

Abstract
We have previously shown that angiotensin II type 1 receptor-associated protein (ATRAP/Agtrap) interacts with the angiotensin II type 1 receptor and promotes constitutive internalization of the receptor so as to inhibit the pathological activation of its downstream signaling but preserve baseline physiological signaling activity. The present study was designed to investigate the role of renal ATRAP in angiotensin II–dependent hypertension. We generated transgenic mice dominantly expressing ATRAP in the renal tubules, including renal distal tubules. The renal ATRAP transgenic mice exhibited no significant change in blood pressure at baseline on normal salt diet. However, in the renal ATRAP transgenic mice compared with wild-type mice, the following took place: (1) the development of high blood pressure in response to angiotensin II infusion was significantly suppressed based on radiotelemetry, (2) the extent of daily positive sodium balance was significantly reduced during angiotensin II infusion in metabolic cage analysis, and (3) the renal Na+-Cl− cotransporter activation and α-subunit of the epithelial sodium channel induction by angiotensin II infusion were inhibited. Furthermore, adenoviral overexpression of ATRAP suppressed the angiotensin II–mediated increase in the expression of α-subunit of the epithelial sodium channel in mouse distal convoluted tubule cells. These results indicate that renal tubule–dominant ATRAP activation provokes no evident effects on blood pressure at baseline but exerts an inhibitory effect on the pathological elevation of blood pressure in response to angiotensin II stimulation, thereby suggesting that ATRAP is a potential target of interest in blood pressure modulation under pathological conditions.
Keywords: angiotensin II, angiotensin receptors, basic science, gene expression/regulation, hypertension (kidney), membrane transport/ion channels, receptors
Activation of angiotensin II (Ang II) type 1 receptor (AT1R) through the tissue renin–angiotensin system plays a pivotal role in the pathogenesis of hypertension and associated end-organ injury. In addition, the activation of renal AT1R signaling plays a key role in the altered renal sodium handling, which occurs in angiotensin-dependent hypertension.1–3 This is consistent with Guyton’s hypothesis that defective handling of sodium by the kidney with a consequent dysregulation of body fluid volume is the requisite final common pathway in the pathogenesis of hypertension.4 The carboxyl (C)-terminal domain of AT1R is involved in the control of AT1R internalization independent of G protein coupling. 5,6 It plays an important role in linking receptor-mediated signal transduction with the specific biological response to Ang II. The AT1R-associated protein (ATRAP/Agtrap) has been identified as the specific binding protein of the C-terminal domain of AT1R.7,8 ATRAP is expressed in many tissues, including the kidney, as is AT1R. Our preceding studies suggest that ATRAP selectively suppresses Ang II–mediated pathological activation of AT1R signaling in cardiovascular cells, and that cardiac ATRAP enhancement ameliorates cardiac hypertrophy in chronic Ang II–infused mice without affecting baseline cardiovascular function including blood pressure (BP).9–13
With respect to the intrarenal distribution of ATRAP, its protein was found to be widely expressed along the renal tubules, with a weak level in the vascular smooth muscle cells of the vasculature, including the interlobular arteries, Bowman capsule, podocytes, and mesangial cells in the glomerulus. 13,14 However, despite there being abundant kidney ATRAP expression and that various pathological stimuli, including Ang II, are reported to downregulate renal ATRAP expression, little is known about actual function of renal ATRAP. 15,16 The present study was designed to obtain in vivo evidence of renal ATRAP, with a special focus on Ang II–dependent hypertension by using transgenic (Tg) mice with a pattern of kidney-dominant ATRAP overexpression.
Materials and Methods
This study was performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All animal studies were reviewed and approved by the Animal Studies Committee of Yokohama City University. Methods are described in detail in the online-only Data Supplement.
Results
Generation of Renal ATRAP Tg Mice
We generated Tg mice with a pattern of kidney-dominant overexpression of ATRAP (Figure S1B and S1C in the online-only Data Supplement). One of 6 lines of ATRAP Tg mice exhibited renal overexpression of the transgene hemagglutinin-tagged mouse ATRAP (HA-ATRAP) in comparison with wild-type (Wt) littermate mice (Figure 1B and Figure S1C) with scant levels of the transgene HA-ATRAP protein in the other tissues examined (Figure 1A). As shown in Figure 1B, the level of renal total ATRAP protein expression detected by the anti-ATRAP antibody was ≈10-fold higher in Tg mice (endogenous ATRAP and transgene HA-ATRAP) than in Wt mice (endogenous ATRAP).
Figure 1.

