

Abstract
Connexin43 (Cx43) plays a critical role in osteoblast function and bone mass accrual, yet the identity of the second messengers communicated by Cx43 gap junctions, the targets of these second messengers and how they regulate osteoblast function remain largely unknown. We have shown that alterations of Cx43 expression in osteoblasts can impact the responsiveness to fibroblast growth factor-2 (FGF2), by modulating the transcriptional activity of Runx2. In this study, we examined the contribution of the phospholipase Cγ1/inositol polyphosphate/PKCδ cascade to the Cx43-dependent transcriptional response of MC3T3 osteoblasts to FGF2. Knockdown of expression and/or inhibition of function of phospholipase Cγ1, inositol polyphosphate multikinase, which generates InsP4 and InsP5, and inositol hexakisphosphate kinase 1/2, which generates inositol pyrophosphates, prevented the ability of Cx43 to potentiate FGF2-induced signaling through Runx2. Conversely, overexpression of phospholipase Cγ1 and inositol hexakisphosphate kinase 1/2 enhanced FGF2 activation of Runx2 and the effect of Cx43 overexpression on this response. Disruption of these pathways blocked the nuclear accumulation of PKCδ and the FGF2-dependent interaction of PKCδ and Runx2, reducing Runx2 transcriptional activity. These data reveal that FGF2-signaling involves the inositol polyphosphate cascade, including IP6K, and demonstrate that IP6K regulates Runx2 and osteoblast gene expression. Additionally, these data implicate the water-soluble inositol polyphosphates as mediators of the Cx43-dependent amplification of the osteoblast response to FGF2, and suggest that these low molecular weight second messengers may be biologically relevant mediators of osteoblast function that are communicated by Cx43-gap junctions.
Keywords: Gap Junction, Bone, Fibroblast Growth Factor 2, Inositol Hexakisphosphate Kinase, Inositol Polyphosphate Multikinase, Inositol Pyrophosphate, Phospholipase C gamma
INTRODUCTION
Gap junctions are intercellular channels formed by hexamers of connexins in one cell that dock with a hexameric array of connexins on an adjacent cell, forming an aqueous pore between the two cells. Gap junctions permit the direct intercellular exchange of ions, small molecules and second messengers. In addition, gap junctions can function as unopposed hemichannels, serving as a direct conduit between the cytosol and extracellular fluid.
In bone, osteoblasts and osteocytes are highly interconnected via gap junctions composed primarily of connexin43 (Cx43). In these cells, Cx43 has been shown to play an important role in transmitting hormonal-, mechanical load- and growth factor-induced signals and, ultimately, in bone mass acquisition via both classic cell-to-cell communication through gap junctions or via hemichannel activity (1,2). Mutations in Gja1, the gene encoding Cx43, result in the pleiotropic disorder oculodentodigital dysplasia, which includes numerous skeletal manifestations (3). Mouse models of oculodentodigital dysplasia and Cx43 genetic ablation (both globally and osteoblast-specific conditional knockout models) have underscored the fundamental importance of Cx43 in skeletal function and bone mass acquisition (4–11). Loss or disruption of Cx43 in these mouse models profoundly impairs osteoblast function and responsiveness to anabolic hormones and mechanical load, typically resulting in osteopenic bone. Indeed, modulation of Cx43 affects signaling transduction cascades, impacting osteoblast and osteocyte survival and/or gene expression (12–19).
Despite the critical role of Cx43 as a regulator of bone mass, the complex molecular mechanisms by which Cx43 regulates osteoblast function are only beginning to emerge. Critical questions remain to be answered, such as what is the identity of the second messengers communicated by Cx43 gap junctions, what are the targets of these second messengers, and how do they regulate osteoblast/osteocyte function?
Ultimately, by defining the molecular pathways by which Cx43 regulates osteoblast function, we can identify the biologically relevant second messengers that are being communicated among osteogenic cells and gain insight into how osteoblasts and osteocytes coordinate their activities to form new bone. Towards this end, we have focused our attention on signaling via the well-defined FGF2 signaling pathway in osteoblasts.
FGF2 is an important regulator of skeletal tissue with complex action, acting at several stages of differentiation to differentially affect osteoblast function (20,21). FGF2 signals through its cognate FGFRs to activate several signaling cascades, including phospholipase Cγ1 (PLCγ1) (22). In osteoblasts, FGF2 signaling converges on Runx2, an important regulator of osteoblast differentiation (23–26).
In our attempts to identify the molecular mechanisms by which Cx43 regulates osteoblasts, we have shown that alterations of Cx43 expression in osteoblasts can impact their responsiveness to FGF2, by modulating the transcriptional activity of Runx2 in a PKCδ and ERK-dependent manner (12,27,28). Further, we have shown that the ability of Cx43 to potentiate Runx2 activity requires gap junctional communication, as gap junction channel blockers and cell culture at low density, where cell-to-cell contacts are kept at a minimum, abrogates the effects of Cx43 overexpression on osteoblast responsiveness to FGF2 (28). In addition we have shown that Cx43 overexpression in MC3T3 cells enhances the percentage of cells responding to FGF2, underscoring that Cx43 is permitting the communication of signals between cells (12,28). In this study, we examined the hypothesis that the amplification of FGF2-regulated signaling cascades by Cx43 occurs as a result of second messengers generated downstream of PLCγ1 that are communicated by Cx43 gap junction channels to affect the expression of osteoblast genes.
MATERIALS AND METHODS
Chemicals, Antibodies and Reagents
N2-(m- (trifluoromethyl) benzyl) N6-(p-nitrobenzyl) purine (TNP), ET-18-OCH3, U73122, RHC-80267 and 2-aminoethoxydiphenyl borate (2-APB) were purchased from Sigma (St. Louis, MO). Human recombinant FGF2 was purchased from Millipore (Billerica, MA). Tissue culture media and fetal bovine serum were purchased from Hyclone (Logan, UT). Antibodies were purchased as follows: anti-PLCγ1 and anti-PKCδ from Santa Cruz Biotechnology (Santa Cruz, CA); anti-phospho-PLCγ1 (Tyr783), anti-phospho-PKCδ (Thr505), anti-phospho-PKCδ (Ser643) and anti-Lamin A/C from Cell Signaling Technology (Danvers, MA); anti-GAPDH from Millipore; anti-IPMK from Abcam (Cambridge, MA); anti-Runx2 from MBL International (Woburn, MA). Scrambled (non-targeting), PLCγ1 and IPMK-directed siRNAs were purchased form Dharmacon (Lafayette, CO). IP6K1 and IP6K2 siRNAs (29) were purchased from Ambion (Grand Island, NY).
Cell Culture and FGF2 Treatments
MC3T3 (clone 4) cells were cultured as has been described (30). Prior to FGF2 treatment, cells were serum-starved overnight in αMEM containing 0.1% fetal bovine serum. Subsequently, the cells were exposed to 10ng/ml FGF2 or the vehicle diluent (phosphate buffered saline + 0.1% bovine serum albumin) for up to 4 hours, unless specified otherwise. For inhibitor studies, cells were pre-treated with DMSO (vehicle) or the inhibitor for 30 minutes prior to FGF2 stimulation. For long term culture osteogenic differentiation media (αMEM + 10% fetal bovine serum supplemented with 50mM ascorbic acid and 10mM β-glycerophosphate) was used. Cell viability was routinely assessed using a CCK-8 cell viability assay (Dojindo Molecular Technologies, Rockville, MD).
