

Abstract
Autophagy is dysfunctional in many degenerative diseases including myopathies. Mutations in valosin-containing protein (VCP) cause inclusion body myopathy (IBM) associated with Paget's disease of the bone, fronto-temporal dementia and amyotrophic lateral sclerosis (IBMPFD/ALS). VCP is necessary for protein degradation via the proteasome and lysosome. IBMPFD/ALS mutations in VCP disrupt autophagosome and endosome maturation resulting in vacuolation, weakness and muscle atrophy. To understand the regulation of autophagy in VCP-IBM muscle, we examined the AKT/FOXO3 and mammalian target of rapamycin (mTOR) pathways. Basal Akt and FOXO3 phosphorylation was normal. In contrast, the phosphorylation of mTOR targets was decreased. Consistent with this, global protein translation was diminished and autophagosome biogenesis was increased in VCP-IBM muscle. Further mTORC1 inhibition with rapamycin hastened weakness, atrophy and vacuolation in VCP-IBM mice. This was accompanied by the accumulation of autophagic substrates such as p62, LC3II and ubiquitinated proteins. The decrease in mTOR signaling was partially rescued by insulin and to a lesser extent by amino acid (AA) stimulation in VCP-IBM muscle. Cells expressing catalytically inactive VCP or treated with a VCP inhibitor also failed to activate mTOR upon nutrient stimulation. Expression of a constitutively active Rheb enhanced mTOR activity and increased the fiber size in VCP-IBM mouse skeletal muscle. These studies suggest that VCP mutations may disrupt mTOR signaling and contribute to IBMPFD/ALS disease pathogenesis. Treatment of some autophagic disorders with mTOR inhibitors such as rapamycin may worsen disease.
INTRODUCTION
Dominantly inherited mutations in valosin-containing protein (VCP) are associated with the multisystem disorder inclusion body myopathy (IBM) associated with Paget's disease of the bone (PDB), fronto-temporal dementia (FTD) and amyotrophic lateral sclerosis (IBMPFD/ALS) or inclusion body myopathy (IBM) associated with PDB, FTD and ALS (1,2). Of these phenotypes, muscle weakness is present in >90% of patients and VCP-associated muscle disease (VCP-IBM) is pathologically the most well characterized (3). VCP-IBM has features of myopathy with vacuolated structures containing proteinaceous and membranous debris (3–5).
Consistent with these pathological changes, VCP was recently found to play a central role in lysosomal protein degradation via autophagy and endocytic pathways (6–10). Chemical or genetic inhibition of VCP function or IBMPFD/ALS mutant VCP expression blocks autophagosome maturation (6,8,11). Impaired autophagosome maturation leads to the accumulation of non-degradative autophagosomes that fail to fuse with lysosomes in VCP-IBM mouse skeletal muscle and IBMPFD/ALS human patients (6,8). VCP also participates in the endolysosomal degradation of plasma membrane proteins by facilitating the sorting of ubiquitinated endocytic cargo (7,9,10). The vacuoles in IBMPFD/ALS patients and skeletal muscle contain autophagic and endocytic markers such as p62, LC3 and LAMP-2 (7,8,12). Endocytic and autophagic dysfunction may underlie the pathogenesis of IBMPFD/ALS and other myopathies such as acid maltase deficiency, Danon's disease and sporadic inclusion body myositis (sIBM) (13). How these changes in lysosomal protein degradation lead to muscle atrophy and weakness is unclear.
IBMPFD/ALS has been modeled in mice via muscle-specific overexpression, systemic overexpression or genetic knock-in of the most common IBMPFD/ALS mutation VCP-R155H (12,14,15). These models recapitulate many of the prominent phenotypes seen in IBMPFD/ALS. One common feature in all of these mouse models is progressive late-onset weakness, muscle degeneration and vacuolar pathology consistent with VCP-IBM. The selective overexpression of VCP-R155H mutation in skeletal muscle (VCP-IBM mice) affords a unique opportunity to study skeletal muscle function and pathology without confounding motor neuron disease, bone abnormalities and dementia that is seen in other IBMPFD/ALS mouse models (15).