Expression and localization of the hemagglutinin angiotensin II type 1 receptor-associated protein (HA-ATRAP) transgene in renal ATRAP transgenic (Tg) mice. A, Representative Western blot analysis of the HA-ATRAP transgene with polyclonal anti-HA antibody. B, Representative Western blot analysis of the total ATRAP protein expression in the kidney of wild-type (Wt) and Tg mice. C, Renal cortical section showing expression of the HA-ATRAP transgene in renal tubules detected by anti-HA antibody (top left). Consecutive sections showing total ATRAP protein expression (transgene HA-ATRAP and endogenous ATRAP) detected by anti-ATRAP antibody (top center). Consecutive sections were also stained with a monoclonal antibody against megalin (top right), a specific marker of proximal tubules, a monoclonal antibody against calbindin-D (bottom left), a specific marker of distal convoluted tubules (DCT) and connecting tubules (CNT), and a polyclonal antibody against aquaporin-2 (AQP2; bottom right), a specific marker of collecting ducts. Original magnification, ×100. D, Representative image of a hematoxylin/eosin-stained section of the proximal (bottom left) and distal (bottom right) tubules in the renal cortex before and after laser microdissection. Original magnification, ×400. Quantitative analysis (top) of mRNA expression in the proximal and distal tubules of the renal cortex. Values are calculated relative to those obtained for ATRAP mRNA expression in extracts from proximal tubules of Wt mice and are expressed as the mean (n=4 in each group).
To determine the expression and distribution of the ATRAP protein in the kidney of Tg mice, we performed an immunohistochemical examination using anti-HA antibody, anti-ATRAP antibody, and antibodies to specific nephron markers (Figure 1C). Although the renal expression of the HA-ATRAP protein prevailed over the endogenous ATRAP protein, histological analysis revealed a similar intrarenal distribution of HA immunostaining (transgene HA-ATRAP protein detected by the anti-HA antibody) and ATRAP immunostaining (transgene HA-ATRAP and endogenous ATRAP proteins detected by the anti-ATRAP antibody), mainly in the cortex.
We next stained consecutive sections with markers specific to the tubular segments. We used a polyclonal antibody against aquaporin-2, which is specifically expressed in the collecting ducts; a monoclonal antibody against calbindin-D, a calcium-binding protein expressed primarily in the distal convoluted tubules (DCT) and connecting tubules; and a monoclonal antibody against megalin, which is specifically expressed in the proximal convoluted tubules; and found that a high level of ATRAP immunostaining was predominantly detected along the renal distal tubules from the DCT to connecting tubules in the renal cortex. As shown in Figure 1D, the distal tubule–dominant expression of HA-ATRAP transgene was quantified by a laser capture microdissection method. The ATRAP mRNA expression in the distal tubules of renal cortex was ≈33.7-fold higher in Tg mice than in Wt mice. However, the ATRAP mRNA expression in the proximal tubules of renal cortex was only 3.5-fold higher in Tg mice than in Wt mice.
Suppression of Ang II–Dependent Hypertension in Renal ATRAP Tg Mice