Plasmid Constructs, Transient Transfection and Luciferase Reporter Assays
Sub-confluent cultures of MC3T3 cells were transiently transfected using X-tremegene9 (Roche, Indianapolis, IN) transfection reagent, as we have published (31). For transfections with multiple plasmids, the amount of transfection reagent was scaled accordingly and the total amount of DNA in each reaction was kept constant by the addition of empty vector (pSFFV-neo). The p6xOSE2-Luc construct, which contains six tandem repeats of the mouse Runx2 binding cognate upstream of a minimal promoter, and its mutated negative control (p6xmutOSE2-Luc) were provided by Dr. Gerard Karsenty (Columbia University, New York) (32). The pSFFV-Cx43 plasmid, which contains the full length Cx43 coding sequence cloned downstream of the Spleen Focus Forming Virus promoter was provided by Dr. Thomas Steinberg (Washington University, St Louis) (33). The empty vector, pSFFV-neo, was provided by Dr. Gabriel Nunez (University of Michigan, Ann Arbor). The wild type PLCγ1 and dominant negative PLCγ1 (Y783F) constructs were provided by Dr. Barbara Rellahan (Food and Drug Administration, Bethesda) (34). The dominant negative PKCδ construct (PKCδ K376R) was from Addgene (Cambridge, MA), as deposited by Dr. I. Bernard Weinstein (35). The IP6K1 and IP6K2 expression plasmids were from Addgene as deposited by Drs. William Hahn and David Root (36) and were subcloned into the pDEST-CMV/V5 backbone vector (Life Technologies, Inc., Grand Island, NY).
For transfection with siRNA constructs, cells plated at 20,000 cells/cm2 were transfected with siRNA at the indicated concentrations using X-tremegene siRNA reagent. After permitting the cells to recover for 24 hours, the cells were sequentially transfected with the required luciferase reporter constructs, as above. Transfection efficiency was monitored by co-transfection with a pRL-TK renilla luciferase reporter (Promega, Madison, WI) and luciferase activity assessed using a dual luciferase assay reagent as reported by Dyer et al (37). Luciferase reporter assays were performed as described previously (31). All luciferase-based experiments were done in triplicate wells and repeated a minimum of three times. Data from a representative experiment are shown. Luciferase data shown on a single graph were from the same experiment so that all other variables are kept constant.
Western Blotting and co-Immunoprecipitation
Samples subjected to SDS-PAGE and western blotting and/or co-immunoprecipitation were done as previously described (27). Nuclear extracts were prepared using the NE-PER kit (Pierce, Rockford, IL) according to manufacturer’s directions. Loading was normalized to the expression levels of GAPDH or Lamin A/C, as appropriate.
Quantitative real time RT-PCR
RNA extraction, reverse transcription and quantitative real time PCR were performed as previously described (38). Relative expression was determined by comparison to the “house keeping” genes, GAPDH and RPL13, using the geNorm software (v3.5, Ghent University Hospital, Ghent, BE). The primer sets used for real time PCR can be found in Table I.
Table I.
Real Time PCR Primers
| Gene | Sequence |
|---|---|
| PLCG1 | CTGTGGTTCCCCTCAAACTATGT (For) GTCCCCCAGTGGGCTGTT (Rev) |
| PLCG2 | AGATCCTGATGACACTCACTGTCAA (For) GGCAATACTCCGCCCTAGTTT (Rev) |
| IP6K1 | ATCGGTCTGGCAGTGGTAGTG (For) CTGGGAGCTCTCAGAGGTCTCA (Rev) |
| IP6K2 | ATGGGCGGAAGCTTTCG (For) CCCATTGTGAAAGAACTGGAAAA (Rev) |
| IP6K3 | CTCTCGGTGGAGGGATTCAG (For) CGGAGGCGGGTTCCAT (Rev) |
| RPL13 | CGAAACAAGTCCACGGAGTCA (For) GAGCTTGGAGCGGTACTCCTT (Rev) |
| GAPDH | CGTGTTCCTACCCCCAATGT (For) TGTCATCATACTTGGCAGGTTTCT (Rev) |
| COL1A1 | CTTCACCTACAGCACCCTTGTG (For) GATGACTGTCTTGCCCCAAGTT (Rev) |
| Osteocalcin | CACAGATGCCACGCCCA (For) TGCCCTCCTGCTTGGACA (Rev) |
| Runx2 | CCGTGGCCTTCAAGGTTGT (For) CGGCCATGACGGTAACCA (Rev) |
| Osterix | TTCTGTCCCCTGCTCCTTCTAG (For) CGTCAACGACGTTATGCTCTTC (Rev) |
Statistics
Unless indicated otherwise, all experiments were repeated a minimum of 3 times with triplicate wells. Graphs depict averages, and error bars indicate standard deviation. Samples were compared by an ANOVA for unpaired samples. Where a statistical difference was indicated (p-value <0.05), we performed a Dunnett’s or Tukey post-hoc test, as appropriate.
RESULTS
PLCγ1 is required for the Cx43-dependent amplification of Runx2 transcriptional activity
Activation of FGF receptors by their cognate ligands is known to activate several signaling cascades, including PLCγ1 (22). Previously, we demonstrated a role for PKCδ, a member of the novel, Ca2+-independent PKC family, in the Cx43-dependent amplification of Runx2 activity in an osteoblast cell line (12,28). Accordingly, we hypothesized that the activation of PLCγ generates a second messenger that can be propagated through Cx43 gap junctions to potentiate PKCδ signaling and Runx2 transcriptional activity. To test this hypothesis, we examined the ability of PLCγ inhibitors to block the Cx43-dependent amplification of Runx2 activity by luciferase reporter assays. MC3T3 osteoblasts were transiently transfected with a Runx2-responsive luciferase reporter construct (p6xOSE2-Luc). The cells were co-transfected with empty vector (pSFFV-neo) or a Cx43 overexpression vector (pSFFV-Cx43), as indicated. Treatment of MC3T3 osteoblasts with the PLC inhibitor U73122 (2μM) reduced the Cx43-dependent potentiation of the FGF2-induced Runx2-mediated transcription relative to vehicle treated controls (Fig. 1A). Notably, U73122 caused the cells to subtly but noticeably “round up” in culture, though no toxicity was detected using a CCK-8 cell viability assay (Supporting Information Fig. S1). Regardless, as cell rounding would impact cell-to-cell contacts and thus gap junctions, we confirmed the involvement of PLC with several other approaches.
Fig. 1.

