by Fontainhas et al. (2011) are consistent with the idea that neural activity in situ may regulate microglial motility. Inhibition of endogenous glutamatergic neurotransmission reduced microglial motility in retinal explants, suggesting that endogenous glutamatergic signaling somehow regulates basal microglial motility. Additionally, inhibition of endogenous GABAergic neurotransmission increased microglial motility, consistent with an earlier report (Nimmerjahn et al., 2005), although the effect was less dramatic than glutamatergic inhibition. Consistent with other reports (Wu and Zhou, 2008; Chen et al., 2010), exogenous neurotransmitter receptor agonists (glutamate, GABA, and AMPA) failed to directly elicit electrophysiological responses in retinal microglia, suggesting that endogenous signals could be acting in an indirect manner, perhaps by modulating extracellular ATP levels.
While evidence is accumulating that microglia respond to neural activity, it has been suggested that microglia may, in turn, regulate neuronal activity. In 2–3 week old acute and 10–14 day old cultured hippocampal slices, inflammatory activation of microglia by LPS increased microglial ATP release and activated astrocytic P2Y1 receptors (Pascual et al., 2012). These receptors, in turn, amplified ATP release, resulting in the modulation of neuronal activity via metabotropic glutamate receptors, implicating an indirect mechanism for microglial control of neuronal activity. Modulation of neuronal networks by LPS-induced microglial activation has been reported in neonatal rats in vivo (Nimmervoll et al., 2012). Although these mechanisms were uncovered during inflammatory stimuli, it presents the intriguing possibility that physiological mechanisms of microglial-neuronal communication during normal neural function are yet to be uncovered.
Indeed a recent study in developing zebrafish brain supports the idea of reciprocal, activity-dependent interactions between microglia and neurons in situ. Using in vivo time-lapse imaging of both microglial morphology and neuronal activity in the optic tectum of larval zebrafish, Li et al. (2012) found that neuronal activity steers resting microglial processes by a Rac-dependent mechanism that facilitates their contact with cell bodies of highly active neurons within a few minutes. This may be mediated by neuronal release of “find-me” signals such as ATP via pannexin-1 hemichannels. In turn, contact by microglia was found to reduce both spontaneous and visually evoked neural activity. It is proposed that these reciprocal relationships play a homeostatic role in regulating neuronal excitability in healthy brains.
In summary, both the extent and mechanisms of physiological microglial-neuronal crosstalk remains to be clarified. Although there is growing evidence that endogenous neurotransmission may regulate microglial basal motility, evidence for direct communication via classical neurotransmitters is lacking since mammalian microglia do not seem to express glutamatergic or GABA-ergic neurotransmitter receptors in the resting state in situ. Non-classical neurotransmitters, including ATP, appear to play a more pronounced role. Nevertheless, microglia upregulate some neurotransmitter receptors upon activation, for example after ischemia (Gottlieb and Matute, 1997), which may enable them to respond to neuronally released neurotransmitters. Finally, inflammatory signals may alter microglial regulation of neuronal activities in the early postnatal and juvenile rodent brain.
Conclusion
Although the first description of microglia occurred almost a century ago, microglial research over the ensuing 60 years was mainly dominated by debate on the nature and embryonic origins of microglia. However, interest in microglia has increased dramatically over the last two decades, overhauling the thought that microglia are simply passive cells waiting for a problem. We have summarized recent evidence suggesting that microglia perform critical functions during development of the nervous system, including roles in angiogenesis, induction of apoptosis, phagocytic clearance of dead cells, and synapse remodeling. Increasing evidence for neuronal-microglial communication raises the likelihood that microglia play active roles in neural communication. Moreover, microglia could serve as powerful cellular targets in pathological conditions such as neuropathic pain. Successful microglial replacement therapy in animal models of Rett syndrome and ischemic stroke provide encouragement that manipulation of microglia may be effective for treating other neurological conditions. Without doubt, much more remains to be discovered in the next century about this enigmatic component of Cajal’s “third element.”