The baseline 24-hour mean systolic BP (SBP), measured by a radiotelemetry method, was comparable between Wt and Tg mice (male, 14–18 weeks of age; 126±2 versus 122±1 mm Hg, unpaired t test; P=0.12; Figure 2A and 2C). However, the SBP elevation by Ang II infusion (1000 ng/kg per min) was significantly suppressed in Tg mice compared with Wt mice (Figure 2A; 2-way repeated measures ANOVA F=7.476; P=0.0257; Figure 2B; unpaired t test; P=0.0023). We also examined the effect of a higher dose of Ang II infusion (2000 ng/kg per min) on the BP of Wt and Tg mice, and the difference in the Ang II-induced SBP elevation between Wt and Tg mice was more prominent at the higher dose (2000 ng/kg per min) of Ang II (Figure 2C; 2-way repeated measures ANOVA F=9.035; P=0.0012) (Figure 2D; unpaired t test; P=0.0017).
Figure 2.
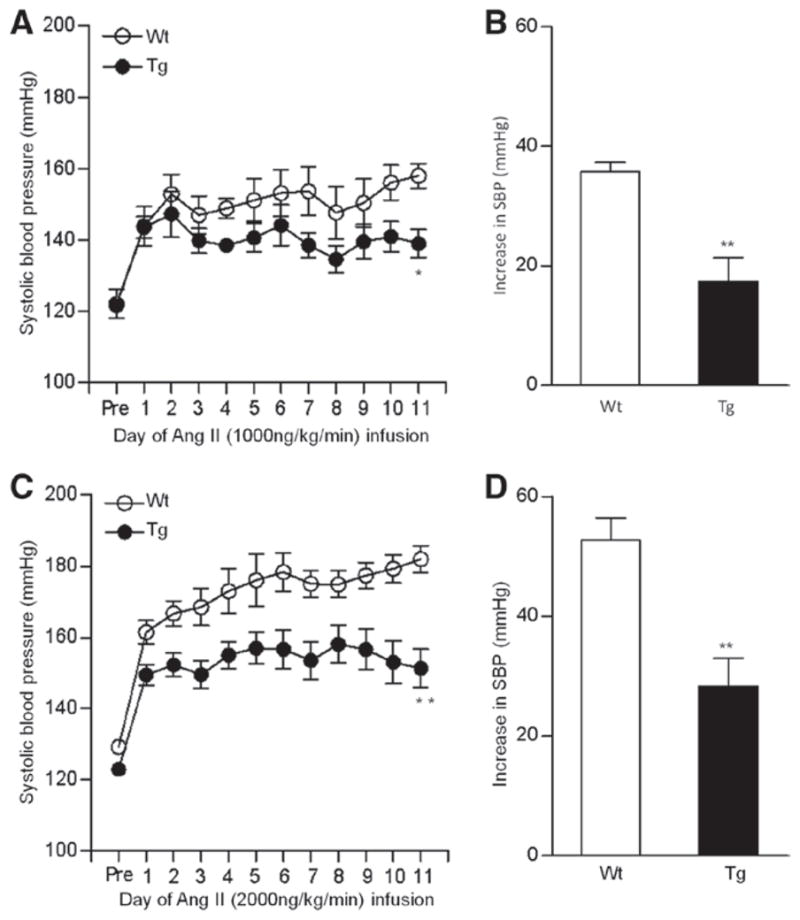
Effects of angiotensin II (Ang II) infusion on blood pressure (BP) analyzed by the radiotelemetric method in wild-type (Wt) and renal angiotensin II type 1 receptor-associated protein transgenic (Tg) mice. A, Daily and 24-hour systolic BP (SBP) in Wt and Tg mice before (pre) and during 11 days of Ang II (1000 ng/kg per min) infusion. Values are expressed as the mean±SE (n=5 in each group), *P<0.05 vs Wt mice. B, The increase in SBP during Ang II (1000 ng/kg per min) infusion was significantly less in Tg mice (17±4 mm Hg) compared with Wt mice (36±2 mm Hg). Values are expressed as the mean±SE (n=5 in each group), **P<0.01 vs Wt mice. C, Daily and 24-hour SBP in Wt and Tg mice before (pre) and during 11 days of Ang II (2000 ng/kg per min) infusion. Values are expressed as the mean±SE (n=6–7 in each group), **P<0.01 vs Wt mice. D, The increase in SBP during the Ang II (2000 ng/kg per min) infusion was significantly less in Tg mice (28±5 mm Hg) compared with Wt mice (53±4 mm Hg). Values are expressed as the mean±SE (n=6–7 in each group), **P<0.01 vs Wt mice.
Increase in Urinary Sodium Excretion in Renal ATRAP Tg Mice
We hypothesized that renal enhancement of ATRAP might suppress angiotensin-dependent hypertension by influencing the handling of renal sodium and performed metabolic cage analysis (Figure S2A–S2D). Because urinary sodium excretion was significantly increased in Tg mice compared with Wt mice during the infusion period (Figure S2D; 2-way repeated measures ANOVA F=12.91; P=0.0029), we analyzed daily sodium balance during Ang II infusion and cumulative sodium balance during the early phase (day 1–6) of Ang II infusion to more exactly compare the status of renal sodium handling between Tg and Wt mice.
As shown in Figure 3A, although sodium balance was comparable in Tg and Wt mice at baseline, the extent of daily positive sodium balance was significantly reduced in Tg mice compared with Wt mice during Ang II infusion (2-way repeated measures ANOVA F=11.37; P=0.0046). Furthermore, the extent of cumulative positive sodium balance during the early phase (day 1–6) was also significantly decreased in Tg mice compared with Wt mice (Figure 3B; 2-way repeated measures ANOVA F=7.04; P=0.043) consistently with facilitated natriuresis as a mechanism for the resistance to hypertension in Tg mice.
Figure 3.
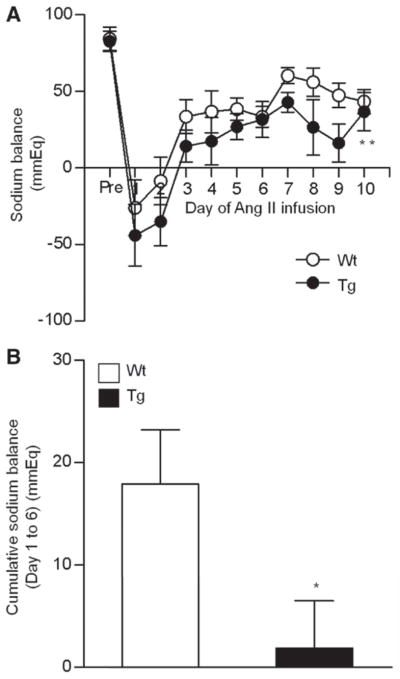
Effects of angiotensin II (Ang II) infusion on sodium balance in wild-type (Wt) and renal Ang II type 1 receptor-associated protein transgenic (Tg) mice. A, Daily and 24-hour sodium balance in Wt and Tg mice before (pre) and during Ang II (2000 ng/kg per min) infusion. Values are expressed as the mean±SE (n=6 in each group), **P<0.01 vs Wt mice. B, Cumulative sodium balance during the 6 days (day 1–6) of Ang II infusion in Wt and Tg mice. Values are expressed as the mean± SE (n=6 in each group), *P<0.05 vs Wt mice.