PLCγ1 mediates the basal and Cx43-dependent potentiation of Runx2 activity by FGF2. Luciferase reporter assays were performed on cells co-transfected with a Runx2-reponsive luciferase reporter (p6xOSE2-Luc) and pSFFV-neo empty vector (grey bars) or pSFFV-Cx43 expression vector (black bars) and treated with vehicle (Veh) or FGF2 (10ng/ml, 4h). Cells were treated with the PLC inhibitor (A) U73122 (2μM), (B) ET-18-OCH3 or (C) transfected with the dominant negative PLCγ1 Y783F or PKCδ K376R constructs. (D) Quantitative real time RT-PCR amplification of PLCγ1 and PLCγ2 from MC3T3 osteoblast RNA. (E) Western blot of whole cell extracts from FGF2 treated cells probed with anti-phospho-PLCγ1 (Tyr783) antibodies. Anti-GAPDH antibodies were used as a load control. (F) Luciferase reporter assays (p6xOSE2-Luc reporter) and western blot (anti-PLCγ1 or anti-GAPDH antibodies) of cells transfected with scrambled siRNA (SCR, 50nM) or PLCγ1 siRNA (PLCG1, 50nM). (G) Luciferase reporter assays (p6xOSE2-Luc or p6xmutOSE2-Luc reporters) of cells co-transfected with pSFFV-neo empty vector (grey bars) or pSFFV-Cx43 expression vector (black bars) along with a wild type PLCγ1 or empty vector construct and treated with vehicle (Veh) or FGF2 (10ng/ml, 4h), as above. For all luciferase assay graphs, cells transfected with pSFFV-neo empty vector are shown with grey bars, cells transfected with pSFFV-Cx43 expression vector are shown with black bars. Graphs depict mean + s.d. for a representative experiment performed in triplicate wells, n ≥ 3. For real time PCR, * p-value < 0.05. For luciferase reporter assays, * p-value < 0.05 relative to the corresponding FGF2-treated control.
The PLC inhibitor ET-18-OCH3 dose-dependently inhibited the basal and Cx43-dependent amplification of transcription from the Runx2-luciferase reporter in response to FGF2 (Fig. 1B). Unlike U73122, we did not observe a change in cell morphology in cells treated with this inhibitor. Further, the inhibitor did not affect cell viability or luciferase activity from a mutated OSE2-luciferase reporter vector (Supporting Information Fig. S1). Also, the specificity of the inhibitors used in these studies was verified by western blotting with phospho-specific antibodies (Supporting Information Fig. S2). Similarly, overexpression of a dominant negative PLCγ1 (Y783F) construct attenuated the basal FGF2 response and completely prevented the Cx43-dependent amplification, mimicking the effectiveness of a dominant negative PKCδ (PKCδ K376R) (Fig. 1C), indicating that the activation of the PLCγ and PKCδ pathways is required for both the basal and Cx43-enhanced responsiveness of a Runx2 reporter.
Next, we assessed the PLC isoform contributing to this effect. PLCγ1 was found to be more than 70-fold more abundant in MC3T3 cells than PLCγ2 as determined by quantitative real time PCR (Fig. 1D). Western blots showed that FGF2 enhanced the phosphorylation of PLCγ1 at Tyr783, indicating the activation of PLCγ1 by FGF2 (Fig. 1E). Furthermore, siRNA-mediated knockdown of PLCγ1 inhibited the basal and Cx43-mediated potentiation of Runx2 activity by FGF2 (Fig. 1F), while overexpression of wild type PLCγ1 enhanced the basal and Cx43-enahnced response of the Runx2-reporter construct to FGF2 (Fig. 1G).
Inositol polyphosphates as mediators of the Cx43-dependent potentiation of Runx2
The classic action of PLCγ1 is to cleave phosphatidylinositol-4,5-bisphosphate generating 1,2-diacyglycerol (DAG) and inositol 1,4,5-triphosphate (InsP3) converging on Ca2+-release from intracellular stores. However, PKCδ is a Ca2+-independent PKC. Consequently, we examined if the Ca2+-independent action of DAG or InsPs were involved in PKCδ activation. The DAG lipase inhibitor RHC-80267 (20μM) failed to potentiate the effects of FGF2 on the Runx2-luciferase reporter, rather a slight inhibition of the Cx43-dependent potentiation of Runx2 transcriptional activity by FGF2 was observed (Fig. 2A). Further, the classic action of InsP3 was investigated using the InsP3 receptor inhibitor 2-APB. 2-APB (50μM) did not significantly affect the basal FGF2 response of a Runx2 reporter or the potentiation of that response by Cx43 overexpression (Fig. 2B). Neither RHC-80267 nor 2-APB affected cell viability or luciferase activity from a mutated OSE2-luciferase reporter vector (Supporting Information Fig. S1).
Fig. 2.

Contribution of DAG lipase or InsP3 Receptors to the basal or Cx43-potentiated FGF2 response of a Runx2 reporter. Luciferase reporter assays were performed on cells co-transfected with p6xOSE2-Luc and pSFFV-neo empty vector (grey bars) or pSFFV-Cx43 expression vector (black bars) and treated with vehicle (Veh) or FGF2 (10ng/ml, 4h) in the presence or absence of (A) the DAG lipase inhibitor RHC-80267 (20μM) or (B) the InsP3 Receptor inhibitor 2-APB (50μM). Graphs depict means + s.d. for a representative experiment performed in triplicate wells, n = 3. * p-value < 0.05 relative to the corresponding FGF2-treated control.
Recently, higher order inositol polyphosphates and pyrophosphates, such as InsP6 and InsP7, have been shown to be important in the control of several cellular processes including apoptosis, signal transduction, vesicular trafficking, exocytosis and gene expression (39–43). These higher order InsPs are produced as a result of the sequential phosphorylation of InsP3 by a series of kinases (Fig. 3A). Accordingly, we tested if these novel inositol polyphosphate and pyrophosphate second messengers may impact the Cx43-dependent amplification of Runx2 by disrupting the expression or function of these kinases. siRNA-mediated knockdown of inositol polyphosphate multikinase (IPMK), which has 3-kinase, 5-kinase and 6-kinase activities on phosphorylated inositol substrates, markedly reduced the activity of a Runx2-luciferase reporter in response to FGF2, as well as the Cx43-dependent enhancement of this response (Fig. 3B).
Fig. 3.

Involvement of higher order inositol polyphosphates in the Cx43-dependent potentiation of Runx2 activity. (A) A schematic of the pathway by which PLC activity can yield higher order inositol polyphosphates and inositol pyrophosphates. (B) Luciferase reporter assays were performed on cells co-transfected with p6xOSE2-Luc and pSFFV-neo empty vector (grey bars) or pSFFV-Cx43 expression vector (black bars) and scrambled siRNA (SCR, 50nM) or IPMK siRNA (IPMK, 50nM) and then treated with vehicle (Veh) or FGF2 (10ng/ml, 4h). Inset, western blot of whole cell extracts from cells transfected with scrambled or IPMK targeted siRNA probed with anti-IPMK and anti-GAPDH antibodies. (C) Luciferase reporter assays were performed as above in cells treated with vehicle (Veh) or FGF2 (10ng/ml, 4h) in the presence or absence of the IP6K inhibitor TNP (5μM). (D) Quantitative real time RT-PCR of IP6K1, IP6K2 and IP6K3 in confluent cultures of MC3T3 cells. (E) Luciferase reporter assays were performed as above in cells treated with 50nM scrambled siRNA, IP6K1 siRNA or IP6K2 siRNA. Quantitative real time RT-PCR (IP6K1 and IP6K2 primers, respectively) of cells transfected with 50nM scrambled siRNA, IP6K1 siRNA or IP6K2 siRNA. For luciferase assay graphs, cells transfected with pSFFV-neo empty vector are shown with grey bars, cells transfected with pSFFV-Cx43 expression vector are shown with black bars. (F) Luciferase reporter assays were performed on cells co-transfected with p6xOSE2-Luc and pSFFV-neo empty vector (grey bars) or pSFFV-Cx43 expression vector (black bars) along with a IP6K1 expression vector, IP6K2 expression vector or an empty vector and treated with vehicle (Veh) or FGF2 (10ng/ml, 4h), as above. For all luciferase assay graphs, cells transfected with pSFFV-neo empty vector are shown with grey bars, cells transfected with pSFFV-Cx43 expression vector are shown with black bars. Graphs depict means + s.d. for a representative experiment performed in triplicate wells, n = 3. For real time PCR, * p-value < 0.05. For luciferase reporter assays, * p-value < 0.05 relative to the corresponding FGF2-treated control.