Supplementary Material
Time-lapse confocal sequence from a live mouse hippocampal tissue slice showing a microglial cell (arrow) extending a branch to contact and phagocytose a nearby injured cell (arrowhead). The tissue slice was derived from a reporter mouse line (CX3CR1+/GPF) in which microglia express green fluorescent protein (GFP). Injured cells were labeled with Sytox, a membrane-impermeant DNA-binding dye that labels the nuclei of dead cells. Original images were taken at 3 min intervals. Time is shown in hr:min from the start of imaging.
Rotation movie showing 3D relationships of microglia and apoptotic cells in the developing mouse hippocampus. Microglia (green) are expressing GFP. Early stage apoptotic cells (blue) are labeled with antibodies to cleaved caspase-3. Late stage apoptotic bodies are labeled with PSVue (red), which binds to phosphatidylserine lipids exposed on the surface of apoptotic cells. Note the wrapping of microglial processes around the soma of an early stage apoptotic pyramidal neuron (upper arrow). Late stage apoptotic bodies are engulfed by processes of phagocytosing microglia (lower arrow).
Time-lapse sequence shows that flufenamic acid (FFA, 100μM), a non-steroidal anti-inflammatory drug (NSAID), modulates microglia motility in a neonatal mouse hippocampal tissue slice. Microglia express GFP in this tissue slice derived from GFP-reporter mouse (CX3CR1+/GPF). Images on the left show raw fluorescence. Images on the right are “difference images,” which depict any changes in cell shape between sequential time points as white. Note the decline in microglial motility upon application of FFA, with slow recovery after washout.
Time-lapse multiphoton imaging sequence shows rapid mobilization of microglia to injured neurons in P2X7 receptor null mice. Focal tissue injury was induced along a line by brief exposure to high intensity laser light (white line between white arrowheads). Within minutes, injured cells begin to take up a membrane-impermeable red fluorescent DNA-binding dye (ToPro3), and nearby microglia extend branches toward the laser damaged cells. Within a couple of hours, activated microglia have migrated and accumulated near the injured cells. Microglia respond to tissue injury even though they lack the P2X7 purinoceptor in these P2X7−/− mice. Time is shown in hr:min.
Acknowledgments
Supported by grants from the American Heart Association (0950160G to MED), the National Institutes of Health (NS064006 to MED), and the Iowa Center for Molecular Auditory Neuroscience (ICMAN) though NIH Grant P30 DC010362. All experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Footnotes
The authors declare that they have no conflict of interest.
References
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Ashwell KW, Hollander H, Streit W, Stone J. The appearance and distribution of microglia in the developing retina of the rat. Vis Neurosci. 1989;2:437–448. doi: 10.1017/s0952523800012335. [DOI] [PubMed] [Google Scholar]
- Barreto G, White RE, Ouyang Y, Xu L, Giffard RG. Astrocytes: targets for neuroprotection in stroke. Cent Nerv Syst Agents Med Chem. 2011;11:164–173. doi: 10.2174/187152411796011303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battisti WP, Wang J, Bozek K, Murray M. Macrophages, microglia, and astrocytes are rapidly activated after crush injury of the goldfish optic nerve: a light and electron microscopic analysis. J Comp Neurol. 1995;354:306–320. doi: 10.1002/cne.903540211. [DOI] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Brockhaus J, Moller T, Kettenmann H. Phagocytozing ameboid microglial cells studied in a mouse corpus callosum slice preparation. Glia. 1996;16:81–90. doi: 10.1002/(SICI)1098-1136(199601)16:1<81::AID-GLIA9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Burek MJ, Oppenheim RW. Programmed cell death in the developing nervous system. Brain Pathol. 1996;6:427–446. doi: 10.1111/j.1750-3639.1996.tb00874.x. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Purinergic nerves. Pharmacol Rev. 1972;24:509–581. [PubMed] [Google Scholar]