With respect to the role of increased natriuresis during the later phase (day 7–9) in the lower BP in Tg mice (Figure 3A and Figure S2D), the difference in SBP between Tg and Wt mice became larger from day 8 to day 11 (Figure 2C; the SBP difference between Tg and Wt mice, 17 mm Hg on day 8 and 31 mm Hg on day 11), which also is consistent with facilitated natriuresis as the mechanism for the resistance to hypertension in Tg mice. However, body weight changes tended to be larger in Tg mice than Wt mice, but the differences did not reach statistical significance (Figure S2E). Accordingly, these results indicate that renal distal tubule–dominant overexpression of ATRAP suppressed Ang II–dependent hypertension, probably via a suppression of sodium reabsorption in vivo.
Suppression of Phosphorylated Na+–Cl− Cotransporter and α-Subunit of the Epithelial Sodium Channel Expression in the Kidneys of Tg Mice
To examine mechanisms involved in the suppression of sodium reabsorption in response to Ang II in Tg mice, we compared renal mRNA expression of the major sodium transporters (sodium-proton antiporter 3, NHE3; sodium-potassium-two-chloride cotransporter, NKCC2; Na+–Cl− cotransporter, NCC; and epithelial sodium channel, ENaC subunits). Age-matched Wt and Tg mice were divided into 4 groups: (1) vehicle-infused Wt mice, (2) Ang II–infused Wt mice, (3) vehicle-infused Tg mice, and (4) Ang II–infused Tg mice. The results of quantitative real time–polymerase chain reaction analysis showed that Ang II infusion for 11 days significantly increased the renal mRNA levels of αENaC by 2.3-fold, and the βENaC and γENaC mRNA levels also tended to increase in response to Ang II infusion, but without statistical significance in Wt mice (Figure S3). On the contrary, the Ang II–mediated upregulation of αENaC mRNA was significantly suppressed in Tg mice.
With respect to protein expression of sodium transporters, the renal NHE3 protein levels were similar in Tg and Wt mice at baseline and decreased to a similar degree after Ang II infusion (Figure 4A). The phosphorylated NKCC2 levels were similar in Tg and Wt mice at baseline and decreased in both groups by Ang II with a tendency to be lower in Tg mice than in Wt mice but without statistical significance (30±6 versus 46±6%, P= 0.086; Figure 4B). However, although expression of phosphorylated NCC, which is the activated form of NCC and plays an important role in sodium reabsorption, was increased by Ang II infusion by 2.2-fold in Wt mice, the Ang II–mediated induction of phosphorylated NCC was significantly suppressed in Tg mice (Figure 4C). Furthermore, the Ang II–mediated increase in the renal αENaC protein expression, which was observed in Wt mice (1.9-fold), was abolished in Tg mice (Figure 4D).
Figure 4.

Suppression of the angiotensin II (Ang II)-mediated renal sodium chloride cotransporter (NCC) activation and α-subunit of the epithelial sodium channel (αENaC) upregulation in renal Ang II type 1 receptor-associated protein transgenic (Tg) mice. Effects of Ang II (2000 ng/kg per min) infusion on protein expression of the major sodium transporters, sodium-proton antiporter 3 (NHE3, A), sodium-potassium-two-chloride cotransporter (NKCC2, B), NCC (C), αENaC (D), βENaC (E), and γENaC (F) in the kidneys of wild-type (Wt) and Tg mice. Values are expressed as the mean±SE (n=6 in each group). *P<0.05 vs vehicle. †P<0.05 vs Wt mice.
To further examine whether the cellular localization of αENaC at the apical membrane is altered in Tg mice, confocal microscopy analysis using anti-αENaC antibody was performed. Under baseline conditions, both Wt and Tg mice exhibited a similar αENaC immunostaining pattern in renal cortex, with a denser staining at the apical membrane of distal tubule cells (Figure 5A). Ang II infusion did not obviously affect the cellular distribution of αENaC immunostaining in either Wt or Tg mice but strongly enhanced αENaC immunostaining intensity only in Wt mice. These findings suggest that the inhibitory effect of distal tubule ATRAP on sodium reabsorption in response to Ang II is not caused by suppressed localization of αENaC to the apical membrane but rather is mediated through downregulation of αENaC expression.
Figure 5.

Suppression of α-subunit of the epithelial sodium channel (αENaC) by renal angiotensin II type 1 receptor-associated protein (ATRAP) is not caused by an effect on the trafficking of αENaC to the apical membrane but through the regulation of αENaC expression levels. A, Representative confocal laser-scanning microscopy image (×80) of the renal cortex from wild-type (Wt) and transgenic (Tg) mice. Phalloidin is green color; DAPI, blue; αENaC, red. Ang II, on day 11 after the start of Ang II infusion. B, Effects of Ang II and adenoviral transfer of recombinant ATRAP on mRNA expression of the α-, β-, and γ-subunits of ENaC in mouse distal convoluted tubule (mDCT) cells. Forty-eight hours after infection with adenoviral vector containing ATRAP cDNA (Ad.HA-ATRAP) or control bacterial β-galactosidase cDNA (Ad.LacZ), cells were stimulated with vehicle or Ang II at 10−6 mol/L for 24 hours. Values are calculated relative to those achieved with extracts from mDCT cells infected with Ad.LacZ and stimulated with vehicle and are expressed as means±SE (n=10–12 in each group). *P<0.05 vs vehicle.