TNP (5μM), a selective chemical inhibitor of inositol hexakisphosphate kinases (IP6Ks) (44), abolished the basal and Cx43-potentiated Runx2 activity in response to FGF2 treatment relative to DMSO treated controls (Fig. 3C). TNP had no effect on cell viability or luciferase activity from a mutated OSE2-luciferase reporter vector (Supporting Information Fig. S1). Three different inositol hexakisphosphate kinases, IP6K1, IP6K2 and IP6K3 have been identified in mouse cells. Quantitative real time RT-PCR revealed that MC3T3 osteoblasts express IP6K1 and IP6K2 mRNA more than twenty-five fold more abundantly than IP6K3 (Fig. 3D). Consequently, we examined the impact of siRNA-mediated knockdown of the two more abundant IP6Ks on Runx2 transcriptional activity. Knockdown of IP6K1 inhibited the FGF2-induced Runx2 activity both basally and in response to Cx43 overexpression more potently than knockdown of IP6K2 (Fig. 3E). These data suggest that IP6K1 and to a lesser extent IP6K2 play a role in the FGF2 response, as well as the signaling cascade by which Cx43 enhances the osteoblast response to FGF2. Notably, in gain of function experiments in which IP6K1 or IP6K2 were overexpressed, they enhanced both the basal and Cx43-potentiated activity of the pOSE2-luciferase reporter following FGF2 stimulation (Fig. 3F), indicating that both IP6K1 and IP6K2 can enhance Runx2 transcriptional activity.
The nuclear translocation and association of PKCδ with Runx2 is dependent upon IP6K1
Next, we examined the molecular mechanisms by which inositol pyrophosphates may impact signaling in osteoblastic cells. Previously we had reported that, upon FGF2 stimulation, PKCδ transiently interacts with Cx43 at the plasma membrane before translocating to the nucleus (12,27). Consistent with the work of others (26,45), co-immunoprecipitaions of Runx2 and PKCδ showed that PKCδ physically interacts with Runx2 after FGF2 exposure (10ng/ml) (Fig. 4A and Supporting Information Fig. S3). Subsequently, we disrupted IP6K expression or function in MC3T3 osteoblasts and examined the nuclear translocation of PKCδ and the physical interaction of PKCδ with Runx2. Disruption of IP6K activity with TNP (5μM) blocked the FGF2-dependent interaction between PKCδ and Runx2 (Fig. 4B). Western blotting analysis of nuclear extracts from FGF2 treated cells revealed that siRNA-mediated knockdown of IP6K1 but not IP6K2 prevented the translocation of PKCδ to the nucleus (Fig. 4C).
Fig. 4.

The Nuclear translocation of PKCδ and the physical interaction of Runx2 and PKCδ require IP6K1. (A) An immunoprecipitation was performed on whole cell extracts from cells treated with 10ng/ml FGF2 for 0, 5 or 30 minutes with anti-total PKCδ, anti-phospho-PKCδ (Thr505) or anti-phospho-PKCδ (Ser643) antibodies. Subsequently, western blots of the bead fractions from these immunoprecipitation were probed with anti-Runx2 antibodies, n=2. (B) An immunoprecipitation with anti-total PKCδ antibodies was performed on whole cell extracts from cells treated with 10ng/ml FGF2 for 0 or 30 minutes in the presence or absence of 5μM TNP. The western blots were then probed with anti-Runx2 antibodies. Top, the eluted bead fraction from the immunoprecipitation; bottom, 1/10th of the input fraction. (C) Western blots of nuclear extracts from cells transfected with 50nM SCR siRNA, IP6K1 siRNA or IP6K2 siRNA and treated with 10ng/ml FGF2 for 0, 5 or 30 minutes. Blots were probed with anti-phospho-PKCδ (Thr505) or Lamin A/C antibodies (load control).
Disruption of IP6K activity suppresses osteoblast gene expression
If inositol pyrophosphates produced downstream of IP6Ks have a physiologic significance in osteoblasts, then disruption of IP6Ks should affect the expression of osteogenic genes whose expression is directly or indirectly regulated by Runx2. To test this hypothesis, we examined by quantitative real time RT-PCR the expression of several osteoblast genes in TNP (5μM, 4h) treated osteoblasts. The expression of collagen Iα1, osteocalcin and Osterix were all decreased by inhibition of IP6K activity, while Runx2 expression was unchanged (Fig. 5). The expression of these genes (collagen Iα1, osteocalcin and Osterix) remained down after treating the cells with TNP (5μM) continuously for 5 days (not shown).
Fig. 5.

Inhibition of IP6Ks reduces the expression of Runx2-regulated osteoblast genes. MC3T3 cells were treated with vehicle (DMSO) or 5μM TNP for 4 hours, prior to isolation of RNA for quantitative real time RT-PCR for collagen Iα1 (Col1a1), osteocalcin, Osterix or Runx2. * p-value < 0.05 relative to the corresponding Veh-treated control, n = 3.
DISCUSSION
In this study, we show that the PLCγ1/IPMK/IP6K/PKCδ pathway is required for the modulation of Runx2 activity by FGF2, as well as for the Cx43-mediated potentiation of that response. These data demonstrate for the first time that higher order InsPs (e.g., InsP5 and InsP7), which are products of IPMK and IP6K activities, are mediators of the FGF2 response in osteoblastic cells. Further, this is the first study to show that IP6Ks regulate Runx2 activity and affects the expression of osteoblast genes.