- Burnstock G, Krugel U, Abbracchio MP, Illes P. Purinergic signalling: from normal behaviour to pathological brain function. Prog Neurobiol. 2011;95:229–274. doi: 10.1016/j.pneurobio.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Caldero J, Brunet N, Ciutat D, Hereu M, Esquerda JE. Development of microglia in the chick embryo spinal cord: implications in the regulation of motoneuronal survival and death. J Neurosci Res. 2009;87:2447–2466. doi: 10.1002/jnr.22084. [DOI] [PubMed] [Google Scholar]
- Carbonell WS, Murase S, Horwitz AF, Mandell JW. Migration of perilesional microglia after focal brain injury and modulation by CC chemokine receptor 5: an in situ time-lapse confocal imaging study. J Neurosci. 2005;25:7040–7047. doi: 10.1523/JNEUROSCI.5171-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchini MG, Dominguez MG, Mocci S, Wetterwald A, Felix R, Fleisch H, Chisholm O, Hofstetter W, Pollard JW, Stanley ER. Role of colony stimulating factor-1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse. Development. 1994;120:1357–1372. doi: 10.1242/dev.120.6.1357. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chen SK, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, Capecchi MR. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell. 2010;141:775–785. doi: 10.1016/j.cell.2010.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Koga K, Li XY, Zhuo M. Spinal microglial motility is independent of neuronal activity and plasticity in adult mice. Mol Pain. 2010;6:19. doi: 10.1186/1744-8069-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, Grahames CB, Casula MA, Yiangou Y, Birch R, Anand P, Buell GN. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–396. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Dailey ME, Waite M. Confocal imaging of microglial cell dynamics in hippocampal slice cultures. Methods. 1999;18:222–30. 177. doi: 10.1006/meth.1999.0775. [DOI] [PubMed] [Google Scholar]
- Dalmau I, Finsen B, Tonder N, Zimmer J, Gonzalez B, Castellano B. Development of microglia in the prenatal rat hippocampus. J Comp Neurol. 1997;377:70–84. [PubMed] [Google Scholar]
- Dalmau I, Finsen B, Zimmer J, Gonzalez B, Castellano B. Development of microglia in the postnatal rat hippocampus. Hippocampus. 1998;8:458–474. doi: 10.1002/(SICI)1098-1063(1998)8:5<458::AID-HIPO6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Davalos D, Lee JK, Smith WB, Brinkman B, Ellisman MH, Zheng B, Akassoglou K. Stable in vivo imaging of densely populated glia, axons and blood vessels in the mouse spinal cord using two-photon microscopy. J Neurosci Methods. 2008;169:1–7. doi: 10.1016/j.jneumeth.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YY, Lu J, Ling EA, Kaur C. Role of microglia in the process of inflammation in the hypoxic developing brain. Front Biosci (Schol Ed) 2011;3:884–900. doi: 10.2741/194. [DOI] [PubMed] [Google Scholar]
- del Rio-Hortega P. El ‘tercer elemento’ de los centros nerviosos. Bol d 1 Soc Esp d Biol. 1919;9:69. [Google Scholar]
- del Rio-Hortega P. La microglia y su transformacion en celulas en basoncito y cuerpos granulo-adiposos. Trab Lab Invest BioL Madrid. 1920;18:37–82. [Google Scholar]
- del Rio-Hortega P. Microglia. In: Penfield W, editor. Cytology and Cellular Pathology of the Nervous System. New York: Hoeber; 1932. 482–1924–534. [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Wu HJ, Li HQ, Qin S, Wang YE, Li J, Lou HF, Chen Z, Li XM, Luo QM, Duan S. Microglial migration mediated by ATP-induced ATP release from lysosomes. Cell Res. 2012;22:1022–1033. doi: 10.1038/cr.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doverhag C, Keller M, Karlsson A, Hedtjarn M, Nilsson U, Kapeller E, Sarkozy G, Klimaschewski L, Humpel C, Hagberg H, Simbruner G, Gressens P, Savman K. Pharmacological and genetic inhibition of NADPH oxidase does not reduce brain damage in different models of perinatal brain injury in newborn mice. Neurobiol Dis. 2008;31:133–144. doi: 10.1016/j.nbd.