ENaC is activated by aldosterone through its binding to the mineralocorticoid receptor. Therefore, to analyze the direct effect of ATRAP on αENaC, we examined whether overexpression of ATRAP would suppress the Ang II–mediated ENaC subunit expression in mouse DCT cells by performing adenoviral transfer of recombinant ATRAP. Although Ang II (10−6 mol/L) treatment of mouse DCT cells infected with control bacterial β-galactosidase cDNA (Ad. LacZ) increased the αENaC mRNA expression, mouse DCT cells infected with adenoviral vector containing ATRAP cDNA (Ad.HA-ATRAP) exhibited an inhibition of the Ang II-induced enhancement (Figure 5B), thereby indicating that ATRAP directly suppressed the Ang II–mediated activation of αENaC expression, independent of the aldosterone-mineralocorticoid receptor pathway.
Discussion
This is the first report, to the best of our knowledge, of an inhibitory function of renal tubular ATRAP in angiotensin-dependent hypertension without an influence on baseline BP. In this study, chronic Ang II infusion was performed at 1000 and 2000 ng/kg per min to examine the effects of distal tubule–dominant overexpression of ATRAP on the Ang II–mediated BP increase. Although the higher dose of Ang II (2000 ng/kg per min) is reported to provoke a reduction in food intake and to cause Ang II–induced wasting and skeletal muscle atrophy, 17 the lower dose of Ang II (1000 ng//kg per min) has been used in many previously performed experiments in mice, 18,19 and suppression of the Ang II–induced BP increase by the distal tubule–dominant overexpression of ATRAP was observed with both the lower and higher doses of Ang II in the present study.
In the present study, the BP at baseline was not affected by renal ATRAP overexpression. On the contrary, the genetic inactivation of other renin–angiotensin system components, such as angiotensinogen, renin, and AT1R, was reported to result in significant decreases in BP, as well as an alteration in renal morphology and function compared with Wt mice even under baseline conditions. 20,21 Thus, ATRAP would be expected to act as a minor player among the renin–angiotensin system components, at least in terms of BP regulation and renal morphological development under physiological conditions. However, the results of present study seem to be consistent with those of our previous studies, which showed that ATRAP is not a general inhibitor of the AT1R signaling as are the clinically available AT1R-specific blockers, but rather specifically inhibits the pathological activation of its downstream signaling with preservation of baseline physiological signaling activity.9–13
With regard to the regulatory role of renal tubule AT1R in renal sodium handling, a previous study reported that Ang II did not affect proximal tubule fluid reabsorption or sodium delivery to distal nephron segments, but sodium reabsorption in distal nephron segments was increased in Ang II–infused mice. 22 Hashimoto et al 23 also observed that disruption of tissue angiotensin-converting enzyme did not alter proximal tubule fluid reabsorption. These results suggest that the distal nephron segments play a role in AT1R signal–mediated renal sodium reabsorption in vivo.
On the contrary, Li et al 24 have shown that a reduction in baseline BP occurred when the proximal tubule AT1R was selectively targeted in the kidney. In addition, Gurley et al 25 examined the effect of proximal tubule–specific AT1R deletion, using mice lacking AT1R only in the renal proximal tubule (PTKO mice), on Ang II–mediated BP elevation and showed that SBP elevation by Ang II (1000 ng/kg per min) was 15 mm Hg lower in PTKO mice than in Wt mice (control) (PTKO versus control, 23 versus 38 mm Hg increase on telemetry) with suppression of antinatriuresis, thereby indicating an important role of proximal tubule AT1R in angiotensin-dependent sodium retention and hypertension. In the present study, SBP elevation by Ang II was 19 mm Hg lower in Tg mice than in Wt mice (Tg versus Wt, 17 versus 36 mm Hg increase on telemetry) on the same dose of Ang II (1000 ng/kg per min) concomitantly with promotion of natriuresis. However, because strain backgrounds of these genetic engineered mice were different among the studies, further studies are needed to examine whether the inhibitory effect of distal tubule–dominant ATRAP activation on angiotensin-dependent sodium retention and BP elevation is comparable with that of proximal tubule–specific AT1R blockade.
