

Abstract
Hepatitis B virus (HBV) genotype C is associated with the development of hepatocellular carcinoma (HCC). In addition, HBV subgenotype C1 is the major subgenotype in Southern China. The aim of this study was to investigate whether there was the specific mutation patterns in HBV/C1 associated with Southern Chinese patients with HCC. Methods: Mutations in HBV basal core promoter (BCP) and their association with HCC were assessed in a matched cross-sectional control study of 102 HCC and 105 chronic hepatitis (CH) patients (from Guangdong, China) infected with HBV/C1. Functional analysis of HBx mutants was performed by the colony formation assay and the luciferase assays. Results: T1762/A1764 double mutations was frequently found in patients infected with HBV/C1, regardless of clinical status (64.7% in HCC and 51.4% in CH, P>0.05). Unexpectedly, the adjacent V1753 or A1768 mutation significantly increased the risk of HCC (P<0.05). Moreover, the prevalence of triple or quadruple mutations in BCP was significantly higher in patients with HCC than those with CH, particularly for HBeAg-positive-carriers (P<0.05). Functional analysis revealed that T1762/A1764 mutation alone did not alter the transcriptional activity and the inhibitory effects on cell proliferation of HBx, but triple or quadruple mutations largely abrogated this effect. Conclusions: Accumulation of mutations involving V1753 or/and A1768 in addition to T1762/A1764 in BCP region were closely related to HCC among the patients infected with HBV/C1, particularly for HBeAg-positive-carriers. The increased risk of HCC caused by BCP variants may be attributable partially to modifying the biological functions of HBx.
Keywords: Hepatitis B virus, basal core promoter, X gene, mutation, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is the fifth most frequent cancer and the third leading cause of cancer-related death in the world [1]. It is accepted that chronic infection with Hepatitis B virus (HBV) is a major risk factor for the development of HCC [2]. The HBV genome consists of a partially double-stranded circular DNA molecule comprising 3200 nucleotides, and exhibits considerable genetic variability because the polymerase lacks a proofreading function. At present, HBV is classified into eight genotypes (A to H) according to a divergence in the entire nucleotide sequence greater than 8% [3]. HBV genotypes have distinct geographical distributions and correlate with the severity of liver disease [3,4]. Genotypes B and C are prevalent in Asia, and genotype C is associated with a more severe course of liver disease and a higher risk of HCC development than genotype B [5,6]. Furthermore, subgenotypes have been identified in different HBV genotypes based on a divergence greater than 4% but less than 8% in the complete nucleotide sequence. There are two subgenotypes of genotype B with distinct geographical distributions, provisionally designated B1 (Bj, Japan) and B2 (Ba, other Asian countries) [7]. Clinical differences between the patients infected with HBV/Ba and HBV/Bj have been demonstrated, indicating a comparatively better prognosis of HBV/Bj-infection in comparison to HBV/Ba [8,9]. Recently, HBV/C has been classified into four subgenotypes (HBV/C1-C4) [10,11]. The two most widely spread subgenotypes are HBV/C1 (Cs), for Southeast Asia, and HBV/C2 (Ce), for East Asia; they not only have different geographic distributions but also different clinical outcomes are becoming clear [12,13].
The HBV core promoter (CP) resides in the overlapping X gene and plays a central role in HBV replication and morphogenesis [14]. Core promoter is composed of the core upstream regulatory sequence (CURS) and the basal core promoter (BCP). Mutations in BCP region at nucleotides (nt) 1762 and 1764 (T1762/A1764) were previously reported in association with HBe antigen seroconversion and viral replication. They were often found in the patients with advanced liver disease and HCC [15-19]. Recently, T1653 mutation in the box α of CURS and V1753 mutation in BCP region has also been reported to increase the risk of HCC in HBeAg-negative patients infected with HBV/C [20,21]. As well, specific mutations in CP region of HBV were differently associated with HCC in the context of HBeAg status among HBV/C1 and HBV/C2 carriers [22]. However, the mechanism underlying CP mutation and hepatocarcinogenesis is largely unknown.
As a highly endemic area for HBV infection, the prevalence of chronic HBV infection in China is between 8 and 20% of the general population, with an estimated 120 million hepatitis B surface antigen (HBsAg) carriers. The prevalence of HBV is uneven in different regions of China: high in some southern provinces (>8%) and low in some western provinces (4-6%) [23]. Genotypes B and C were identified as the most common HBV strains and account for approximately 95% of Chinese patients. Further study confirmed all genotype B strains belong to subgenotype Ba. Two of genotype C subgenotypes, C1 and C2, were found in China and they showed different geographic distributions. HBV/C1 was the major subgenotype in Southern China and C2 was predominant in Northern China [23-26]. Previous reports indicated a significantly higher incidence of HCC in Southern China [27,28]. However, it is unclear whether there are the specific mutation patterns in CP of HBV responsible for the development of HCC in Southern China.