Autophagy is essential for normal skeletal muscle homeostasis. Mice lacking skeletal muscle autophagy due to muscle-specific deletion of ATG7 are viable but develop age-associated weakness, myopathic features, ubiquitinated inclusions and aberrant mitochondria (16). The regulation of autophagy in skeletal muscle is mediated via several pathways. FOXO3 induces coordinated transcription of proteolytic effectors such as the E3 ligases, Atrogin-1/muscle atrophy F-box protein and Murf-1 and the autophagy inducers beclin-1 and BNIP3 in the setting of starvation, disuse and denervation-induced muscle atrophy (17,18). Autophagy is activated in skeletal muscle during exercise in a BCL2-beclin-1 dependent manner (19). The metabolic regulator mammalian target of rapamycin (mTOR) also mediates skeletal muscle autophagy. Inhibition of mTOR with drugs such as rapamycin induces autophagy similar to that seen following nutrient deprivation in mouse skeletal muscle (20).
We reasoned that pathways which regulate autophagy may contribute to IBMPFD/ALS pathogenesis. One possible candidate is mTOR. mTOR is responsible for integrating multiple metabolic signaling pathways to promote protein synthesis, cell growth and autophagy (21). Enhanced mTOR activity leads to muscle hypertrophy (22). The consequence of diminished mTOR function in skeletal muscle is less clear; however, genetic deletion of mTOR in skeletal muscle leads to a degenerative myopathy (23). Modulation of mTOR-mediated autophagy is an attractive therapy in skeletal muscle disease since many drugs such as rapamycin are currently available (24,25). In order to identify a potential therapy for autophagic myopathies, we evaluated mTOR signaling and autophagic regulation in VCP-IBM mice that have a hereditary form of IBM due to mutations in VCP.
RESULTS
mTOR activity is diminished in VCP-IBM mice
To evaluate autophagic regulation in VCP-IBM, we immunoblotted skeletal muscle lysates from presymptomatic 5-month-old control, VCP-WT expressing mice or one of the two VCP-IBM mouse lines that express the VCP-R155H mutation (VCP-RH9 or VCP-RH12) with antibodies specific for the phosphorylation states of AKT, FOXO3 and the S6 ribosomal subunit. Basal levels of phosphorylated AktSer473 (pAkt) and phosphorylated FOXO3Ser253 (pFOXO3) were similar in control, VCP-WT, VCP-RH9, and VCP-RH12 muscle lysates (Fig. 1A). In addition, expression of downstream FOXO3 targets such as the E3 ligase Atrogin-1 and Murf-1 or the levels of Beclin-1 and BNIP3 was unchanged (Fig. 1B and C). This was in contrast to the levels of phosphorylated S6Ser240/244 (pS6) which were reduced in both VCP-IBM mouse lines when compared with control and VCP-WT expressing mice (Fig. 1A, D and E). This suggested that mTOR signaling independent of Akt activity was decreased in VCP-RH expressing mouse muscle. To further evaluate mTOR activity, we measured the phosphorylation states of two direct targets of mTOR, 4EBP1 and p70S6 kinase. Phosphorylated 4EBP1Thr37/46 (p4EBP1) and phosphorylated p70S6 kinase-1Thr389 (pp70S6K) were decreased in VCP-RH expressing muscle when compared with VCP-WT muscle (Fig. 1D and E). This change was similar to that seen in skeletal muscle when mice were treated with 10 mg/kg intraperitoneal (i.p.) rapamycin for 12 h (Fig. 1D).
Figure 1.