In the proximal tubules, NHE3 plays an important role in sodium reabsorption, and previous in vitro studies reported that Ang II stimulation increases NHE3 expression to increase sodium reabsorption. 26,27 In addition, in the medullary thick ascending limb, NKCC2 is a major sodium transporter and is involved in sodium reabsorption. The present study showed that abundance of NHE3 and activation of NKCC2 were equivalent at baseline in the 2 groups and fell to a similar extent by Ang II infusion. The downregulation of these renal sodium transporters in response to Ang II–mediated hypertension may be 1 mechanism facilitating natriuresis as pressure increases, 28 which is consistent with the results observed by other group in the same Ang II–mediated hypertensive mice. 25
In the distal nephron, the modulation of sodium reabsorption in response to stimuli, such as Ang II, is mediated by NCC and ENaCs. 22,29–33 The results of recent studies showed that Ang II induces phosphorylation of the renal NCC through with-no-lysine kinase 4–dependent pathway, independent of aldosterone. 30,32,33 However, the ENaCs consist of 3 homologous subunits (α, β, and γ), and αENaC is reported to play an essential role in the formation of a functional ion channel among the ENaC subunits. 34,35 Previous studies also showed a regulatory role of AT1R signaling in the renal αENaC expression and an antihypertensive effect of ENaC blockade in angiotensin-dependent hypertension. 36,37 In the present study, we demonstrated that the enhancement of ATRAP in the distal nephron significantly suppressed the activation of NCC and the upregulation of αENaC by Ang II stimulation in vivo and, further, that overexpression of ATRAP completely suppressed Ang II–mediated activation of αENaC expression using mouse DCT cells. These results suggest that inhibition of NCC activity and downregulation of αENaC expression are likely to be involved in the suppression of angiotensin-dependent hypertension in renal ATRAP Tg mice.
Nevertheless, a limitation of the present study is that the results do not allow us to completely distinguish ATRAP functions in the distal tubules of the kidney. Tg mice with distal tubule–dominant overexpression of ATRAP were unexpectedly and fortuitously obtained on screening for cellular expression in these Tg animals. This model is not a specifically targeted cellular overexpression model, but rather a model in which there is variation in ATRAP expression within the nephron. Although the distal tubule is a predominant ATRAP expression site, other nephron segments, including the proximal tubules, do overexpress ATRAP to some degree in Tg mice. Therefore, it is necessary to further investigate the role of renal ATRAP in angiotensin-dependent hypertension in vivo using cellular-targeted models. Another limitation is the lack of functional data with regard to the activity of NCC and NKCC2, such as diuretic tests or clearance experiments. In addition, because 33.7-fold increase in ATRAP mRNA expression in the distal tubules of Tg mice compared with Wt mice could not completely inhibit the Ang II–mediated NCC activation, the effect of ATRAP seems to be, at best, minor in the present study.
Supplementary Material
Perspectives.
Hypertension is the most common chronic disease worldwide. It is a multifactorial disease in which genetic and environmental factors are intricately intertwined. Understanding the mechanism underlying hypertension is thus extremely complex, and caution should be used in interpreting the findings of this study in Tg mice in terms of the pathophysiology of human hypertension. Nevertheless, the findings of the present study do provide a useful basis for the further investigation of the in vivo functional roles of ATRAP in angiotensin-dependent hypertension and also suggest the potential benefit of an ATRAP activation strategy.
Novelty and Significance.
What Is New?
Angiotensin II (Ang II) type 1 receptor–associated protein (ATRAP), a specific binding molecule to Ang II type 1 receptor, inhibits pathological activation of Ang II type 1 receptor in local tissues but is downregulated in the kidney by Ang II. In transgenic mice dominantly expressing ATRAP in renal distal tubules, Ang II–induced hypertension was found to be attenuated with a concomitant increase in natriuresis via a suppression of the epithelial sodium channel.
What Is Relevant?
A potential therapeutic effect of ATRAP activation in the renal distal tubule on Ang II–mediated salt-sensitive hypertension was implicated. This observation suggests that ATRAP is a target of interest in hypertension.
Summary
The findings in this study suggest a possible role for renal distal tubule ATRAP in blood pressure regulation.
Acknowledgments
We thank Yuichi Koide, Tomoaki Ishigami, Toru Dejima, Tomohiko Kanaoka, Machiko Yabana (Yokohama City University), and Yoshihiro Noda (Tokyo Metropolitan Institute of Gerontology) for help in the experiments. We also thank Yutaka Kakizoe (Kumamoto University) and Shih-Hua Lin (Tri-Service General Hospital) for kindly providing antibodies. We thank Tetsuya Fujikawa (Yokohama City University) for statistical analysis.
Sources of Funding
This work was supported by a Health and Labor Sciences Research grant and by grants from the Japanese Ministry of Education, Science, Sports and Culture, the Salt Science Research Foundation (No. 1134), the Kidney Foundation, Japan (JKFB11-25), and the Novartis Foundation for Gerontological Research (2012).
Footnotes
The online-only Data Supplement is available with this article at http://hyper.ahajournals.org/lookup/suppl/doi:10.1161/HYPERTENSIONAHA.111.00572/-/DC1.
Disclosures
None.