The aim of this study was to determine the HCC-associated mutations of the HBV genome in the entire CP and the overlapping X gene region in Southern Chinese patients infected with HBV/C1, and analyze the hotspot mutations through a cross-sectional control study.
Materials and methods
Subjects
A total of 209 serum samples were obtained from a cohort of patients with chronic hepatitis B, including 102 from chronic hepatitis (CH) patients and 102 from HCC patients, who had been recruited at the First affiliated Hospital of Guangzhou Medical College, Guangzhou China between June 2005 and December 2009. The diagnosis of HCC was made pathologically. All patients were chronic HBV C1 carriers, as identified by PCR-restriction fragment length polymorphism. None of the patients had co-infection with hepatitis C virus or other agents that are causes of liver disease. All serum samples were stored at -40°C until analysis.
Amplification and sequencing of CP region of HBV
HBV DNA was extracted from 100 μl serum using a QIAamp DNA blood mini kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer’s instructions. In order to detect the genetic variations in CP region, HBV DNA sequence (nt. 1176-1838) bearing the core promoter region and the overlapping X gene was amplified by PCR with hemi-nested primers. The first-round PCR was performed with the sense primer HBVF (1176-1195) and the antisense primer HBVR (1823-1804). The second-round PCR was performed with the sense primer HBVF (1266-1285) and the antisense primer HBVR (1823-1804) (Table 1). Amplified products were directly sequenced with Prism BigDye (Applied Biosystems, Foster City, CA) in the ABI 3100 DNA automated sequencer. Sequences were compared using the Claustral W program.
Table 1.
Oligonucleotide primers used in the study

|
The underlined letters, restriction enzyme site (HindIII and XhoI); the solid letters, X gene mutant sites.
Site-directed mutagenesis and plasmid construction
The mutations in CP region were introduced by overlap extension PCR, by PCR-mediated site-directed mutagenesis with a set of designed mutagenetic primers (Table 1), as reported previously [29]. Briefly, single base changes were introduced by PCR using internal primers containing the desired point mutation(s): HBVF2-2 (1739-1773), HBVF2-3.1 (1739-1773), HBVF2-3.2 (1739-1773) and HBVF2-4 (1739-1773), paired with the outside primer HBV1883R2. Primer set HBVF1 (1374-1393)/HBVR1 (1728-1756) was used to generate the products with the homologue ends overlapped with the above mutated fragments. The products of the first round PCR were purified and served as the templates for the second round PCR with the outside primer set HBVF1 (1374-1393)/HBVR2 (1813-1838) to generate a full-length HBx. The PCR products were cloned into the HindIII-XhoI sites of pcDNA3.1 (Invitrogen, Carlsbad, CA) to obtain the expression vectors of wild type or mutated HBx, respectively. All constructs were sequenced to confirm the desired sequences.
The NF-κB luciferase reporter plasmids, NF-κB-Luc and mtNF-κB-Luc have been previously described [30]. NF-κB-Luc encodes consensus NF-κB sites upstream of a luciferase reporter; mtNF-κB-Luc is identical to NF-κB-Luc except that the NF-κB consensus sites are mutated so that NF-κB cannot bind and stimulate transcription.
Cell culture and transfection
HepG2 cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen) at 37°C in a 5% CO2 environment. For transfection, cells were seeded in 96-well plates at 1.5×105 cells/ml, grown to 50-80% confluency and transfected with the plasmids described above using Lipofectamine2000 (Invitrogen) according to the manufacturer’s protocol. Following transfection, cells were incubated for 24 h before being harvested for the luciferase assay or gene expression assay.
Western blot analysis
Total cell lysates were prepared in RIPA buffer [50 mmol/L TrisHCl, pH 8.0, 150 mmol/L NaCl, 1% (vol/vol) Nonidet P-40, 0.5% (wt/vol) sodium desoxycholate, 0.1% (wt/vol) SDS] containing the complete protease inhibitor cocktail. Western blot analysis was performed as described with the following primary antibodies: monoclonal mouse anti-HBx (Abcam, CA, USA) and mouse anti-β-actin (Sigma, MO, USA). Experiments were repeated in triplicate.
Luciferase assays
Transfected cells were harvested in Passive Lysis Buffer (Promega) and the cell lysates were analyzed for luciferase activity by the Dual Luciferase Reporter Assay System (Promega, CA USA) according to the manufacturer’s directions.
Colony formation assay
After cells were transfected for 48 h, they were transferred to a 60 mm plate and cultured in G418 (Invitrogen) selective medium for 2 weeks (G418 700 μg/ml). The cell culture medium was changed every 3 or 4 days until the colonies formed. Drug-resistant colonies were fixed with cold methanol, stained with Giemsa, and then scored.
Statistical analysis
Mutations between the two groups of samples were analyzed with relative risk test (OR value) and chi-square test (χ 2 analysis). Statistical comparisons between other indexes were analyzed by Student’s t-test. Regression and correlation were done by Pearson method. A P-value of less than 0.05 was taken as statistically significant.
