

Case Summary
History
A 42-year old Caucasian male was referred to the National Institutes of Health for evaluation. His symptoms began approximately 10 years prior to referral with pituitary dysfunction that manifested as diabetes insipidus, secondary hypogonadism and hyperprolactinemia, treated with desmopressin and testosterone gel. Eight years later, he began to experience vertigo, dizziness, diplopia, and difficulty focusing and tracking. This was followed by progressive left-sided weakness, poor coordination and balance difficulties. Magnetic resonance imaging (MRI) without contrast revealed multiple non-specific white matter lesions. Subsequent evaluation failed to identify an underlying infection or inflammatory etiology. Systemic corticosteroid therapy was initiated with minimal improvement. The patient was tapered off corticosteroids two months later and noted new asymptomatic skin lesions on his bilateral upper and lower extremities, leading to re-initiation of systemic steroids and introduction of interferon alpha. He denied fevers, chills, weight loss or difficulty breathing.
Physical Examination
The patient was a well appearing Caucasian male. Cutaneous examination was notable for approximately twenty 2-3 mm, firm, reddish-brown papules scattered on the trunk and extremities, with greater density on the anterior thighs (Fig 1). Diascopy revealed a yellowish hue. Ophthalmologic examination was notable for nystagmus, saccadic pursuit and mild dysmetria. Neurologic examination revealed unsteady tandem gait, abnormal finger-to-nose pointing and left-sided dysdiadochokinesia. Left arm strength was diminished (4/5). The oral mucosa was unremarkable. No lymphadenopathy was appreciated.
Figure 1.


Cutaneous Erdheim-Chester disease. Isolated red-brown papule on the thigh (A); close-up image (B)
Histopathology
A punch skin biopsy of a papule on the left thigh was obtained, and histology revealed a moderately dense dermal infiltrate of epithelioid histiocytes characterized by rounded to slightly elongated nuclei with inconspicuous nucleoli and moderate to abundant eosinophilic cytoplasm (Fig 2A). CD68 and CD163 immunochemistry highlighted the lesional histiocytes (Fig 2B). Lesional cells also showed strong, diffuse staining for Factor XIIIa (Fig 2C). CD1a and S-100 immunostains were negative.
Figure 2.



Histology findings in cutaneous Erdheim-Chester disease. H&E stained tissue section revealed a moderately dense infiltrate of epithelioid histiocytes in the dermis. A mild superficial perivascular lymphocytic inflammatory infiltrate is present (A) (original magnification, 200x). CD163 immunohistochemistry (B) (original magnification, 200x). Factor XIIIa immohistochemistry (C) (original magnification, 200x).
Other significant diagnostic tests
Radiographs of the upper and lower extremities revealed sclerotic lesions involving the metaphyseal and diaphyseal regions of the long bones with sparing of the epiphyses. Positron emission tomography - computed tomography (PET/CT) scan showed multiple foci of increased uptake in the lower extremity, including the bilateral femurs and tibiae corresponding to areas of sclerosis on CT scan. Multiple similar areas of abnormal uptake were detected on whole body bone scan (Fig 3). MRI of the brain showed small focal white matter hyperintensities of the cerebral hemispheres, as well as signal abnormalities in the pons and cerebellum (Fig 4). Chest CT did not show evidence of pulmonary disease. Transthoracic echocardiogram and serum lipid panel were unremarkable.
Figure 3.