References
- 1.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 2.Coffman TM, Crowley SD. Kidney in hypertension: guyton redux. Hypertension. 2008;51:811–816. doi: 10.1161/HYPERTENSIONAHA.105.063636. [DOI] [PubMed] [Google Scholar]
- 3.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension. 2011;57:355–362. doi: 10.1161/HYPERTENSIONAHA.110.163519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guyton AC. Blood pressure control–special role of the kidneys and body fluids. Science. 1991;252:1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 5.Hein L, Meinel L, Pratt RE, Dzau VJ, Kobilka BK. Intracellular trafficking of angiotensin II and its AT1 and AT2 receptors: evidence for selective sorting of receptor and ligand. Mol Endocrinol. 1997;11:1266–1277. doi: 10.1210/mend.11.9.9975. [DOI] [PubMed] [Google Scholar]
- 6.Miura S, Saku K, Karnik SS. Molecular analysis of the structure and function of the angiotensin II type 1 receptor. Hypertens Res. 2003;26:937–943. doi: 10.1291/hypres.26.937. [DOI] [PubMed] [Google Scholar]
- 7.Daviet L, Lehtonen JY, Tamura K, Griese DP, Horiuchi M, Dzau VJ. Cloning and characterization of ATRAP, a novel protein that interacts with the angiotensin II type 1 receptor. J Biol Chem. 1999;274:17058–17062. doi: 10.1074/jbc.274.24.17058. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Ilasaca M, Liu X, Tamura K, Dzau VJ. The angiotensin II type I receptor-associated protein, ATRAP, is a transmembrane protein and a modulator of angiotensin II signaling. Mol Biol Cell. 2003;14:5038–5050. doi: 10.1091/mbc.E03-06-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka Y, Tamura K, Koide Y, Sakai M, Tsurumi Y, Noda Y, Umemura M, Ishigami T, Uchino K, Kimura K, Horiuchi M, Umemura S. The novel angiotensin II type 1 receptor (AT1R)-associated protein ATRAP down-regulates AT1R and ameliorates cardiomyocyte hypertrophy. FEBS Lett. 2005;579:1579–1586. doi: 10.1016/j.febslet.2005.01.068. [DOI] [PubMed] [Google Scholar]
- 10.Azuma K, Tamura K, Shigenaga A, Wakui H, Masuda S, Tsurumi-Ikeya Y, Tanaka Y, Sakai M, Matsuda M, Hashimoto T, Ishigami T, Lopez-Ilasaca M, Umemura S. Novel regulatory effect of angiotensin II type 1 receptor-interacting molecule on vascular smooth muscle cells. Hypertension. 2007;50:926–932. doi: 10.1161/HYPERTENSIONAHA.107.096115. [DOI] [PubMed] [Google Scholar]
- 11.Tamura K, Tanaka Y, Tsurumi Y, Azuma K, Shigenaga A, Wakui H, Masuda S, Matsuda M. The role of angiotensin AT1 receptor-associated protein in renin-angiotensin system regulation and function. Curr Hypertens Rep. 2007;9:121–127. doi: 10.1007/s11906-007-0022-6. [DOI] [PubMed] [Google Scholar]
- 12.Wakui H, Tamura K, Tanaka Y, et al. Cardiac-specific activation of angio-tensin II type 1 receptor-associated protein completely suppresses cardiac hypertrophy in chronic angiotensin II-infused mice. Hypertension. 2010;55:1157–1164. doi: 10.1161/HYPERTENSIONAHA.109.147207. [DOI] [PubMed] [Google Scholar]
- 13.Masuda S, Tamura K, Wakui H, et al. Expression of angiotensin II type 1 receptor-interacting molecule in normal human kidney and IgA nephropathy. Am J Physiol Renal Physiol. 2010;299:F720–F731. doi: 10.1152/ajprenal.00667.2009. [DOI] [PubMed] [Google Scholar]
- 14.Tsurumi Y, Tamura K, Tanaka Y, Koide Y, Sakai M, Yabana M, Noda Y, Hashimoto T, Kihara M, Hirawa N, Toya Y, Kiuchi Y, Iwai M, Horiuchi M, Umemura S. Interacting molecule of AT1 receptor, ATRAP, is colocalized with AT1 receptor in the mouse renal tubules. Kidney Int. 2006;69:488–494. doi: 10.1038/sj.ki.5000130. [DOI] [PubMed] [Google Scholar]
- 15.Wakui H, Tamura K, Matsuda M, Bai Y, Dejima T, Shigenaga A, Masuda S, Azuma K, Maeda A, Hirose T, Ishigami T, Toya Y, Yabana M, Minamisawa S, Umemura S. Intrarenal suppression of angiotensin II type 1 receptor binding molecule in angiotensin II-infused mice. Am J Physiol Renal Physiol. 2010;299:F991–F1003. doi: 10.1152/ajprenal.00738.2009. [DOI] [PubMed] [Google Scholar]
- 16.Matsuda M, Tamura K, Wakui H, Dejima T, Maeda A, Ohsawa M, Kanaoka T, Haku S, Azushima K, Yamasaki H, Saito D, Hirose T, Maeshima Y, Nagashima Y, Umemura S. Involvement of Runx3 in the basal transcriptional activation of the mouse angiotensin II type 1 receptor-associated protein gene. Physiol Genomics. 2011;43:884–894. doi: 10.1152/physiolgenomics.00005.2011. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida T, Semprun-Prieto L, Wainford RD, Sukhanov S, Kapusta DR, Delafontaine P. Angiotensin II reduces food intake by altering orexigenic neuropeptide expression in the mouse hypothalamus. Endocrinology. 2012;153:1411–1420. doi: 10.1210/en.2011-1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sparks MA, Parsons KK, Stegbauer J, Gurley SB, Vivekanandan-Giri A, Fortner CN, Snouwaert J, Raasch EW, Griffiths RC, Haystead TA, Le TH, Pennathur S, Koller B, Coffman TM. Angiotensin II type 1A receptors in vascular smooth muscle cells do not influence aortic remodeling in hypertension. Hypertension. 