Results
Mutations in the core promoter of HBV/C1 in patients with HCC and CH
In order to detect the genetic variations in CP region, DNA fragment from HBV nt 1176-1838 was amplified by hemi-nested PCR. As expected, the most frequently occurring mutation in CP was T1762/A1764, which could be found in 57.9% of all HBV isolates. However, there was no difference in the prevalence of T1762/A1764 between patients with HCC and CH (64.7% vs. 51.4%, P>0.05). Interestingly, the adjacent T1766 and A1768 mutations in BCP were significantly higher for the HCC group than for the CH group (18.6% vs. 6.7% and 24.5% vs. 8.6%, P<0.05, respectively) (Table 2).
Table 2.
Prevalence of mutations in the core promoter region between HCC and CH patients infected with HBV/C1
| Mutation | Total (n=207) | HCC (n=102) | CH (n=105) | P value | OR (95% CI) |
|---|---|---|---|---|---|
| T1653 | 27 (13.0%) | 18 (17.6%) | 9 (8.5%) | 0.103 | … |
| V1753 | 43 (29.5%) | 43 (42.1%)* | 18 (17.1%) | 0.034 | 3.071 (1.68–5.86) |
| T1762/A1764 | 135 (65.2%) | 77 (75.5%) | 58 (55.1%) | 0.843 | … |
| T1766 | 26 (25.5%) | 19 (18.6%)* | 7 (6.7%) | 0.044 | 2.146 (0.96–7.12) |
| A1768 | 35 (16.9%) | 26 (24.5%)* | 9 (8.6%) | 0.021 | 2.869 (1.21–6.49) |
HCC, hepatocellular carcinoma; CH, chronic heptitis; OR, odds ratio; CI, confidence interval.
P<0.05 comparing the frequency of triple mutation or quadruple mutation in HCC vs. CH.
Since T1653 and V1753 mutations have been reported to be more closely associated with the progression of liver disease from chronic hepatitis to cirrhosis and/or HCC, we also compared the prevalence of these two mutations between HCC and CH patients. The prevalence of T1653 was relatively lower, with 17.6% in HCC group and 8.5% in CH group, but no statistical difference was found in this study (P>0.05). However, the frequency of V1753 mutation was significantly higher in patients with HCC than those with CH (42.1% vs. 17.1%, P<0.05) (Table 2).
The patterns of BCP mutation in patients infected with HBV/C1 are presented in Table 3. Single mutation was rare, which was only found in two cases for V1753. Interestingly, the prevalence of V1753 or A1768 mutation always occurred along with T1762/A1764. Although previously reports that T1762/A1764 double mutation increased the risk of HCC4-7, no the correlation was observed between T1762/A1764 mutation and clinical diagnosis in this study. However, mutations at position 1762/1764 in addition to V1753 or A1768 exhibited a close association to HCC. The frequencies of 1753/1762/1764, 1762/1764/1768, or 1753/1762/1764/1768 mutations were significantly higher in patients with HCC than those with CH (11.8% vs. 3.8%, 6.9% vs. 2.9%, or 2.9% vs. 0.95%, P<0.05, respectively).
Table 3.
Mutation patterns of BCP in HBV isolated from patients infected with HBV/C1
| Type | Patterns | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| V1753 | T1762 | A1764 | T1766 | A1768 | HCC (n=102) | CH (n=105) | P value | |
| wild-type | - | - | - | - | - | 2 (2.0%) | 25 (23.8%) | 0.026 |
| single mutation | + | - | - | - | - | 0 (0%) | 13 (12.4%) | |
| double mutation | - | + | + | - | - | 66 (64.7%) | 54 (51.4%) | 0.802 |
| - | - | - | + | + | 4 (3.9%) | 2 (1.9%) | 0.064 | |
| triple mutation | + | + | + | - | - | 12 (11.8%)* | 4 (3.8%) | 0.022 |
| - | + | + | + | - | 6 (5.9%) | 5 (4.8%) | 0.092 | |
| - | + | + | - | + | 7 (6.9%)* | 3 (2.9%) | 0.039 | |
| quadruple mutation | + | + | + | - | + | 3 (2.9%)* | 1 (0.95%) | 0.041 |
| - | + | + | + | + | 2 (2.0%) | 0 (0%) | ||
HCC, hepatocellular carcinoma; CH, chronic heptitis;
P<0.05 comparing the frequency of triple mutation or quadruple mutation in HCC vs. CH.