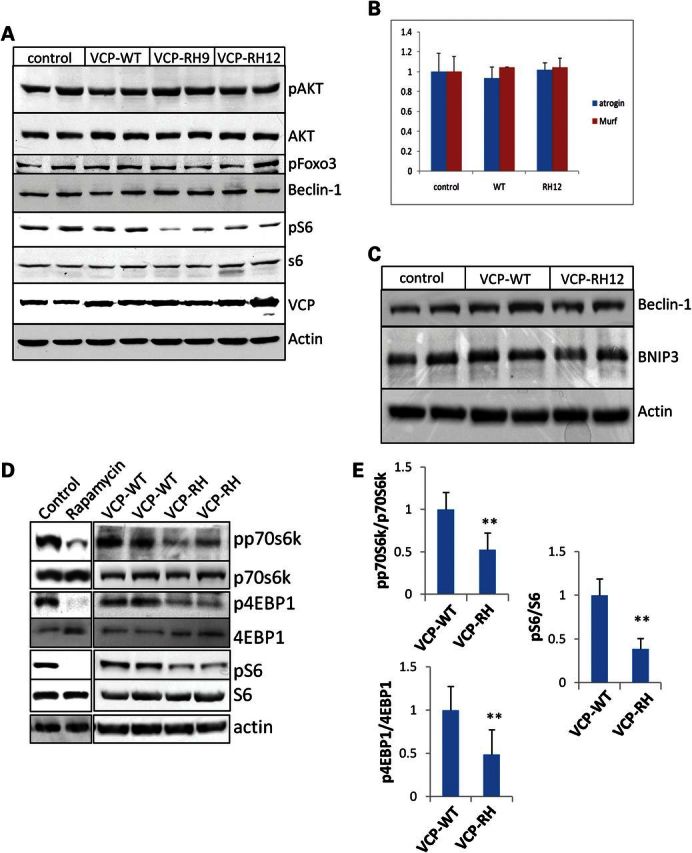
mTOR activity is diminished in VCP-IBM mouse muscle. (A) Immunoblot analysis of phosphorylated AktSer473 (pAkt), total Akt, phosphorylated FOXO3Ser253 (pFOXO3), phosphorylated S6Ser240/244 (pS6), total S6, VCP or actin in the TA muscle of control, VCP-WT or one of the two independent VCP-R155H expressing mouse lines (VCP-RH9 or VCP-RH12). (B) Quantitative PCR analysis of Atrogin-1 and Murf-1 transcript levels in TA of control, VCP-WT or VCP-RH12 mice. (C) Immunoblot of beclin-1, BNIP3 or actin in the TA of control, VCP-WT or VCP-RH12 mice. (D) Immunoblot of phosphorylated p70S6 kinase-1Thr389 (pp70s6k), total p70S6 kinase-1 (p70s6k), phosphorylated 4EBP1Thr37/46 (p4EBP1), total 4EBP1, pS6, S6 and actin in the TA muscle of control, VCP-WT, VCP-RH or control mice treated with 10 mg/kg i.p. rapamycin for 12 h. (E) Quantitation of the densitometric levels of the ratio of pp70S6K/p70S6K (n = 6 mice/condition); p4EBP1/4EBP1 (n = 8 mice/condition) and pS6/S6 (n = 4 mice/condition) in the skeletal muscle of VCP-WT and VCP-RH mice. **P < 0.001. All blots are representative of at least two independent experiments.
Many downstream targets of mTOR such as p70s6k and 4EBP1 modulate protein translation (21). Therefore, to test the amount of protein translation in mouse muscle, we performed in vivo surface sensing of translation (26,27). This technique administers puromycin to live animals. Puromycin is a structural analog of aminoacyl transfer RNAs, which is incorporated into the nascent polypeptide chain, and prevents elongation. Following i.p. administration of puromycin, mouse muscle is harvested and the level of puromycin incorporation into the translating polypeptides is measured with an antibody to puromycin via immunoblot (27). A decrease in anti-puromycin immunoreactivity indicates a decrease in protein translation. VCP-RH expressing mice had a decrease in puromycin incorporation consistent with a decrease in protein translation (Fig. 2A). The changes in puromycin immunoreactivity were similar to that seen when mice were treated with rapamycin (Fig. 2A).
Figure 2.
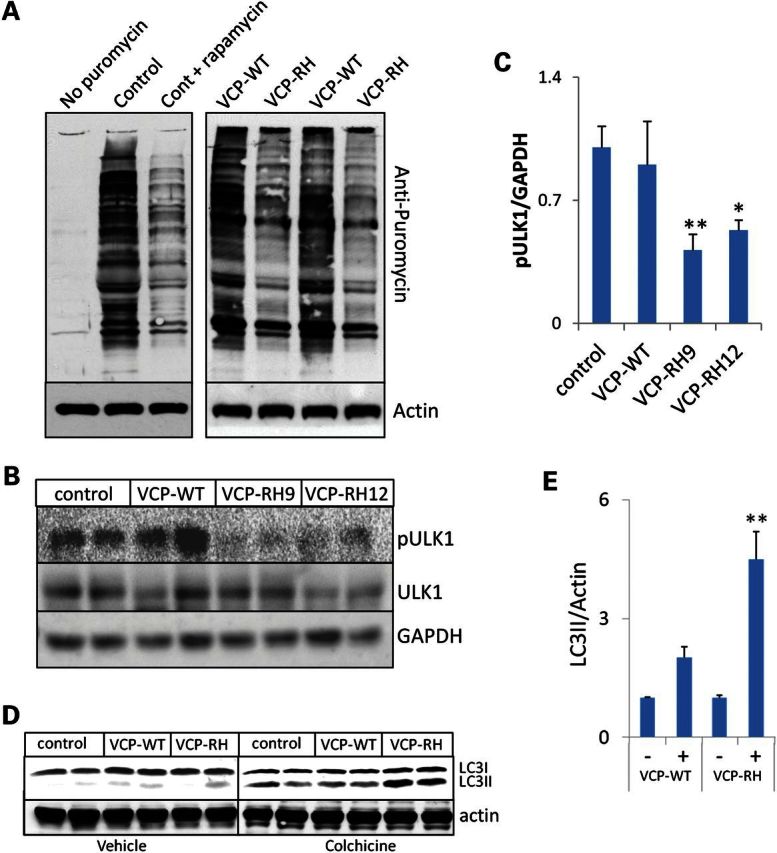
Protein translation is decreased and autophagosome biogenesis is increased in VCP-IBM mouse muscle. (A) Immunoblot analysis for incorporated puromycin and actin in TA of control, VCP-WT, VCP-RH or control mice treated with 10 mg/kg i.p. rapamycin for 12 h prior to the administration of i.p. puromycin. (B) Immunoblot for phosphorylated ULK1Ser757 (pULK1), total ULK1 and GAPDH from TA of control, VCP-WT, VCP-RH9 or RH12 mice. (C) Quantitation of the densitometric levels of the ratio of pULK1/GAPDH (n = 4 mice/condition) of control, VCP-WT, VCP-RH9 and VCP-RH12 mice. *P < 0.01 and **P < 0.001. (D) Immunoblot for LC3 and actin in TA of vehicle treated or 0.4 mg/kg i.p. colchicine for 24 h prior to harvesting skeletal muscle of control, VCP-WT and VCP-RH mice. (E) Quantitation of the densitometric levels of LC3II to actin ratio in TA of vehicle-treated (−) or 24 h of 0.4 mg/kg i.p. colchicine treatment (+) in VCP-WT and VCP-RH mice. **P < 0.001 and n = 6 mice/condition.
Autophagosome biogenesis is increased in VCP-IBM mouse muscle