2011;57:577–585. doi: 10.1161/HYPERTENSIONAHA.110.165274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ong FS, Lin CX, Campbell DJ, Okwan-Duodu D, Chen X, Blackwell WL, Shah KH, Gonzalez-Villalobos RA, Shen XZ, Fuchs S, Bernstein KE. Increased angiotensin II-induced hypertension and inflammatory cytokines in mice lacking angiotensin-converting enzyme N domain activity. Hypertension. 2012;59:283–290. doi: 10.1161/HYPERTENSIONAHA.111.180844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niimura F, Labosky PA, Kakuchi J, Okubo S, Yoshida H, Oikawa T, Ichiki T, Naftilan AJ, Fogo A, Inagami T. Gene targeting in mice reveals a requirement for angiotensin in the development and maintenance of kidney morphology and growth factor regulation. J Clin Invest. 1995;96:2947–2954. doi: 10.1172/JCI118366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oliverio MI, Kim HS, Ito M, Le T, Audoly L, Best CF, Hiller S, Kluckman K, Maeda N, Smithies O, Coffman TM. Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci USA. 1998;95:15496–15501. doi: 10.1073/pnas.95.26.15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao D, Seth DM, Navar LG. Enhanced distal nephron sodium reabsorption in chronic angiotensin II-infused mice. Hypertension. 2009;54:120–126. doi: 10.1161/HYPERTENSIONAHA.109.133785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hashimoto S, Adams JW, Bernstein KE, Schnermann J. Micropuncture determination of nephron function in mice without tissue angiotensin-converting enzyme. Am J Physiol Renal Physiol. 2005;288:F445–F452. doi: 10.1152/ajprenal.00297.2004. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Weatherford ET, Davis DR, Keen HL, Grobe JL, Daugherty A, Cassis LA, Allen AM, Sigmund CD. Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1067–R1077. doi: 10.1152/ajpregu.00124.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurley SB, Riquier-Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13:469–475. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He P, Klein J, Yun CC. Activation of Na+/H+ exchanger NHE3 by angiotensin II is mediated by inositol 1,4,5-triphosphate (IP3) receptor-binding protein released with IP3 (IRBIT) and Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2010;285:27869–27878. doi: 10.1074/jbc.M110.133066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li XC, Hopfer U, Zhuo JL. AT1 receptor-mediated uptake of angiotensin II and NHE-3 expression in proximal tubule cells through a microtubule-dependent endocytic pathway. Am J Physiol Renal Physiol. 2009;297:F1342–F1352. doi: 10.1152/ajprenal.90734.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonough AA, Leong PK, Yang LE. Mechanisms of pressure natriuresis: how blood pressure regulates renal sodium transport. Ann N Y Acad Sci. 2003;986:669–677. doi: 10.1111/j.1749-6632.2003.tb07281.x. [DOI] [PubMed] [Google Scholar]
- 29.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT(1) receptors. J Am Soc Nephrol. 2002;13:1131–1135. doi: 10.1097/01.asn.0000013292.78621.fd. [DOI] [PubMed] [Google Scholar]
- 30.San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, Gamba G. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci USA. 2009;106:4384–4389. doi: 10.1073/pnas.0813238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christensen BM, Perrier R, Wang Q, Zuber AM, Maillard M, Mordasini D, Malsure S, Ronzaud C, Stehle JC, Rossier BC, Hummler E. Sodium and potassium balance depends on αENaC expression in connecting tubule. J Am Soc Nephrol. 2010;21:1942–1951. doi: 10.1681/ASN.2009101077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Lubbe N, Lim CH, Fenton RA, Meima ME, Jan Danser AH, Zietse R, Hoorn EJ. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int. 2011;79:66–76. doi: 10.1038/ki.2010.290. [DOI] [PubMed] [Google Scholar]
- 33.Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA. 2012;109:7929–7934. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 35.Pratt JH. Central role for ENaC in development of hypertension. J Am Soc Nephrol. 2005;16:3154–3159. doi: 10.1681/ASN.2005050460. [DOI] [PubMed] [Google Scholar]
- 36.Brooks HL, Allred AJ, Beutler KT, Coffman TM, Knepper MA. Targeted proteomic profiling of renal Na(+) transporter and channel abundances in angiotensin II type 1a receptor knockout mice. Hypertension. 2002;39(2 pt 2):470–473. doi: 10.1161/hy02t2.102959. [DOI] [PubMed] [Google Scholar]
- 37.Beutler KT, Masilamani S, Turban S, Nielsen J, Brooks HL, Ageloff S, Fenton RA, Packer RK, Knepper MA. Long-term regulation of ENaC expression in kidney by angiotensin II. Hypertension. 2003;41:1143–1150. doi: 10.1161/01.HYP.0000066129.12106.E2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.