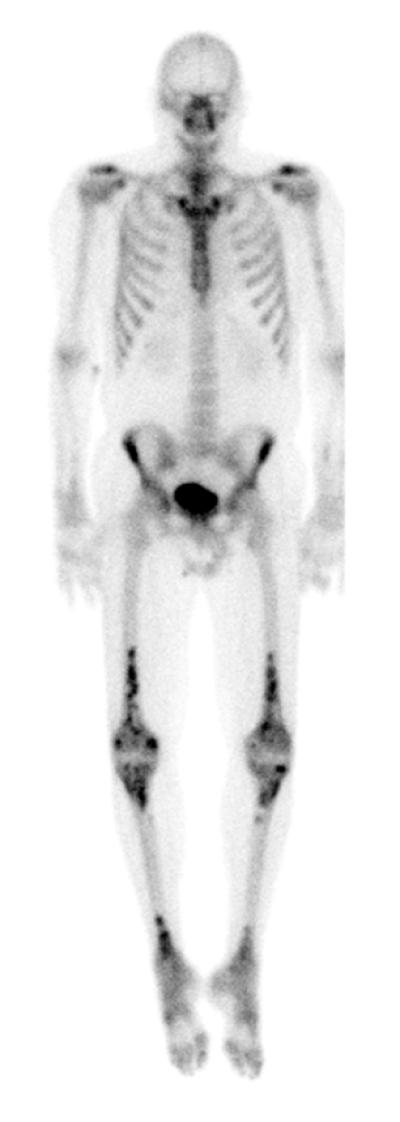
Bony involvement in Erdheim-Chester disease. Whole body bone scan after the administration of Technitium-99 showing several areas of abnormal increased uptake in the skeleton. Most prominent is bilateral heterogeneous increased uptake in the distal half of both femurs extending to the condyles, in the proximal portion of the tibias, and in the distal portion of the tibias.
Figure 4.

CNS involvement in Erdheim-Chester disease. Brain MRI showing include extensive patchy signal abnormality throughout the pons extending into the middle cerebellar peduncles and numerous small foci of signal abnormality throughout the white matter (arrows).
Diagnosis
Erdheim-Chester Disease (ECD) with cutaneous, skeletal, and central nervous system (CNS) involvement
Follow-up
The patient’s medical condition continued to deteriorate over the next several months despite treatment with peginterferon alfa 2a 135 mcg/week. Whereas he was walking independently during his first visit, one year later the patient required the use of a wheelchair or walker at all times. Neuro-ophthalmologic evaluation revealed saccadic dysmetria and end gaze nystagmus consistent with cerebellar disease as well as vestibule-ocular reflex deficit which correlated with brainstem involvement. Disease progression in the pons, brainstem and cerebellum was confirmed by radiographic imaging. All other organ systems involved were stable in comparison to the patient’s first visit to the NIH.
Subsequent to the patient’s first evaluation at the NIH, Haroche et al. reported the BRAF V600E mutation in 54% of patients with Erdheim-Chester disease, but not in other forms of non-Langerhans cell histiocytosis.1 Based on this new finding, molecular testing was performed on the patient’s biopsy specimen. DNA was extracted from paraffin-embedded tissue sections using the Qiagen QIAamp DNA FFPE Tissue Kit. The percentage of tumor cells was ~20%. The DNA was subjected to COLD-PCR using a single primer set encompassing codons 599-601 of the BRAF gene. The product was subjected to pyrosequencing on a Qiagen PyroMark Q24 system. A c.1799 T>A (p.V600E) mutation was detected. All positive and negative control reactions were adequate.
Discussion
ECD is a rare multisystem disorder of non-Langerhans cell histiocytes described by Jacob Erdheim and William Chester in 1930.2, 3 The disease course is highly variable, ranging from asymptomatic bone lesions to life-threatening multi-organ involvement2, 3 and is associated with an overall 5-year survival of 68%.4 Due to the varied clinical presentation, the differential diagnosis is broad (Table I) and diagnosis requires a high index of suspicion and integration of clinical, radiographic and histologic findings.2, 4,5, 6 Skeletal lesions primarily affecting the lower limbs are the most common manifestation of the disease,3, 4 leading to bone pain in approximately one-half of patients.3 The radiographic features of bone involvement are specific and demonstrate symmetric sclerosis involving the distal portion of the long bones.3, 7 Bone scintigraphy, PET and MRI may be used to evaluate osseous structures as well.2 In 5-8% of patients, lytic bone lesions develop which simulates Langerhans cell histiocytosis (LCH), myeloma or metastatic disease if a complete skeletal survey is not performed.3
Table I.