mTOR phosphorylates ULK1, thus inhibiting autophagosome formation (28). We evaluated the level and phosphorylation of ULK1 using antibodies specific for ULK1 and phosphorylated ULK1Ser757 (pULK1) (Fig. 2B). Consistent with a decrease in mTOR activity, VCP-RH expressing mice had a decrease in the levels of pULK1 when compared with control or VCP-WT mouse muscle (Fig. 2B and C). We reasoned that the decrease in pULK1 identified above would favor autophagosome biogenesis in VCP-IBM mouse muscle. At 5 months of age, prior to the onset of weakness and obvious muscle pathology, the basal levels of LC3II were similar in control, VCP-WT and VCP-RH mouse muscle (Fig. 2D and E). In order to block the degradation of autophagosomes so that we could identify the amount of LC3II produced in 24 h, we treated mice with 0.4 mg/kg i.p. colchicine for 24 h prior to harvesting skeletal muscle. The levels of LC3II increased in all animals but increased more in VCP-RH mice, suggesting that they have an increase in autophagosome biogenesis (Fig. 2D and E).
Rapamycin exacerbates weakness and muscle pathology in VCP-IBM mice
To evaluate whether further mTOR inhibition would be detrimental to VCP-RH mice, we treated pre-symptomatic 5-month-old control, VCP-WT and VCP-RH expressing mice with 10 mg/kg i.p. rapamycin every other day and quantified muscle strength and rotarod performance. The 5-month-old VCP-RH mice were used as the greatest decline in muscle strength occurs between 5 and 9 months of age (15). Surprisingly after 21 days of treatment, VCP-RH mice had a significant decrease in grip strength and rotarod performance when compared with vehicle-treated VCP-RH mice (Fig. 3A and B). There was also a selective increase in serum creatine kinase levels in vehicle-treated VCP-RH mice which was further increased with rapamycin treatment when compared with VCP-WT mice (Fig. 3C). Rapamycin treated control and VCP-WT mice had no change in myofiber structure or size as measured by routine hematoxylin and eosin, congo red and gomori trichrome staining of the quadriceps or tibialis anterior (TA) muscles. VCP-RH mice do not develop vacuolar changes consistent with IBM in the quadriceps and TA muscles until 9 months of age (8,15) and 5-month-old vehicle-treated VCP-RH mice had unremarkable routine histopathology (Fig. 3D). In contrast, VCP-RH mice treated with every other day rapamycin for 21 days had a significant increase in the number of atrophic and vacuolated fibers (Fig. 3D–F). Control and VCP-WT mice have no evidence of vacuolar pathology at 5 months of age treated with or without rapamycin and thus, the number of vacuoles was not quantified. Some vacuoles from VCP-RH mice treated with 21 days of rapamycin were positive for LC3 via immunofluorescence, suggesting that they were indeed autophagic in origin (Fig. 3D). To further explore the mechanism of rapamycin-induced muscle weakness in VCP-RH expressing mouse, we evaluated the levels of p62, LC3, ubiquitinated proteins and BNIP3 via immunoblot in control, VCP-WT or VCP-RH mouse muscle following 21 days of treatment with rapamycin. Interestingly, these autophagic markers accumulated in VCP-RH mice treated with rapamycin when compared with VCP-WT and control mice (Fig. 4A and B). An increase in p62 was further visualized via immunohistochemistry of VCP-RH mice treated with vehicle or rapamycin for 21 days (Fig. 4C). In vehicle-treated VCP-RH mouse muscle, p62 is present diffusely throughout myofibers. In contrast, p62 is more granular in scattered fibers and accumulated in the sarcoplasm at structures consistent with vacuoles in VCP-RH mice treated for 21 days with rapamycin (Fig. 4C). There was no change seen in the intensity or distribution of p62 in VCP-WT treated with rapamycin for 21 days (Fig. 4C).