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Eyo U, Dailey ME. Effects of oxygen-glucose deprivation on microglial mobility and viability in developing mouse hippocampal tissues. Glia. 2012;60:1747–1760. doi: 10.1002/glia.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, Peri F, Wilson SW, Ruhrberg C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. 2010;116:829–840. doi: 10.1182/blood-2009-12-257832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino JV, Wang X, Johnson CE, Klibanov A, Derugin N, Wendland MF, Vexler ZS. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci. 2011;31:12992–13001. doi: 10.1523/JNEUROSCI.2102-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Bernet E, Soriano E, del Rio T, Fonseca M. Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience. 1990;39:451–458. doi: 10.1016/0306-4522(90)90281-8. [DOI] [PubMed] [Google Scholar]
- Fontainhas AM, Wang M, Liang KJ, Chen S, Mettu P, Damani M, Fariss RN, Li W, Wong WT. Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS One. 2011;6:e15973. doi: 10.1371/journal.pone.0015973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade JM, Barde YA. Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron. 1998;20:35–41. doi: 10.1016/s0896-6273(00)80432-8. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb M, Matute C. Expression of ionotropic glutamate receptor subunits in glial cells of the hippocampal CA1 area following transient forebrain ischemia. J Cereb Blood Flow Metab. 1997;17:290–300. doi: 10.1097/00004647-199703000-00006. [DOI] [PubMed] [Google Scholar]
- Graeber MB. Changing face of microglia. Science. 2010;330:783–788. doi: 10.1126/science.1190929. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol. 2010;119:89–105. doi: 10.1007/s00401-009-0622-0. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ, Kreutzberg GW. Identity of ED2-positive perivascular cells in rat brain. J Neurosci Res. 1989;22:103–106. doi: 10.1002/jnr.490220114. [DOI] [PubMed] [Google Scholar]
- Greter M, Merad M. Regulation of microglia development and homeostasis. Glia. 2013;61:121–127. doi: 10.1002/glia.22408. [DOI] [PubMed] [Google Scholar]
- Grossmann R, Stence N, Carr J, Fuller L, Waite M, Dailey ME. Juxtavascular microglia migrate along brain microvessels following activation during early postnatal development. Glia. 2002;37:229–240. [PubMed] [Google Scholar]
- Guo JM, Liu AJ, Su DF. Genetics of stroke. Acta Pharmacol Sin. 2010;31:1055–1064. doi: 10.1038/aps.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas S, Brockhaus J, Verkhratsky A, Kettenmann H. ATP-induced membrane currents in ameboid microglia acutely isolated from mouse brain slices. Neuroscience. 1996;75:257–261. doi: 10.1016/0306-4522(96)00270-9. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Tomimatsu Y, Suzuki H, Yamada J, Wu Z, Yao H, Kagamiishi Y, Tateishi N, Sawada M, Nakanishi H. The intra-arterial injection of microglia protects hippocampal CA1 neurons against global ischemia-induced functional deficits in rats. Neuroscience. 2006;142:87–96. doi: 10.1016/j.neuroscience.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- He WJ, Cui J, Du L, Zhao YD, Burnstock G, Zhou HD, Ruan HZ. Spinal P2X(7) receptor mediates microglia activation-induced neuropathic pain in the sciatic nerve injury rat model. Behav Brain Res. 2012;226:163–170. doi: 10.1016/j.bbr.2011.09.015. [DOI] [PubMed] [Google Scholar]
- Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci. 2001;21:1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore P, Donnelly-Roberts D, Namovic MT, Hsieh G, Zhu CZ, Mikusa JP, Hernandez G, Zhong C, Gauvin DM, Chandran P, Harris R, Medrano AP, Carroll W, Marsh K, Sullivan JP, Faltynek CR, Jarvis MF. A-740003 [N-(1-{[(cyanoimino)(5-quinolinylamino) methyl]amino}-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide], a novel and selective P2X7 receptor antagonist, dose-dependently reduces neuropathic pain in the rat. J Pharmacol Exp Ther. 2006;319:1376–1385. doi: 10.1124/jpet.106.111559. [DOI] [PubMed] [Google Scholar]