Differential Diagnosis of Erdheim-Chester disease
| Xanthoma disseminatum |
| Juvenile Xanthogranuloma |
| Langerhans cell histiocytosis |
| Sinus histiocytosis with massive lymphadenopathy |
| Sarcoidosis |
| Amyloidosis |
| Gaucher disease |
| Niemann-Pick disease |
| Mastocytosis |
| Paget’s disease |
| Fluoride intoxication |
| Adult progressive diaphysial dysplasia |
| Whipple disease |
| Mucopolysaccharidoses |
| Hermansky-Pudlak syndrome |
| Malakoplakia |
Table from Caputo et al. J Am Acad Dermatol. 20076
Almost any organ system is susceptible to histiocytic infiltration and nearly all (98%) patients with ECD exhibit extraskeletal manifestations.4 Cutaneous manifestations develop in approximately one-third of patients but are often non-specific. The most commonly reported skin findings are xanthomatous lesions 3 which may be clinically indistinguishable from the lesions of xanthoma disseminatum and papular xanthoma.6 Other reported cutaneous signs are pruritic eruptions, cutaneous masses,3 yellow-red papules, nodules, indurated erythematous plaques5 and infiltration of the vulva and clitoris.2 In addition to the skin, the heart, lungs, central nervous system, endocrine glands2 and gastrointestinal system8 may be affected. Infiltration of the retroperitoneum can lead to bilateral hydronephrosis4 and sheathing of the aorta.2 The most common causes of death are heart failure, respiratory distress, pulmonary fibrosis3 and CNS disease.4
Neurologic involvement can lead to significant functional impairment and is an important prognostic factor since it has been shown to be an independent predictor of mortality.4 Diabetes insipidus and exophthalmos are two characteristic signs of intracranial involvement that occur secondary to histiocytic infiltration of the pituitary and retro-orbital tissues, respectively, and are found in approximately one-third of patients.3 Non-LCH should be considered in the differential diagnosis of any patient with unexplained diabetes insipidus.
Histology is essential and is characterized by xanthomatous or xanthogranulomatous infiltration of various tissues with lipid-laden macrophages3 that display immunoreactivity with CD68 and factor XIIIa.9, 10 ECD can be differentiated from LCH by the lack of S100 and CD1a staining.2, 3 Ultrastructural examination is notable for the absence of Birbeck granules, a cardinal feature of Langerhans cells.3
The pathogenesis of ECD is poorly understood and studies are limited by small numbers of patients.2 Tran et al. reported elevated circulating levels of interleukin-1beta, IL-6 and tumor necrosis factor-alpha (TNFα) in a pediatric patient with ECD.11 Dagna et al. assessed spontaneous and stimulated cytokine release in isolated mononuclear cells from a biopsy specimen of a patient with ECD and found spontaneous secretion of IL-6 and IL-8, as well as stimulated production of TNFα.10 The presence of IL-8, a chemoattractant for polymorphonuclear cells and monocytes, was a novel finding. Similarly, Haroche et al. described immune activation involving IL-1 and IL-6 in addition to interferon-alpha and monocyte chemotactic protein-1, implicating disruption of the Th1 immune response.2
Currently, there is no established first-line therapeutic regimen for ECD. Corticosteroids are the most commonly reported treatment. More recently, therapy with IFNα was shown to confer a greater likelihood of survival.4 Other reported treatment regimens include chemotherapy (vinca alkaloids, anthracyclines and cyclophosphamide), imatinib mesylate, sunitinib, anakinra, methotrexate, bisphosphonates, autologous hematopoetic stem cell transplantation and radiation therapy. 3, 4, 12 The recent discovery of a BRAFV600E mutation in 54% of patients with ECD suggest the possibly utility of targeted therapy with vemurafenib, a BRAF inhibitor recently approved for treatment of metastatic melanoma.13
KEY TEACHING POINTS
ECD is a multisystem non-Langerhans cell histiocytosis characterized by histiocytic infiltration of the bones, viscera, CNS and skin requiring comprehensive skeletal, visceral and neurologic evaluation. Hallmark skeletal lesions are symmetric distal osteosclerosis of the long bones with sparing of the epiphyses.
Cutaneous involvement most commonly consists of xanthelasma and xanthoma-like lesions. Histology reveals lipid-laden macrophages expressing CD68 and factor XIIIa, and lacking S100 and CD1a expression.