Figure 3.

Rapamycin treatment exacerbates weakness and muscle pathology in VCP-IBM mice. Five-month-old control, VCP-WT or VCP-RH mice were treated with vehicle or every other day dosing of 10 mg/kg i.p. rapamycin for 21 days. Graphs represent change in grip strength (A) or rotarod performance (B) from day 1. Results are obtained from four mice per group and three independent experiments for grip strength analysis and four mice per group and two independent experiments for rotarod performance. *P < 0.01. (C) Serum creatine phosphokinase (CPK) levels from mice VCP-WT or VCP-RH mice treated with vehicle or every other day 10 mg/kg i.p. rapamycin. (D) Congo red staining or LC3 immunofluorescence of TA muscle from 5-month-old VCP-RH mice treated with vehicle or every other day rapamycin for 21 days. Open arrows denote vacuolated fibers and closed arrows denote angular and atrophic fibers. Bar is 100 microns. (E) Quantitation of the percentage of vacuolated fibers and (F) percentage of fibers having a CSA of <200 µm from TA muscle of 5-month-old VCP-WT or VCP-RH mice treated with vehicle or every other day rapamycin (+) for 21 days. **P < 0.001.
Figure 4.

Autophagic substrates accumulate in VCP-IBM mice following chronic rapamycin treatment. (A) Immunoblot analysis for ubiquitinated proteins (FK2), BNIP3, p62, LC3 and GAPDH in TA from control, VCP-WT or VCP-RH mice treated with vehicle or every other day dosing of 10 mg/kg i.p. rapamycin for 21 days. (B) Quantitation of the densitometric levels of the ratio of p62/GAPDH or LC3II/GAPDH (n = 4 mice/condition) from the skeletal muscle of VCP-RH12 mice treated with vehicle or rapamycin for 21 days. (C) Immunohistochemistry for p62 (red) from VCP-WT or VCP-RH mice treated with vehicle or every other day dosing of 10 mg/kg i.p. rapamycin for 21 days. Arrows denote p62-positive vacuolar structures. Rightmost image is an enlargement of box in the middle image. Bar is 50 microns.
VCP-IBM mice fail to activate mTOR in response to nutrients
mTOR activity can be manipulated via activation of Akt following IGF1 or insulin stimulation (21). To confirm that the decrease in mTOR activity was not related to a defect in insulin signaling pathways through Akt, we treated VCP-WT and VCP-RH mice with i.p. insulin for 5 min and then harvested mouse muscle. Following insulin treatment, VCP-WT and VCP-RH mice had an increase in pAKT and pS6 levels (Fig. 5A). However, the levels of pS6 did not increase to the same levels as VCP-WT mice. mTOR activity can also be modulated via AMP-activated protein kinase (AMPK) (21). Therefore, we evaluated the phosphorylation of the α1 subunit of AMPKThr172 (pAMPK) and the phosphorylation of its downstream target acetyl-CoA carboxylase (pACC). pAMPK and pACC levels were similar in VCP-WT and VCP-RH mouse muscle (Fig. 5B). mTOR activity is also responsive to AAs. In particular, branched chain AAs, such as leucine, are able to activate mTOR independent of Akt or AMPK (21). To test this, we starved mice overnight and then treated mice with i.p. leucine 1 h prior to muscle harvesting. Sixteen hours of food deprivation decreased the levels of pS6 and treatment with leucine increased the pS6 levels in VCP-WT mice (Fig. 5C). VCP-RH mice failed to increase the levels of pS6 when compared with VCP-WT mice (Fig. 5C). Diminished activation of mTOR with i.p. leucine treatment was also detected when lysates were probed with antibodies to pp70S6k and p4EBP1 (Fig. 5D).