Accumulation of BCP mutations and HBeAg status in patients with HCC and CH
BCP mutations were reported to be able to down-regulate the expression of HBeAg in cell culture system13. We have compared the rates of accumulation of BCP mutations and HBeAg status in patients with HCC and CH (Table 4). Among HBeAg-positive patients, there was no difference in the rate of T1762/A1764 double mutation between HCC group and CH group (65.7% vs. 54.1%, P>0.05). However, the rates of accumulative BCP mutations, such as 1753/1762/1764, 1762/1764/1768, or 1753/1762/1764/1768 mutations were significantly higher for the HCC group than for the CH group (14.3% vs. 4.1%, 8.6% vs. 2.7%, or 4.3% vs. 1.4%, P<0.05, respectively).
Table 4.
Accumulation of BCP mutations among patients with HCC and CH in context of HBeAg
| Mutation | HBeAg-positive group | HBeAg-negative group | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| HCC (n=70) | CH (n=74) | P value | HCC (n=32) | CH (n=31) | P value | |
| T1762/A1764 | 46 (65.7%) | 40 (54.1%) | 0.891 | 20 (62.5%) | 14 (45.2%) | 0.634 |
| V1753/T1762/A1764 | 10 (14.3%)* | 3 (4.1%) | 0.027 | 2 (6.3%) | 1 (3.2%) | 0.062 |
| T1762/A1764/A1768 | 6 (8.6%)* | 2 (2.7%) | 0.031 | 1 (3.1%) | 1 (3.2%) | 0.961 |
| V1753/T1762/A1764/A1768 | 3 (4.3%)* | 1 (1.4%) | 0.041 | 0 (0%) | 0 (0%) | |
HCC vs. CH, P<0.05 (Yates corrected chi-square).
Significant data are shown in bold.
Effect of BCP mutations on transactivation activity of HBx
V1753, T1762, A1764 and A1768 mutation in BCP result in four amino acid (aa) substitutions change in HBx, i.e., aa 127 (Isoleucine to asparagine/serine/threonine), 130 (lysine-to-methionine), 131 (valine-to-isoleucine) and 132 (phenylalanine-to-tyrosin). T1766 mutation does not cause aa change (Figure 1). In order to define the biological impact of BCP mutations on HBx, we introduced a series of artificial mutants into a wild type HBx template. pcDNA3.1-Xwt and pcDNA3.1-Xmt-2, pcDNA3.1-Xmt-3.1, pcDNA-3.1-Xmt-3.2 and pcDNA3.1-Xmt-4 were constructed to express wild type HBx and HBx mutants. Transfection efficiency was monitored by comparing the relative amount of cellular X protein using Western blot analysis (Figure 2B). To monitor the transactivation activity of different HBx constructs, a luciferase assay using the reporter construct of NF-κB-Luc or mtNF-κB-Luc was performed. HepG2 cells were transiently cotransfected with HBx and NF-κB-Luc or mtNF-κB-Luc plasmids. Like the wild type HBx, 1762/1764 double mutation induced a 4-5-fold increase in luciferase activity, but 1753/1762/1764, 1762/1764/1768 or 1753/1762/1764/1768 mutations revealed largely inhibition of transactivation activity (Figure 2A). In contrast, we found that none of HBx constructs could activate mtNF-κB-Luc (Figure 2A). This result indicated that accumulative BCP mutations could inhibit the transcriptional activity of HBx.
Figure 1.
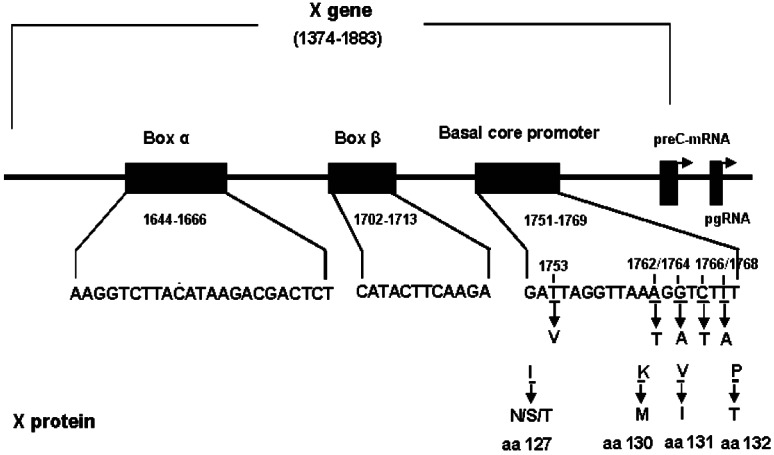
T1753V, A1762T/G1764A and C1766T/T1768A mutations in the X region, including the basal core promoter region.
Figure 2.

Transactivation activity of HBx mutants. A. HepG2 cells were co-transfected with the HBx artificial mutants constructed according to the naturally occurring mutation pattern of BCP and the NF-κB-Luc or mtNF-κB-Luc construct. Cells were harvested for luciferase assay 48 h after transfection. Relative luciferase activities indicate the mean of three independent transfections. B. expression of wild-type HBx and mutants HBx in HepG2 48 h after transfection. Xwt: expression vector containing wild-type X gene; Xmt-2, Xmt-3.1, Xmt-3.2 and Xmt-4: expression vector containing mutants X gene, bearing 1762/A1764 double mutant, V1753/A1764/T1762 triple mutant, T1762/A1764/A1768 triple mutant and V1753/T1762/A1764/A1768 quadruple mutant, respectively. *P<0.05; **P<0.01 vs. controls, such as pcDNA3 empty vector.