- Hughes V. Microglia: The constant gardeners. Nature. 2012;485:570–572. doi: 10.1038/485570a. [DOI] [PubMed] [Google Scholar]
- Inoue K. Purinergic systems in microglia. Cell Mol Life Sci. 2008;65:3074–3080. doi: 10.1007/s00018-008-8210-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Tsuda M. Purinergic systems, neuropathic pain and the role of microglia. Exp Neurol. 2012;234:293–301. doi: 10.1016/j.expneurol.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Imai F, Suzuki H, Oda J, Ninomiya T, Ono K, Sano H, Sawada M. Neuroprotective effect of exogenous microglia in global brain ischemia. J Cereb Blood Flow Metab. 2007;27:488–500. doi: 10.1038/sj.jcbfm.9600362. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kim JV, Dustin ML. Innate response to focal necrotic injury inside the blood-brain barrier. J Immunol. 2006;177:5269–5277. doi: 10.4049/jimmunol.177.8.5269. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Takata K, Inden M, Tsuchiya D, Yanagisawa D, Nakata J, Taniguchi T. Intracerebroventricular injection of microglia protects against focal brain ischemia. J Pharmacol Sci. 2004;94:203–206. doi: 10.1254/jphs.94.203. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Yanagisawa D, Inden M, Takata K, Tsuchiya D, Kawasaki T, Taniguchi T, Shimohama S. Recovery of focal brain ischemia-induced behavioral dysfunction by intracerebroventricular injection of microglia. J Pharmacol Sci. 2005;97:289–293. doi: 10.1254/jphs.sc0040129. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Takahashi E, Miyagawa Y, Yamanaka H, Noguchi K. Induction of the P2X7 receptor in spinal microglia in a neuropathic pain model. Neurosci Lett. 2011;504:57–61. doi: 10.1016/j.neulet.2011.08.058. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci. 2008;28:2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel I, Elliott A, Chen HJ, Mansuy IM, Kennedy MB. A role for synGAP in regulating neuronal apoptosis. Eur J Neurosci. 2005;21:611–621. doi: 10.1111/j.1460-9568.2005.03908.x. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Takubo K, Shimizu T, Ohno H, Kishi K, Shibuya M, Saya H, Suda T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med. 2009;206:1089–1102. doi: 10.1084/jem.20081605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurpius D, Nolley EP, Dailey ME. Purines induce directed migration and rapid homing of microglia to injured pyramidal neurons in developing hippocampus. Glia. 2007;55:873–884. doi: 10.1002/glia.20509. [DOI] [PubMed] [Google Scholar]
- Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84:49–59. doi: 10.1139/Y05-143. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Du X, Liu C, Wen Z, Du J. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Developmental Cell. 2012;23(6):1189–1202. doi: 10.1016/j.devcel.2012.10.027. [DOI] [PubMed] [Google Scholar]
- Linnartz B, Kopatz J, Tenner AJ, Neumann H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J Neurosci. 2012;32:946–952. doi: 10.1523/JNEUROSCI.3830-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GJ, Nagarajah R, Banati RB, Bennett MR. Glutamate induces directed chemotaxis of microglia. Eur J Neurosci. 2009;29:1108–1118. doi: 10.1111/j.1460-9568.2009.06659.x. [DOI] [PubMed] [Google Scholar]
- Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009;29:5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41:535–547. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Chu KL, Namovic MT, Donnelly-Roberts DL, Harris RR, Zhang XF, Shieh CC, Wismer CT, Zhu CZ, Gauvin DM, Fabiyi AC, Honore P, Gregg RJ, Kort ME, Nelson DW, Carroll WA, Marsh K, Faltynek CR, Jarvis MF. P2X7-related modulation of pathological nociception in rats. Neuroscience. 2007;146:1817–1828. doi: 10.1016/j.neuroscience.2007.03.035. [DOI] [PubMed] [Google Scholar]
- McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, Maki RA. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- Milner R, Campbell IL. Cytokines regulate microglial adhesion to laminin and astrocyte extracellular matrix via protein kinase C-dependent activation of the alpha6beta1 integrin. J Neurosci. 2002;22(5):1562–72. doi: 10.1523/JNEUROSCI.22-05-01562.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner R, Campbell IL. The extracellular matrix and cytokines regulate microglial integrin expression and activation. J Immunol. 2003;170(7):3850–8. doi: 10.4049/jimmunol.170.7.3850. [DOI] [PubMed] [Google Scholar]
- Murase S, Poser SW, Joseph J, McKay RD. p53 controls neuronal death in the CA3 region of the newborn mouse hippocampus. Eur J Neurosci. 2011;34:374–381. doi: 10.1111/j.1460-9568.2011.07758.x. [DOI] [PubMed] [Google Scholar]
- Napoli I, Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009;158:1030–1038. doi: 10.1016/j.neuroscience.2008.06.046. [DOI] [PubMed] [Google Scholar]
- Narantuya D, Nagai A, Sheikh AM, Masuda J, Kobayashi S, Yamaguchi S, Kim SU. Human microglia transplanted in rat focal ischemia brain induce neuroprotection and behavioral improvement. PLoS One. 2010;5:e11746. doi: 10.1371/journal.pone.0011746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Dirnagl U. Role of glial cells in cerebral ischemia. Glia. 2005;50:281–286. doi: 10.1002/glia.20205. [DOI] [PubMed] [Google Scholar]
- Nelson KB, Lynch JK. Stroke in newborn infants. Lancet Neurol. 2004;3:150–158. doi: 10.1016/S1474-4422(04)00679-9. [DOI] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K. Microglia provide neuroprotection after ischemia. FASEB J. 2006;20:714–716. doi: 10.1096/fj.05-4882fje. [DOI] [PubMed] [Google Scholar]
- Ngu EM, Sahley CL, Muller KJ. Reduced axon sprouting after treatment that diminishes microglia accumulation at lesions in the leech CNS. J Comp Neurol. 2007;503:101–109. doi: 10.1002/cne.21386. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Nimmervoll B, White R, Yang JW, An S, Henn C, Sun JJ, Luhmann HJ. LPS-Induced Microglial Secretion of TNFalpha Increases Activity-Dependent Neuronal Apoptosis in the Neonatal Cerebral Cortex. Cereb Cortex. 2012 doi: 10.1093/cercor/bhs156. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Irino Y, Nakamura Y, Akazawa C, Inoue K, Kohsaka S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia. 2007;55:604–616. doi: 10.1002/glia.20489. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Gross CT. Microglia in development: linking brain wiring to brain environment. Neuron Glia Biol. 2011;7:77–83. doi: 10.1017/S1740925X12000105. [DOI] [PubMed] [Google Scholar]
- Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A. 2012;109:E197–205. doi: 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfield W. Microglia and the process of phagocytosis in gliomas. Am J Pathol. 1925;1:77–90.15. [PMC free article] [PubMed] [Google Scholar]
- Peri F, Nusslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008;133:916–927. doi: 10.1016/j.cell.2008.04.037. [DOI] [PubMed] [Google Scholar]
- Petersen MA, Dailey ME. Diverse microglial motility behaviors during clearance of dead cells in hippocampal slices. Glia. 2004;46:195–206. doi: 10.1002/glia.10362. [DOI] [PubMed] [Google Scholar]
- Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005 Nov;28(11):571–3. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- Rigato C, Buckinx R, Le-Corronc H, Rigo JM, Legendre P. Pattern of invasion of the embryonic mouse spinal cord by microglial cells at the time of the onset of functional neuronal networks. Glia. 2011;59:675–695. doi: 10.1002/glia.21140. [DOI] [PubMed] [Google Scholar]