Notably, the impact of disruption of the PLCγ1/IPMK/IP6K/PKCδ pathway consistently has a stronger effect on the Cx43 amplified response (greater fold inhibition) that it does on the response in the absence of overexpressed Cx43, strongly suggesting that a factor in this pathway mediates the effects of Cx43 on Runx2. Further, since we have previously shown that the effects of Cx43 on FGF2-signaling requires functional gap junctional communication (28), this hints that inositol polyphosphates produced by the action of IPMK and IP6K1 may be biologically relevant second messengers communicated by Cx43. Alternately, these inositol polyphosphates may simply lie along the pathway in which gap junctional communication participates without necessarily being directly communicated. Future studies will need to be done to directly test the ability of higher order InsPs to transverse gap junctions and stimulate signaling in a coupled cell. That said, InsPs like InsP7 do meet the necessary criteria for a molecule capable of passing through a Cx43 channel. InsPs are soluble in cytoplasm and could pass through the aqueous pore of a gap junction channel. Indeed, the molecular weight and charge of InsPs are consistent with the known permeability of Cx43-containing gap junctions, which permit the passage of molecules under ~1000 molecular weight with a preference for negatively charged molecules (46). The molecular weight of the backbone inositol (C6H12O6) is ~180, while the pyrophosphorylated InsP7 has a molecular weight of ~740, making them capable of being transmitted through Cx43 gap junctions. Indeed, InsP3 is communicated through Cx43 gap junctions (47).
Related to the permeability and size of a gap junction communicated second messenger, the synergistic activities of Cx43 on FGF2 signaling cannot be recapitulated by Cx45, which has the opposing effect of Cx43 on signaling, gene transcription and Runx2 activity in osteoblasts (12–14,48,49). Cx45 forms a gap junction with a smaller pore size, reduced permeability and greater selectively for positively charged molecules (50,51) and, thus, is unlikely to support communication of the large, negatively charged higher order InsPs. This reinforces the hypothesis that InsPs are a biologically relevant second messenger that is propagated by Cx43 channels.
It is worth mentioning that we do not intend to suggest a lack of importance of other inositol polyphosphates (or other second messengers) in gap junction communication, but our current data support a role for IP6Ks and specifically IP6K1. It is unclear if the minimal effect of IP6K2 knockdown on FGF2 signaling and the Cx43-dependent regulation of Runx2 transcriptional activity is because IP6K2 plays only a minor role in this cascade or if knockdown was insufficient to expose an effect on Runx2. Notably, overexpression of IP6K2 enhanced both the FGF2 response of Runx2 and the potentiation of this response by Cx43 overexpression, indicating that IP6K2 can contribute to Runx2 regulation even if it may not required for this action. Further, it is possible that IP6K2 (and IP6K1) may play important roles in Cx43 signaling independent of Runx2 activity. Indeed, IP6K2 is known to be an inducer of apoptosis (52–54), an outcome associated with loss of Cx43 function in bone cells (9,16,55). Future studies will require examination of these effects in primary osteoblasts isolated form IP6K1 and IP6K2 null mice, as we were unable to achieve greater than 50% knockdown of IP6K1 or IP6K2 with various siRNAs.
Based on these data and our other published data, we propose a speculative model (Fig. 6) in which FGF2 binds to its cognate FGFR in one cell, activating PLCγ1, leading to DAG and InsP3 generation. The action of IMPK and IP6Ks convert the InsP3 into higher order InsPs. The action of IP6K1 and the second messenger InsP7 are required for the translocation of PKCδ into the nucleus, where it interacts with Runx2, promoting the expression of Runx2-responsive osteogenic genes. Further, InsP7 can diffuse through Cx43 gap junctions to initiate PKCδ activation and Runx2-dependent transcription in an adjacent cell. In fact, we have shown that PKCδ is transiently recruited to the Cx43 channel prior to nuclear translocation following FGF2 treatment (27). Thus, Cx43 is a docking platform for the signal complex that responds to the communicated second messenger. The cell-to-cell communication of the second messengers causes the population of osteoblasts to respond more robustly to the extracellular cue (FGF2) than would happen in the absence of gap junctional communication. In contrast, when Cx43 expression is reduced, such as in Cx43 knockout models, the response of a population of cells is blunted, as information is not shared among the cells. As a result, there is a reduction in osteoblast activation, gene expression and bone formation by the cell population. Such an effect may underlie the skeletal phenotype of the Cx43 deletion models. These concepts are consistent with our previous data, revealing an increase in the number of osteoblastic cells responding to a cue in the presence of overexpressed Cx43 (12,28) and the need for Cx43 function and direct cell-to-cell contacts for the potentiation of FGF2 responses by these cells (28). As mentioned above, future studies will be needed to definitively establish that InsP7 is passed through Cx43 gap junction channels, and that it is required to re-activate signaling in the adjacent, gap junction coupled cell.
Fig. 6.
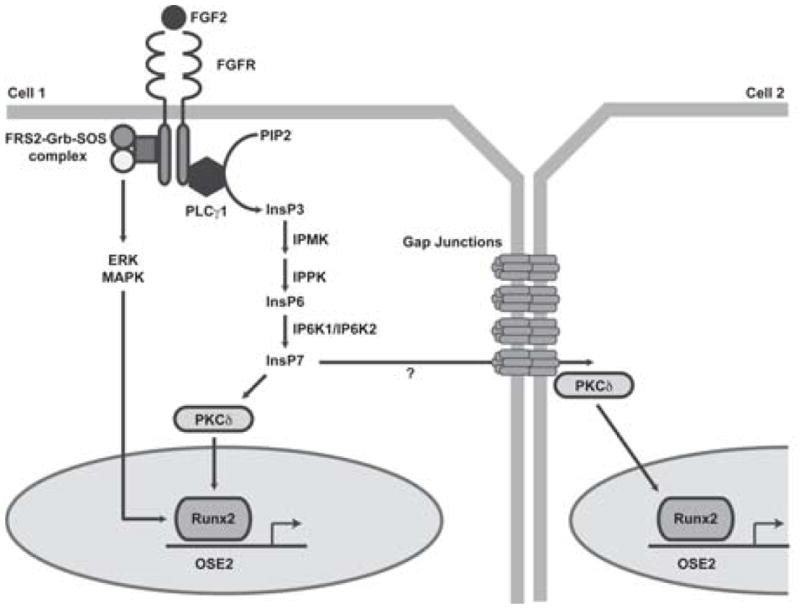
Model of Cx43 potentiated Runx2 activity via an inositol pyrophosphate second messenger following FGF2 stimulation. Upon binding to its receptor (FGFR) in one cell, FGF2 activates PLCγ1, generating DAG and InsP3 (IP3). Subsequently, the activity of IPMK and IP6K1 leads to the production of inositol polyphosphates and pyrophosphates, such as InsP6 (IP6) and InsP7 (IP7). The InsPs activate PKCδ, which translocates to the nucleus where it interacts with Runx2, increasing its transcriptional activity and driving the expression of osteoblast genes. In addition, we speculate that these small, water soluble second messengers may be communicated to adjacent cells via Cx43 containing gap junctions. In the coupled cell, PKCδ, which is locally recruited to the Cx43-containing gap junction channel, can re-initiate signaling in this cell, independent of direct stimulation by FGF2, resulting in a potentiation of the response among coupled cells.
It is possible that this mechanism can be extrapolated to other extracellular cues, such as the responsiveness to mechanical strain, growth factor and hormonal signaling. Indeed, the conditional genetic ablation of Cx43 in cells of the osteoblast lineage makes these mice refractory to the anabolic effects of intermittent parathyroid hormone (5), as well as mechanical loading (56) and unloading (7). Notably, a similar knockout model has reported an increase in the osteoanabolic response to mechanical loading in hOCN-Cre driven Cx43 conditional knockout mice (10), suggesting considerable complexity in the signals communicated by Cx43. As we have mentioned, it is likely that multiple second messengers and signaling pathways lie downstream of Cx43 activity, some anabolic and some catabolic to bone. Further, these second messengers and signaling pathways will undoubtedly be dependent upon the nature of the extracellular cue. InsP signaling likely represents only a subset of these pathways.