Impact of BCP mutations on growth-suppressive effect of HBx
Because HBx can induce cell apoptosis, we next investigated the proapoptotic ability of different HBx mutants, using a colony formation assay. HepG2 cells were transfected with the different HBx expression plasmids, and 2 weeks after G418 selection, the number of colonies was scored. As expected, the expression of HBx caused a substantial reduction (5-fold) in the number of colonies compared with the control cells transfected with the empty vector pcDNA3.1. A degree of colony formation inhibition similar to that of HBx was observed in 1762/1764 double mutation. Strikingly, 1753/1762/1764, 1762/1764/1768, or 1753/1762/1764/1768 mutations exhibited abrogation of the growth-suppressive effect (Figure 3). The result indicated that BCP mutations affected the growth-suppressive effect of HBx in an accumulative manner.
Figure 3.
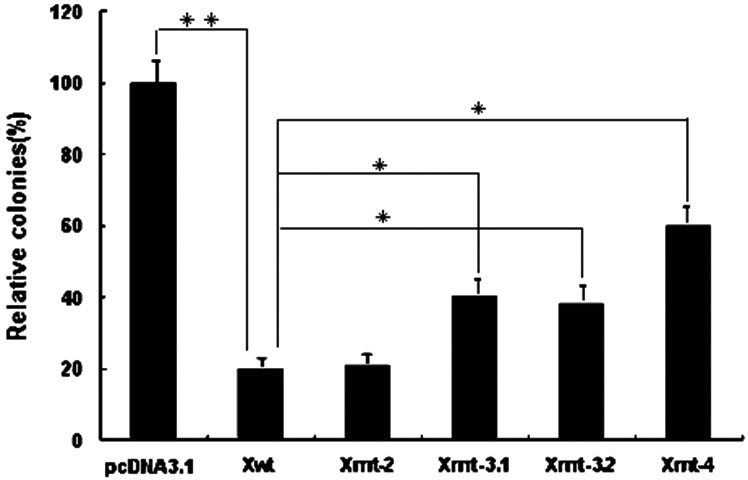
Growth-suppressive effect of HBx mutants. HepG2 cells were transfected with the HBx artificial mutants constructed according to the naturally occurring mutation pattern of BCP. After G418 selection for 2 weeks, drug-resistant clones were stained and scored. The results show the mean of two independent transfections. *P<0.05; **P<0.01.
Discussion
The infection of HBV is a major factor contributing to the development of HCC in China and the mutation in HBx plays an important role in this process. However, the impact of BCP mutations on HBx activity was unclear. We characterized CP mutations based on the serum samples taken from patients infected with HBV/C1 from the Southern China (Guangdong province) with a higher prevalence of HCC. We showed that although the frequency of T1762/A1764 double mutation was high in HCC patients, but there was no difference compared with that of CH patients. In contrast, the adjacent V1753 or A1768 mutation significantly increased the risk of HCC. Recently, a longitudinal study demonstrated that T1762/A1764 mutation could be detected 4-7 years prior to HCC in most cases, while T1766/A1768 mutation occurred only at or near the stage of HCC, suggesting T1766/A1768 may be a more valuable predictive marker for HCC [31]. Because several reports demonstrated that T1766/A1768 mutation enhanced viral replication [29,32], high viral load caused by this mutation might contribute to hepatocarcinogenesis. The V1753 mutation had been reported was specifically associated with HBV C1 among Chinese HCC patients [33], particularly for HCC among HBeAg-positive-C1-carriers [22]. Our study also confirmed that the V1753 mutation increases the risk of HCC in Chinese Guangdong with HBV/C1.
It is noteworthy that T1762/A1764 double mutation always accompanied with V1753 or A1768 point mutation. We found that the rates of V1753/T1762/A1764, T1762/A1764/A1768 or V1753/T1762/A1764/A1768 mutations were significantly higher in HCC patients compared with that in CH patients. The accumulation of BCP mu-tations involving V1753 or/and A1768 in addition to T1762/A1764 were closely related to HCC among the patients infected with HBV/C1, particularly for HBeAg-positive-carriers. Although T1762/A1764 mutation alone was not a triggering factors of HCC, T1762/A1764 with the additional V1753 or/and A1768 mutations in BCP region could be appreciably higher risk for hepatocarcinogenesis. Some in vitro study showed that the T1762/A1764 double mutation had no significant effect on replication capacity in comparison to the wild type [32]. However, all CP mutants bearing additional mutations in this region, i.e., mutations at the position of V1753/T1762/A1764, T1762/A1764/A1768 or V1753/T1762/A1764/A1768 exhibited to markedly enhance viral replication than the wild type [32,34], supporting a carcinogenetic potential of this mutations pattern. Thus, it is reasonable to think these mutations in BCP may play a synergistic role on enhancing HBV carcinogenesis.