- Rigato C, Swinnen N, Buckinx R, Couillin I, Mangin JM, Rigo JM, Legendre P, Le Corronc H. Microglia proliferation is controlled by P2X7 receptors in a Pannexin-1-independent manner during early embryonic spinal cord invasion. J Neurosci. 2012;32:11559–11573. doi: 10.1523/JNEUROSCI.1042-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymo SF, Gerhardt H, Wolfhagen Sand F, Lang R, Uv A, Betsholtz C. A two-way communication between microglial cells and angiogenic sprouts regulates angiogenesis in aortic ring cultures. PLoS One. 2011;6:e15846. doi: 10.1371/journal.pone.0015846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago MF, Veliskova J, Patel NK, Lutz SE, Caille D, Charollais A, Meda P, Scemes E. Targeting pannexin1 improves seizure outcome. PLoS One. 2011;6:e25178. doi: 10.1371/journal.pone.0025178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Stevens B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia. 2013;61:24–36. doi: 10.1002/glia.22389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui TA, Lively S, Vincent C, Schlichter LC. Regulation of podosome formation, microglial migration and invasion by Ca2+-signaling molecules expressed in podosomes. J Neuroinflammation. 2012 Nov 17;9(1):250. doi: 10.1186/1742-2094-9-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieger D, Moritz C, Ziegenhals T, Prykhozhij S, Peri F. Long-range Ca2+ waves transmit brain-damage signals to microglia. Dev Cell. 2012;22:1138–1148. doi: 10.1016/j.devcel.2012.04.012. [DOI] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SJ, Cooper M, Waxman A. Laser microscopy of subcellular structure in living neocortex: Can one see dendritic spines twitch? In: Squire LR, Lindenlaub E, editors. XXIII Symposia Medica Hoechst: The Biology of Memory. Vol. 23. Stuttgart: Schattauer Verlag; 1990. pp. 49–71. [Google Scholar]
- Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33:256–266. [PubMed] [Google Scholar]
- Svahn AJ, Graeber MB, Ellett F, Lieschke GJ, Rinkwitz S, Bennett MR, Becker TS. Development of ramified microglia from early macrophages in the zebrafish optic tectum. Dev Neurobiol. 2012;73:60–71. doi: 10.1002/dneu.22039. [DOI] [PubMed] [Google Scholar]
- Swinnen N, Smolders S, Avila A, Notelaers K, Paesen R, Ameloot M, Brone B, Legendre P, Rigo JM. Complex invasion pattern of the cerebral cortex by microglial cells during development of the mouse embryo. Glia. 2013;61:150–63. doi: 10.1002/glia.22421. [DOI] [PubMed] [Google Scholar]
- Thomas WE. Brain macrophages: on the role of pericytes and perivascular cells. Brain Res Brain Res Rev. 1999;31:42–57. doi: 10.1016/s0165-0173(99)00024-7. [DOI] [PubMed] [Google Scholar]
- Tozaki-Saitoh H, Tsuda M, Miyata H, Ueda K, Kohsaka S, Inoue K. P2Y12 receptors in spinal microglia are required for neuropathic pain after peripheral nerve injury. J Neurosci. 2008;28:4949–4956. doi: 10.1523/JNEUROSCI.0323-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang T, Beggs S, Salter MW. Brain-derived neurotrophic factor from microglia: a molecular substrate for neuropathic pain. Neuron Glia Biol. 2011 Feb;7(1):99–108. doi: 10.1017/S1740925X12000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8:e1000527. doi: 10.1371/journal.pbio.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Beggs S, Salter MW, Inoue K. Microglia and intractable chronic pain. Glia. 2013 Jan;61(1):55–61. doi: 10.1002/glia.22379. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Kuboyama K, Inoue T, Nagata K, Tozaki-Saitoh H, Inoue K. Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Mol Pain. 2009;5:28. doi: 10.1186/1744-8069-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008;28:11263–11268. doi: 10.1523/JNEUROSCI.2308-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent C, Siddiqui TA, Schlichter LC. Podosomes in migrating microglia: components and matrix degradation. J Neuroinflammation. 2012;9 doi: 10.1186/1742-2094-9-190. 190-2094-9-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt A, Hormuzdi SG, Monyer H. Pannexin1 and Pannexin2 expression in the developing and mature rat brain. Brain Res Mol Brain Res. 2005;141(1):113–20. doi: 10.1016/j.molbrainres.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakselman S, Bechade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. 2008;28:8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw JM, Murray V, Berge E, Del Zoppo GJ. Thrombolysis for acute ischaemic stroke. Cochrane Database Syst Rev. 2009;(4):CD000213. doi: 10.1002/14651858.CD000213.pub2. [DOI] [PubMed] [Google Scholar]