In total, these data extend our understanding of FGF2 signaling and the convergence of the PLCγ1/IPMK/IP6K/PKCδ pathway on Runx2 activity in osteoblasts. Also, these data indicate a role for inositol polyphosphates in osteogenic differentiation. Further, these data show that IPMK and IP6K1 are required for the Cx43-dependent potentiation of Runx2 activity in response to FGF2, suggesting that higher order InsPs, like InsP7, are likely biologically relevant second messengers that are communicated by Cx43 gap junctions in osteoblasts to regulate the expression of osteoblast genes. This role may extend to osteogenic cues beyond FGF2 and could explain the defective osteogenesis and skeletal phenotypes observed in many of the Cx43 genetic deletion or mutation models.
Supplementary Material
Fig. S1. (A) Cx43 and FGF2 regulated transcription from an intact (pOSE2) but not a mutated (mutpOSE2) luciferase reported construct. Likewise, neither U73122 (5μM) ET-18-OCH3 (20μM), 2-APB (50μM), RHC-80267 (20μM), Rottlerin (5μM) or TNP (5μM) regulated transcription from the mutated Runx2 reporter (mutpOSE2). (B) While there was a mild effect of FGF2 on relative cell number/viability, as determined by CCK-8 assay, this effect was not significantly impacted by treatment with any of the inhibitors used in this study nor by Cx43 overexpression.
Fig. S2. Assessment of Pathway inhibitor specificity. Western blots were performed on whole cell extracts of MC3T3 cells treated with PMA (100nM) for 15 minutes to stimulate the PKC pathway in the presence of the chemical inhibitors of the classic and novel PKC pathway and the IP6K inhibitor (TNP) or the vehicle diluent (DMSO), as indicated. Western blots were probed with antibodies to p-panPKC (Ser 660), p-PKCδ (Thr 505), p-ERK1/2 (Thr202/Tyr204), as indicated. GAPDH antibodies were used as a load control. Each panel is from a single membrane and single exposure, though not all lanes were contiguous. As expected, ET-18-OCH3, Rottlerin and TNP reduced phosphorylation of PKCs (pan) including PKCδ, while RHC-80267 maintained or even enhanced p-panPKC and p-PKCδ levels, respectively. 2-APB did not affect the Ca2+-independent PKCδ phosphorylation, but did reduce pan-PKC phosphorylation levels. Conversely, minimal impact of the any of the inhibitors was seen on the ERK MAPK cascade, suggesting specificity of the inhibitors for the PKC pathway.
Fig. S3. Co-immunoprecipitation of PKCδ and Runx2. An immunoprecipitation with non-immune IgG, PKCδ or Runx2 antibodies was performed in whole cell extracts of osteoblasts treated for 0, 5 or 30 minutes with FGF2 (10ng/ml). Subsequently the bead and input fractions were subjected to western blotting and probed with anti-PKCδ antibodies. Note, the FGF2-dependent co-precipitation of PKCδ with Runx2 antibodies in the last three lanes.
Acknowledgments
This work was funded in part by grants from the National Institute of Health (NIAMS/R01 AR05719 and AR063631) awarded to J.P.S.
Footnotes
DISCLOSURES
All authors state that they have no conflicts of interest.
References
- 1.Civitelli R. Cell-cell communication in the osteoblast/osteocyte lineage. Arch Biochem Biophys. 2008;473(2):188–92. doi: 10.1016/j.abb.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batra N, Kar R, Jiang JX. Gap junctions and hemichannels in signal transmission, function and development of bone. Biochim Biophys Acta. 2012;1818(8):1909–18. doi: 10.1016/j.bbamem.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, Jabs EW. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72(2):408–18. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lecanda F, Warlow PM, Sheikh S, Furlan F, Steinberg TH, Civitelli R. Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J Cell Biol. 2000;151(4):931–44. doi: 10.1083/jcb.151.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung DJ, Castro CH, Watkins M, Stains JP, Chung MY, Szejnfeld VL, Willecke K, Theis M, Civitelli R. Low peak bone mass and attenuated anabolic response to parathyroid hormone in mice with an osteoblast-specific deletion of connexin43. J Cell Sci. 2006;119(Pt 20):4187–98. doi: 10.1242/jcs.03162. [DOI] [PubMed] [Google Scholar]
- 6.Dobrowolski R, Sasse P, Schrickel JW, Watkins M, Kim JS, Rackauskas M, Troatz C, Ghanem A, Tiemann K, Degen J, Bukauskas FF, Civitelli R, Lewalter T, Fleischmann BK, Willecke K. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum Mol Genet. 2008;17(4):539–54. doi: 10.1093/hmg/ddm329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimston SK, Goldberg DB, Watkins M, Brodt MD, Silva MJ, Civitelli R. Connexin43 deficiency reduces the sensitivity of cortical bone to the effects of muscle paralysis. J Bone Miner Res. 2011;26(9):2151–60. doi: 10.1002/jbmr.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watkins M, Grimston SK, Norris JY, Guillotin B, Shaw A, Beniash E, Civitelli R. Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol Biol Cell. 2011;22(8):1240–51. doi: 10.1091/mbc.E10-07-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bivi N, Condon KW, Allen MR, Farlow N, Passeri G, Brun LR, Rhee Y, Bellido T, Plotkin LI. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J Bone Miner Res. 2011 doi: 10.1002/jbmr.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Paul EM, Sathyendra V, Davison A, Sharkey N, Bronson S, Srinivasan S, Gross TS, Donahue HJ. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS One. 2011;6(8):e23516. doi: 10.1371/journal.pone.0023516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flenniken AM, Osborne LR, Anderson N, Ciliberti N, Fleming C, Gittens JE, Gong XQ, Kelsey LB, Lounsbury C, Moreno L, Nieman BJ, Peterson K, Qu D, Roscoe W, Shao Q, Tong D, Veitch GI, Voronina I, Vukobradovic I, Wood GA, Zhu Y, Zirngibl RA, Aubin JE, Bai D, Bruneau BG, Grynpas M, Henderson JE, Henkelman RM, McKerlie C, Sled JG, Stanford WL, Laird DW, Kidder GM, Adamson SL, Rossant J. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development. 2005;132(19):4375–86. doi: 10.1242/dev.02011. [DOI] [PubMed] [Google Scholar]
- 12.Lima F, Niger C, Hebert C, Stains JP. Connexin43 potentiates osteoblast responsiveness to fibroblast growth factor 2 via a protein kinase C-delta/Runx2-dependent mechanism. Mol Biol Cell. 2009;20(11):2697–708. doi: 10.1091/mbc.E08-10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stains JP, Lecanda F, Screen J, Towler DA, Civitelli R. Gap junctional communication modulates gene transcription by altering the recruitment of Sp1 and Sp3 to connexin-response elements in osteoblast promoters. J Biol Chem. 2003;278(27):24377–87. doi: 10.1074/jbc.M212554200. [DOI] [PubMed] [Google Scholar]