Another feature of our study is that we try to understand the hepatocarcinogenesis potential of BCP mutations through HBV X protein (HBx) function. HBx is a multi-functional viral protein. It can modulate gene transcription, apoptosis, cell cycle, cell responses to genotoxic stress, protein degradation, and numerous signal transduction pathways [35-37] and, as a result, has been shown to induce cell transformation leading to HCC in a select strain of transgenic mice [38]. Functional study revealed that the COOH-terminus of HBx plays a key role in regulating its transcriptional activity and cells proliferation [39]. Since V1753, T1762, A1764 and A1768 mutations cause four amino acid (aa) substitutions changes at HBx COOH-terminus, i.e., 127 (I-to-N/S/T), 130 (K-to-M), 131 (V-to-I) and 132 (P-to-T), it is of interest to test whether BCP mutations affect the biological activity of HBx. We introduced a series of artificial mutants into a wild type HBx and constructed different HBx mutants. Transfection efficiency was monitored by comparing the relative amount of HBx by Western blot analysis. First, we analyzed the transactivation activity of different HBx through a luciferase assays. HepG2 cells were transiently co-transfected with NF-κB-Luc or mtNF-κB-Luc and HBx, and luciferase activity were examined. Like wild type HBx, T1762/A1764 double mutation induced a 4-5-fold increase in luciferase activity. This result indicated that T1762/A1764 double mutation alone did not impact the effect of HBx. However, the mutations at positions V1753/T1762/A1764, T1762/A1764/A1768 or V1753/T1762/A1764/A1768 largely inhibited the transactivation activity of HBx. In contrast, we found that none of HBx constructs could activate mtNF-κB-Luc. We suggest that the number of BCP variants may be involved in the transactivation function of HBx, and contributed to the development of HCC. However, the relationship between transcriptional activity and oncogenic potential of mutated HBx in hepatocarcinogenesis needs to be carefully analyzed in each mutation.
We next investigated the proapoptotic ability of different HBx by a colony formation assay. We noted that the transactivation function of HBx is generally linked to its proapoptotic function. As expected, the expression of HBx caused a substantial reduction (5-fold) in the number of colonies compared with the control cells transfected with pcDNA3.1. A degree of colony formation inhibition similar to that of HBx was observed in T1762/A1764 double mutation. Strikingly, V1753/T1762/A1764, T1762/A1764/A1768 or V1753/T1762/A1764/A1768 mutations exhibited abrogation of the growth-suppressive effect. These data further confirmed a synergistic role of BCP mutations could affect the function of HBx. However, the true impact of BCP mutations on the regulation of the biological function of HBx needs to be further evaluated.
Acknowledgements
We thank Dr. Yun Li for her technical help in luciferase assays. This study was supported by Guangdong Province Nature Science Founda-tion (9151040701000037) and Research Project of Guangzhou Science and Technology (2009J1-C321-2).
References
- 1.Parkin DM. International variation. Oncogene. 2004;23:6329–6340. doi: 10.1038/sj.onc.1207726. [DOI] [PubMed] [Google Scholar]
- 2.Yu MC, Yuan JM, Govindarajan S, Ross RK. Epidemiology of hepatocellular carcinoma. Can J Gastroenterol. 2000;14:703–709. doi: 10.1155/2000/371801. [DOI] [PubMed] [Google Scholar]
- 3.Miyakawa Y, Mizokami M. Classifying hepatitis B virus genotypes. Intervirology. 2003;46:329–338. doi: 10.1159/000074988. [DOI] [PubMed] [Google Scholar]
- 4.Kidd-Ljunggren K, Miyakawa Y, Kidd AH. Genetic variability in hepatitis B viruses. J Gen Virol. 2002;83:1267–1280. doi: 10.1099/0022-1317-83-6-1267. [DOI] [PubMed] [Google Scholar]
- 5.Chan HL, Hui AY, Wong ML, Tse AM, Hung LC, Wong VW, Sung JJ. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut. 2004;53:1494–1498. doi: 10.1136/gut.2003.033324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orito E, Mizokami M, Sakugawa H, Michitaka K, Ishikawa K, Ichida T. A case-control study for clinical and molecular biological differences between hepatitis B viruses of genotypes B and C. Japan HBV Genotype Research Group. Hepatology. 2001;33:218–223. doi: 10.1053/jhep.2001.20532. [DOI] [PubMed] [Google Scholar]