The recent report of BRAF V600E mutations in 54% of patient with Erdheim Chester disease suggests a potential new therapeutic approach. However, further research is needed to better understand the pathophysiology of this rare disease.
Acknowledgments
Funding Sources: This research was supported by the Intramural Program of NIH, Center for Cancer Research, National Cancer Institute and the National Human Genome Research Institute.
Abbreviations
- ECD
Erdheim-Chester disease
- LCH
Langerhans cell histiocytosis
- MRI
Magnetic resonance imaging
- PET/CT
Positron emission tomography - computed tomography
- TNFα
Tumor necrosis factor-alpha
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
“The authors have no conflicts of interest to disclose.”
“This article has not been presented previously.
Note: Dr. Kornik evaluated this patient during an elective rotation at the NIH
REFERENCES
- 1.Haroche J, Charlotte F, Arnaud L, von Deimling A, Helias-Rodzewicz Z, Hervier B, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120:2700–3. doi: 10.1182/blood-2012-05-430140. [DOI] [PubMed] [Google Scholar]
- 2.Haroche J, Arnaud L, Amoura Z. Erdheim-Chester disease. Curr Opin Rheumatol. 2012;24:53–9. doi: 10.1097/BOR.0b013e32834d861d. [DOI] [PubMed] [Google Scholar]
- 3.Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, Wechsler J, Brun B, Remy M, et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996;75:157–69. doi: 10.1097/00005792-199605000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Arnaud L, Hervier B, Neel A, Hamidou MA, Kahn JE, Wechsler B, et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood. 2011;117:2778–82. doi: 10.1182/blood-2010-06-294108. [DOI] [PubMed] [Google Scholar]
- 5.Skinner M, Briant M, Morgan MB. Erdheim-Chester disease: a histiocytic disorder more than skin deep. Am J Dermatopathol. 2011;33:e24–6. doi: 10.1097/DAD.0b013e3181f2bf06. [DOI] [PubMed] [Google Scholar]
- 6.Caputo R, Marzano AV, Passoni E, Berti E. Unusual variants of non-Langerhans cell histiocytoses. J Am Acad Dermatol. 2007;57:1031–45. doi: 10.1016/j.jaad.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 7.Al-Quran S, Reith J, Bradley J, Rimsza L. Erdheim-Chester disease: case report, PCR-based analysis of clonality, and review of literature. Mod Pathol. 2002;15:666–72. doi: 10.1038/modpathol.3880583. [DOI] [PubMed] [Google Scholar]
- 8.Pan A, Doyle T, Schlup M, Lubcke R, Schultz M. Unusual manifestation of Erdheim-Chester disease. BMC Gastroenterol. 2011;11:77. doi: 10.1186/1471-230X-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen TC, Chevez-Barrios P, Shetlar DJ, Cagle PT. Pulmonary and ophthalmic involvement with Erdheim-Chester disease: a case report and review of the literature. Arch Pathol Lab Med. 2004;128:1428–31. doi: 10.5858/2004-128-1428-PAOIWE. [DOI] [PubMed] [Google Scholar]
- 10.Dagna L, Girlanda S, Langheim S, Rizzo N, Bozzolo EP, Sabbadini MG, et al. Erdheim-Chester disease: report on a case and new insights on its immunopathogenesis. Rheumatology (Oxford) 2010;49:1203–6. doi: 10.1093/rheumatology/kep461. [DOI] [PubMed] [Google Scholar]
- 11.Tran TA, Pariente D, Lecron JC, Delwail A, Taoufik Y, Meinzer U. Treatment of pediatric Erdheim-Chester disease with interleukin-1-targeting drugs. Arthritis Rheum. 2011;63:4031–2. doi: 10.1002/art.30638. [DOI] [PubMed] [Google Scholar]
- 12.Wilejto M, Abla O. Langerhans cell histiocytosis and Erdheim-Chester disease. Curr Opin Rheumatol. 2012;24:90–6. doi: 10.1097/BOR.0b013e32834db53e. [DOI] [PubMed] [Google Scholar]
- 13.Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2012 Dec 20; doi: 10.1182/blood-2012-07-446286. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]