- 14.Lecanda F, Towler DA, Ziambaras K, Cheng SL, Koval M, Steinberg TH, Civitelli R. Gap junctional communication modulates gene expression in osteoblastic cells. Mol Biol Cell. 1998;9(8):2249–58. doi: 10.1091/mbc.9.8.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bivi N, Lezcano V, Romanello M, Bellido T, Plotkin LI. Connexin43 interacts with betaarrestin: A pre-requisite for osteoblast survival induced by parathyroid hormone. J Cell Biochem. 2011;112(10):2920–30. doi: 10.1002/jcb.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plotkin LI, Manolagas SC, Bellido T. Transduction of cell survival signals by connexin-43 hemichannels. J Biol Chem. 2002;277(10):8648–57. doi: 10.1074/jbc.M108625200. [DOI] [PubMed] [Google Scholar]
- 17.Li Z, Zhou Z, Saunders MM, Donahue HJ. Modulation of connexin43 alters expression of osteoblastic differentiation markers. Am J Physiol Cell Physiol. 2006;290(4):C1248–55. doi: 10.1152/ajpcell.00428.2005. [DOI] [PubMed] [Google Scholar]
- 18.McLachlan E, Plante I, Shao Q, Tong D, Kidder GM, Bernier SM, Laird DW. ODDD-linked Cx43 mutants reduce endogenous Cx43 expression and function in osteoblasts and inhibit late stage differentiation. J Bone Miner Res. 2008;23(6):928–38. doi: 10.1359/jbmr.080217. [DOI] [PubMed] [Google Scholar]
- 19.Gramsch B, Gabriel HD, Wiemann M, Grummer R, Winterhager E, Bingmann D, Schirrmacher K. Enhancement of connexin 43 expression increases proliferation and differentiation of an osteoblast-like cell line. Exp Cell Res. 2001;264(2):397–407. doi: 10.1006/excr.2000.5145. [DOI] [PubMed] [Google Scholar]
- 20.Marie PJ. Fibroblast growth factor signaling controlling bone formation: an update. Gene. 2012;498(1):1–4. doi: 10.1016/j.gene.2012.01.086. [DOI] [PubMed] [Google Scholar]
- 21.Marie PJ, Coffin JD, Hurley MM. FGF and FGFR signaling in chondrodysplasias and craniosynostosis. J Cell Biochem. 2005;96(5):888–96. doi: 10.1002/jcb.20582. [DOI] [PubMed] [Google Scholar]
- 22.Mohammadi M, Honegger AM, Rotin D, Fischer R, Bellot F, Li W, Dionne CA, Jaye M, Rubinstein M, Schlessinger J. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-gamma 1. Mol Cell Biol. 1991;11(10):5068–78. doi: 10.1128/mcb.11.10.5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sierra OL, Cheng SL, Loewy AP, Charlton-Kachigian N, Towler DA. MINT, the Msx2 interacting nuclear matrix target, enhances Runx2-dependent activation of the osteocalcin fibroblast growth factor response element. J Biol Chem. 2004;279(31):32913–23. doi: 10.1074/jbc.M314098200. [DOI] [PubMed] [Google Scholar]
- 24.Willis DM, Loewy AP, Charlton-Kachigian N, Shao JS, Ornitz DM, Towler DA. Regulation of osteocalcin gene expression by a novel Ku antigen transcription factor complex. J Biol Chem. 2002;277(40):37280–91. doi: 10.1074/jbc.M206482200. [DOI] [PubMed] [Google Scholar]
- 25.Xiao G, Jiang D, Gopalakrishnan R, Franceschi RT. Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J Biol Chem. 2002;277(39):36181–7. doi: 10.1074/jbc.M206057200. [DOI] [PubMed] [Google Scholar]
- 26.Kim HJ, Kim JH, Bae SC, Choi JY, Kim HJ, Ryoo HM. The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. J Biol Chem. 2003;278(1):319–26. doi: 10.1074/jbc.M203750200. [DOI] [PubMed] [Google Scholar]
- 27.Niger C, Hebert C, Stains JP. Interaction of connexin43 and protein kinase C-delta during FGF2 signaling. BMC Biochem. 2010;11:14. doi: 10.1186/1471-2091-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niger C, Buo AM, Hebert C, Duggan BT, Williams MS, Stains JP. ERK acts in parallel to PKCdelta to mediate the connexin43-dependent potentiation of Runx2 activity by FGF2 in MC3T3 osteoblasts. Am J Physiol Cell Physiol. 2012;302(7):C1035–44. doi: 10.1152/ajpcell.00262.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Illies C, Gromada J, Fiume R, Leibiger B, Yu J, Juhl K, Yang SN, Barma DK, Falck JR, Saiardi A, Barker CJ, Berggren PO. Requirement of inositol pyrophosphates for full exocytotic capacity in pancreatic beta cells. Science. 2007;318(5854):1299–302. doi: 10.1126/science.1146824. [DOI] [PubMed] [Google Scholar]
- 30.Gupta RR, Yoo DJ, Hebert C, Niger C, Stains JP. Induction of an osteocyte-like phenotype by fibroblast growth factor-2. Biochem Biophys Res Commun. 2010;402(2):258–64. doi: 10.1016/j.bbrc.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niger C, Lima F, Yoo DJ, Gupta RR, Buo AM, Hebert C, Stains JP. The transcriptional activity of osterix requires the recruitment of Sp1 to the osteocalcin proximal promoter. Bone. 2011;49(4):683–92. doi: 10.1016/j.bone.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol. 1995;15(4):1858–69. doi: 10.1128/mcb.15.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beyer EC, Paul DL, Goodenough DA. Connexin43: a protein from rat heart homologous to a gap junction protein from liver. J Cell Biol. 1987;105(6 Pt 1):2621–9. doi: 10.1083/jcb.105.6.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeBell K, Graham L, Reischl I, Serrano C, Bonvini E, Rellahan B. Intramolecular regulation of phospholipase C-gamma1 by its C-terminal Src homology 2 domain. Mol Cell Biol. 2007;27(3):854–63. doi: 10.1128/MCB.01400-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soh JW, Weinstein IB. Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J Biol Chem. 2003;278(36):34709–16. doi: 10.1074/jbc.M302016200. [DOI] [PubMed] [Google Scholar]
- 36.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, Zhao JJ, Yang X, Alkan O, Kim S, Harris JL, Wilson CJ, Myer VE, Finan PM, Root DE, Roberts TM, Golub T, Flaherty KT, Dummer R, Weber BL, Sellers WR, Schlegel R, Wargo JA, Hahn WC, Garraway LA. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dyer BW, Ferrer FA, Klinedinst DK, Rodriguez R. A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal Biochem. 2000;282(1):158–61. doi: 10.1006/abio.2000.4605. [DOI] [PubMed] [Google Scholar]
- 38.Niger C, Howell FD, Stains JP. Interleukin-1beta increases gap junctional communication among synovial fibroblasts via the extracellular-signal-regulated kinase pathway. Biol Cell. 2010;102(1):37–49. doi: 10.1042/BC20090056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakraborty A, Kim S, Snyder SH. Inositol pyrophosphates as mammalian cell signals. Sci Signal. 2011;4(188):re1. doi: 10.1126/scisignal.2001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsui MM, York JD. Roles of inositol phosphates and inositol pyrophosphates in development, cell signaling and nuclear processes. Adv Enzyme Regul. 2010;50(1):324–37. doi: 10.1016/j.advenzreg.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alcazar-Roman AR, Wente SR. Inositol polyphosphates: a new frontier for regulating gene expression. Chromosoma. 2008;117(1):1–13. doi: 10.1007/s00412-007-0126-4. [DOI] [PubMed] [Google Scholar]