- 7.Sugauchi F, Orito E, Ichida T, Kato H, Sakugawa H, Kakumu S. Hepatitis B virus of genotype B with or without recombination with genotype C over the precore region plus the core gene. J Virol. 2002;76:5985–5992. doi: 10.1128/JVI.76.12.5985-5992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orito E, Sugauchi F, Tanaka Y, Ichida T, Sata M, Tanaka E, Okanoue T, Sakugawa H, Watanabe H, Miyakawa H, Nishiguchi S, Kumada H, Ueda R, Mizokami M. Differences of hepatocellular carcinoma patients with hepatitis B virus genotypes of Ba, Bj or C in Japan. Intervirology. 2005;48:239–245. doi: 10.1159/000084601. [DOI] [PubMed] [Google Scholar]
- 9.Sugauchi F, Orito E, Ichida T, Kato H, Sakugawa H, Kakumu S, Ishida T, Chutaputti A, Lai CL, Gish RG, Ueda R, Miyakawa Y, Mizokami M. Epidemiologic and virologic characteristics of hepatitis B virus genotype B having the recombination with genotype C. Gastroenterology. 2003;124:925–932. doi: 10.1053/gast.2003.50140. [DOI] [PubMed] [Google Scholar]
- 10.Huy TT, Ushijima H, Quang VX, Win KM, Luengrojanakul P, Kikuchi K, Sata T, Abe K. Genotype C of hepatitis B virus can be classified into at least two subgroups. J Gen Virol. 2004;85:283–292. doi: 10.1099/vir.0.19633-0. [DOI] [PubMed] [Google Scholar]
- 11.Norder H, Couroucé AM, Coursaget P, Echevarria JM, Lee SD, Mushahwar IK, Robertson BH, Locarnini S, Magnius LO. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology. 2004;47:289–299. doi: 10.1159/000080872. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka Y, Orito E, Yuen MF, Mukaide M, Sugauchi F, Ito K, Ozasa A, Sakamoto T, Kurbanov F, Lai CL, Mizokami M. Two subtypes (subgenotypes) of hepatitis B virus genotype C: a novel subtyping assay based on restriction fragment length polymorphism. Hepatol Res. 2005;33:216–224. doi: 10.1016/j.hepres.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Chan HL, Tsui SK, Tse CH, Ng EY, Au TC, Yuen L, Bartholomeusz A, Leung KS, Lee KH, Locarnini S, Sung JJ. Epidemiological and virological characteristics of 2 subgroups of hepatitis B virus genotype C. J Infect Dis. 2005;191:2022–2032. doi: 10.1086/430324. [DOI] [PubMed] [Google Scholar]
- 14.Kramvis A, Kew MC. The core promoter of hepatitis B virus. J Viral Hepat. 1999;6:415–427. doi: 10.1046/j.1365-2893.1999.00189.x. [DOI] [PubMed] [Google Scholar]
- 15.Baptista M, Kramvis A, Kew MC. High prevalence of 1762(T) 1764(A) mutations in the basic core promoter of hepatitis B virus isolated from black Africans with hepatocellular carcinoma compared with asymptomatic carriers. Hepatology. 1999;29:946–953. doi: 10.1002/hep.510290336. [DOI] [PubMed] [Google Scholar]
- 16.Chou YC, Yu MW, Wu CF, Yang SY, Lin CL, Liu CJ, Shih WL, Chen PJ, Liaw YF, Chen CJ. Temporal relationship between hepatitis B virus enhancer II/basal core promoter sequence variation and risk of hepatocellular carcinoma. Gut. 2008;57:91–97. doi: 10.1136/gut.2006.114066. [DOI] [PubMed] [Google Scholar]
- 17.Kao JH, Chen PJ, Lai MY, Chen DS. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology. 2003;124:327–334. doi: 10.1053/gast.2003.50053. [DOI] [PubMed] [Google Scholar]
- 18.Liu CJ, Chen BF, Chen PJ, Lai MY, Huang WL, Kao JH, Chen DS. Role of hepatitis B virus precore/core promoter mutations and serum viral load on noncirrhotic hepatocellular carcinoma: a casecontrol study. J Infect Dis. 2006;194:594–599. doi: 10.1086/505883. [DOI] [PubMed] [Google Scholar]
- 19.Yuen MF, Tanaka Y, Shinkai N, Poon RT, But DY, Fong DY, Fung J, Wong DK, Yuen JC, Mizokami M, Lai CL. Risk for hepatocellular carcinoma with respect to hepatitis B virus genotypes B/C, specific mutations of enhancer II/core promoter/precore regions and HBV DNA levels. Gut. 2008;57:98–102. doi: 10.1136/gut.2007.119859. [DOI] [PubMed] [Google Scholar]