- Weinstein JR, Koerner IP, Moller T. Microglia in ischemic brain injury. Future Neurol. 2010;5:227–246. doi: 10.2217/fnl.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LJ, Vadakkan KI, Zhuo M. ATP-induced chemotaxis of microglial processes requires P2Y receptor-activated initiation of outward potassium currents. Glia. 2007;55(8):810–21. doi: 10.1002/glia.20500. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Zhuo M. Resting microglial motility is independent of synaptic plasticity in mammalian brain. J Neurophysiol. 2008;99:2026–2032. doi: 10.1152/jn.01210.2007. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Wu G, Sharif MRA, Baker A, Jia Y, Fahey FH, Luo HR, Feener EP, Clapham DE. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci. 2012;15:565–573. doi: 10.1038/nn.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics. 2010;7:378–391. doi: 10.1016/j.nurt.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Wu G, Wu LJ. Neuronal and microglial mechanisms of neuropathic pain. Mol Brain. 2011 Jul 30;4:31. doi: 10.1186/1756-6606-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse confocal sequence from a live mouse hippocampal tissue slice showing a microglial cell (arrow) extending a branch to contact and phagocytose a nearby injured cell (arrowhead). The tissue slice was derived from a reporter mouse line (CX3CR1+/GPF) in which microglia express green fluorescent protein (GFP). Injured cells were labeled with Sytox, a membrane-impermeant DNA-binding dye that labels the nuclei of dead cells. Original images were taken at 3 min intervals. Time is shown in hr:min from the start of imaging.
Rotation movie showing 3D relationships of microglia and apoptotic cells in the developing mouse hippocampus. Microglia (green) are expressing GFP. Early stage apoptotic cells (blue) are labeled with antibodies to cleaved caspase-3. Late stage apoptotic bodies are labeled with PSVue (red), which binds to phosphatidylserine lipids exposed on the surface of apoptotic cells. Note the wrapping of microglial processes around the soma of an early stage apoptotic pyramidal neuron (upper arrow). Late stage apoptotic bodies are engulfed by processes of phagocytosing microglia (lower arrow).
Time-lapse sequence shows that flufenamic acid (FFA, 100μM), a non-steroidal anti-inflammatory drug (NSAID), modulates microglia motility in a neonatal mouse hippocampal tissue slice. Microglia express GFP in this tissue slice derived from GFP-reporter mouse (CX3CR1+/GPF). Images on the left show raw fluorescence. Images on the right are “difference images,” which depict any changes in cell shape between sequential time points as white. Note the decline in microglial motility upon application of FFA, with slow recovery after washout.
Time-lapse multiphoton imaging sequence shows rapid mobilization of microglia to injured neurons in P2X7 receptor null mice. Focal tissue injury was induced along a line by brief exposure to high intensity laser light (white line between white arrowheads). Within minutes, injured cells begin to take up a membrane-impermeable red fluorescent DNA-binding dye (ToPro3), and nearby microglia extend branches toward the laser damaged cells. Within a couple of hours, activated microglia have migrated and accumulated near the injured cells. Microglia respond to tissue injury even though they lack the P2X7 purinoceptor in these P2X7−/− mice. Time is shown in hr:min.