- 42.Azevedo C, Szijgyarto Z, Saiardi A. The signaling role of inositol hexakisphosphate kinases (IP6Ks) Adv Enzyme Regul. 2011;51(1):74–82. doi: 10.1016/j.advenzreg.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 43.Barker CJ, Illies C, Gaboardi GC, Berggren PO. Inositol pyrophosphates: structure, enzymology and function. Cell Mol Life Sci. 2009;66(24):3851–71. doi: 10.1007/s00018-009-0115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Padmanabhan U, Dollins DE, Fridy PC, York JD, Downes CP. Characterization of a selective inhibitor of inositol hexakisphosphate kinases: use in defining biological roles and metabolic relationships of inositol pyrophosphates. J Biol Chem. 2009;284(16):10571–82. doi: 10.1074/jbc.M900752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim BG, Kim HJ, Park HJ, Kim YJ, Yoon WJ, Lee SJ, Ryoo HM, Cho JY. Runx2 phosphorylation induced by fibroblast growth factor-2/protein kinase C pathways. Proteomics. 2006;6(4):1166–74. doi: 10.1002/pmic.200500289. [DOI] [PubMed] [Google Scholar]
- 46.Elfgang C, Eckert R, Lichtenberg-Frate H, Butterweck A, Traub O, Klein RA, Hulser DF, Willecke K. Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J Cell Biol. 1995;129(3):805–17. doi: 10.1083/jcb.129.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niessen H, Harz H, Bedner P, Kramer K, Willecke K. Selective permeability of different connexin channels to the second messenger inositol 1,4,5-trisphosphate. J Cell Sci. 2000;113 ( Pt 8):1365–72. doi: 10.1242/jcs.113.8.1365. [DOI] [PubMed] [Google Scholar]
- 48.Stains JP, Civitelli R. Gap junctions regulate extracellular signal-regulated kinase signaling to affect gene transcription. Mol Biol Cell. 2005;16(1):64–72. doi: 10.1091/mbc.E04-04-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Zhou Z, Yellowley CE, Donahue HJ. Inhibiting gap junctional intercellular communication alters expression of differentiation markers in osteoblastic cells. Bone. 1999;25(6):661–6. doi: 10.1016/s8756-3282(99)00227-6. [DOI] [PubMed] [Google Scholar]
- 50.Veenstra RD. Size and selectivity of gap junction channels formed from different connexins. J Bioenerg Biomembr. 1996;28(4):327–37. doi: 10.1007/BF02110109. [DOI] [PubMed] [Google Scholar]
- 51.Steinberg TH, Civitelli R, Geist ST, Robertson AJ, Hick E, Veenstra RD, Wang HZ, Warlow PM, Westphale EM, Laing JG, et al. Connexin43 and connexin45 form gap junctions with different molecular permeabilities in osteoblastic cells. EMBO J. 1994;13(4):744–50. doi: 10.1002/j.1460-2075.1994.tb06316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chakraborty A, Koldobskiy MA, Sixt KM, Juluri KR, Mustafa AK, Snowman AM, van Rossum DB, Patterson RL, Snyder SH. HSP90 regulates cell survival via inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A. 2008;105(4):1134–9. doi: 10.1073/pnas.0711168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koldobskiy MA, Chakraborty A, Werner JK, Jr, Snowman AM, Juluri KR, Vandiver MS, Kim S, Heletz S, Snyder SH. p53-mediated apoptosis requires inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A. 2010;107(49):20947–51. doi: 10.1073/pnas.1015671107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagata E, Luo HR, Saiardi A, Bae BI, Suzuki N, Snyder SH. Inositol hexakisphosphate kinase-2, a physiologic mediator of cell death. J Biol Chem. 2005;280(2):1634–40. doi: 10.1074/jbc.M409416200. [DOI] [PubMed] [Google Scholar]
- 55.Plotkin LI, Lezcano V, Thostenson J, Weinstein RS, Manolagas SC, Bellido T. Connexin 43 is required for the anti-apoptotic effect of bisphosphonates on osteocytes and osteoblasts in vivo. J Bone Miner Res. 2008;23(11):1712–21. doi: 10.1359/JBMR.080617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grimston SK, Brodt MD, Silva MJ, Civitelli R. Attenuated response to in vivo mechanical loading in mice with conditional osteoblast ablation of the connexin43 gene (Gja1) J Bone Miner Res. 2008;23(6):879–86. doi: 10.1359/JBMR.080222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (A) Cx43 and FGF2 regulated transcription from an intact (pOSE2) but not a mutated (mutpOSE2) luciferase reported construct. Likewise, neither U73122 (5μM) ET-18-OCH3 (20μM), 2-APB (50μM), RHC-80267 (20μM), Rottlerin (5μM) or TNP (5μM) regulated transcription from the mutated Runx2 reporter (mutpOSE2). (B) While there was a mild effect of FGF2 on relative cell number/viability, as determined by CCK-8 assay, this effect was not significantly impacted by treatment with any of the inhibitors used in this study nor by Cx43 overexpression.
Fig. S2. Assessment of Pathway inhibitor specificity. Western blots were performed on whole cell extracts of MC3T3 cells treated with PMA (100nM) for 15 minutes to stimulate the PKC pathway in the presence of the chemical inhibitors of the classic and novel PKC pathway and the IP6K inhibitor (TNP) or the vehicle diluent (DMSO), as indicated. Western blots were probed with antibodies to p-panPKC (Ser 660), p-PKCδ (Thr 505), p-ERK1/2 (Thr202/Tyr204), as indicated. GAPDH antibodies were used as a load control. Each panel is from a single membrane and single exposure, though not all lanes were contiguous. As expected, ET-18-OCH3, Rottlerin and TNP reduced phosphorylation of PKCs (pan) including PKCδ, while RHC-80267 maintained or even enhanced p-panPKC and p-PKCδ levels, respectively. 2-APB did not affect the Ca2+-independent PKCδ phosphorylation, but did reduce pan-PKC phosphorylation levels. Conversely, minimal impact of the any of the inhibitors was seen on the ERK MAPK cascade, suggesting specificity of the inhibitors for the PKC pathway.
Fig. S3. Co-immunoprecipitation of PKCδ and Runx2. An immunoprecipitation with non-immune IgG, PKCδ or Runx2 antibodies was performed in whole cell extracts of osteoblasts treated for 0, 5 or 30 minutes with FGF2 (10ng/ml). Subsequently the bead and input fractions were subjected to western blotting and probed with anti-PKCδ antibodies. Note, the FGF2-dependent co-precipitation of PKCδ with Runx2 antibodies in the last three lanes.