- 20.Ito K, Tanaka Y, Orito E, Sugiyama M, Fujiwara K, Sugauchi F. T1653 mutation in the box alpha increases the risk of hepatocellular carcinoma in patients with chronic hepatitis B virus genotype C infection. Clin Infect Dis. 2006;42:1–7. doi: 10.1086/498522. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi K, Ohta Y, Kanai K, Akahane Y, Iwasa Y, Hino K. Clinical implications of mutations C-to-T1653 and T-to-C/A/G1753 of hepatitis B virus genotype C genome in chronic liver disease. Arch Virol. 1999;144:1299–1308. doi: 10.1007/s007050050588. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka Y, Mukaide M, Orito E, Yuen MF, Ito K, Kurbanov F. Specific mutations in enhancer II/core promoter of hepatitis B virus subgenotypes C1/C2 increase the risk of hepatocellular carcinoma. J Hepatol. 2006;45:646–653. doi: 10.1016/j.jhep.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 23.Wang ZH, Huang YH, Wen SJ, Zhou B, Hou JL. Hepatitis B virus genotypes and subgenotypes in China. Hepatology Research. 2007;37:S36–S41. doi: 10.1111/j.1872-034X.2007.00102.x. [DOI] [PubMed] [Google Scholar]
- 24.Wang ZH, Tanaka Y, Huang YH, Kurbanov F, Chen JJ, Zeng GB. Clinical and Virological Characteristics of Hepatitis B Virus Subgenotypes Ba, C1, and C2 in China. J Clin Microbiol. 2007;45:1491–1496. doi: 10.1128/JCM.02157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng G, Wang Z, Wen S. Geographic distribution, virologic and clinical characteristics of hepatitis B virus genotypes in China. J Viral Hepat. 2005;12:609–617. doi: 10.1111/j.1365-2893.2005.00657.x. [DOI] [PubMed] [Google Scholar]
- 26.Xia G, Nainan OV, Jia Z. Characterization and distribution of hepatitis B virus genotypes and subtypes in 4 provinces of China. Zhonghua Liu Xing Bing Xue Za Zhi. 2001;22:348–351. (in Chinese) [PubMed] [Google Scholar]
- 27.Wang ZJ, Zhou YP, Cheng B. An epidemiologic study on the aetiological factors of primary liver cancer in Shunde City of Guangdong province. Zhonghua Liu Xing Bing Xue Za Zhi. 1996;17:141–144. (in Chinese) [PubMed] [Google Scholar]
- 28.Zhang S, Li L, Lu F. Mortality of primary liver cancer in China from 1990 through 1999. Zhonghua Zhong Liu Za Zhi. 1999;21:245–249. (in Chinese) [PubMed] [Google Scholar]
- 29.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 30.Puro R, Schneider R. Tumor necrosis factor activates a conserved innate antiviral response to hepatitis B virus that destabilizes nucleocapsids and reduces nuclear viral DNA. J Virol. 2007;81:7351–7362. doi: 10.1128/JVI.00554-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo X, Jin Y, Qian GS, Tu H. Sequential accumulation of the mutations in core promoter of hepatitis B virus is associated with the development of hepatocellular carcinoma in Qidong, China. J Hepatol. 2008;49:718–725. doi: 10.1016/j.jhep.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 32.Parekh S, Zoulim F, Ahn SH, Tsai A, Li J, Kawai S, Khan N, Trépo C, Wands J, Tong S. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J Virol. 2003;77:6601–6612. doi: 10.1128/JVI.77.12.6601-6612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang ZH, Tanaka Y, Huang YH, Kurbanov F, Chen JJ, Zeng GB. Clinical and Virological Characteristics of Hepatitis B Virus Subgenotypes Ba, C1, and C2 in China. J Clin Microbiol. 2007;45:1491–1496. doi: 10.1128/JCM.02157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jammeh S, Tavner F, Watson R, Thomas HC, Karayiannis P. Effect of basal core promoter and pre-core mutations on hepatitis B virus replication. J Gen Virol. 2008;89:901–909. doi: 10.1099/vir.0.83468-0. [DOI] [PubMed] [Google Scholar]
- 35.Block TM, Mehta AS, Fimmel CJ, Jordan R. Molecular viral oncology of hepatocellular carcinoma. Oncogene. 2003;22:5093–5107. doi: 10.1038/sj.onc.1206557. [DOI] [PubMed] [Google Scholar]
- 36.Brechot C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: old and new paradigms. Gastroenterology. 2004;127:S56–S61. doi: 10.1053/j.gastro.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka Y, Kanai F, Ichimura T, Tateishi K, Asaoka Y, Guleng B. The hepatitis B virus X protein enhances AP-1 activation through interaction with Jab1. Oncogene. 2006;25:633–642. doi: 10.1038/sj.onc.1209093. [DOI] [PubMed] [Google Scholar]
- 38.Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317–320. doi: 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- 39.Tu H, Bonura C, Giannini C, Mouly H, Soussan P, Kew M, Paterlini-Bréchot P, Bréchot C, Kremsdorf D. Biological impact of natural COOH-terminal deletions of hepatitis B virus X protein in hepatocellular carcinoma tissues. Cancer Res. 2001;61:7803–7811. [PubMed] [Google Scholar]