Figure 5.

VCP-IBM mice have a diminished mTOR response. (A) VCP-WT or VCP-RH12 (RH) mice were treated with saline or 0.75 units/kg i.p. insulin 5 min prior to sacrifice. TA muscle was subjected to immunoblot for pAKT, AKT, pS6 or S6. (B) Levels of pAMPK, AMPK or pACC, ACC were measured in VCP-WT and VCP-RH12 mice. (C) Untreated VCP-WT or VCP-RH mice were starved for 16 h or starved and then treated with 360 µm/100 g l-leucine 1 h prior to sacrifice. TA muscle was immunoblotted for pS6 and S6. Blots are representative of at least two animals from three separate experiments. (D) Immunoblot of pp70S6K, p70S6K, p4EBP1, 4EBP1 and actin from VCP-WT or VCP-RH mice starved for 16 h and then treated with i.p. l-leucine 1 h prior to sacrifice.
VCP activity is required for mTOR activation
To see if a decrease in VCP activity can explain the inhibition of mTOR function seen in VCP-IBM mouse muscle, we treated U20S cells with rapamycin or increasing concentrations of a potent VCP inhibitor, N2,N4-dibenzylquinazoline-2,4-diamine (DBeQ) (11), for 15 h prior to harvesting cells. Basal levels of pS6 and phosphorylated 4EBP1 were decreased in cells treated with rapamycin or 10 µm DBeQ (Fig. 6A). DBeQ (10 µm) has been shown to be the EC50 for VCP inhibition in cell culture (11). To see if DBeQ was able to inhibit serum-stimulated mTOR activation, we serum starved U20S cells for 15 h and then stimulated with 10% fetal bovine serum (FBS) for 1 h prior to harvesting cells. Some cells were also co-treated with rapamycin or 10 µm DBeQ and subjected to serum stimulation. DBeQ was able to potently inhibit serum-stimulated mTOR activity similar to rapamycin treatment as demonstrated by a decrease in pS6 levels (Fig. 6B). Similarly, U20S cells expressing a dominant-negative VCP (VCP-E578Q) that is unable to hydrolyze ATP or expressing the VCP-R155H disease mutation also had a decrease in their ability to activate mTOR as shown by decreased pS6 levels (Fig. 6C). The addition of DBeQ to VCP-E578Q and VCP-R155H expressing cells further decreased pS6 levels (Fig. 6C). This is likely explained by the potent nature of DBeQ which inactivates all VCPs, whereas in VCP-E578Q and VCP-R155H expressing cells there is still active endogenous VCP-WT. mTOR activity can be enhanced by the expression of Rheb (22). In some settings, Rheb expression can overcome a genetic or chemical inhibition of mTOR activity. To see whether a constitutively active Rheb (caRheb-N153T-Flag) could increase serum-stimulated mTOR activity in the setting of DBeQ treatment, VCP-E578Q or VCP-R155H expression, we transfected GFP or caRheb-N153T-flag into U20S cells. As expected, caRheb increased the levels of pS6 in VCP-WT expressing cells (Fig. 6C). This increase was less robust in VCP-E578Q expressing cells and absent in cells treated with DBeQ during serum stimulation (Fig. 6C). In contrast, VCP-R155H cells that expressed caRheb had an increase in pS6 levels similar to that seen in VCP-WT cells (Fig. 6C).
Figure 6.

VCP activity is required for mTOR activation. (A) Immunoblot analysis of pS6, S6, 4EBP1 or actin from U20S cells treated for 18 h with DMSO, rapamycin or DBeQ. 4EBP1 immunoblots have multiple bands. The higher molecular weight bands represent phosphorylated 4EBP1 (double arrows). Rapamycin treatment and 10 µm DBeQ decrease the levels of pS6 and lead to a shift in 4EBP1 to lower molecular weight non-phosphorylated bands consistent with reduced mTOR activity. (B) Immunoblot for pS6, S6 or actin from serum-starved U20S cells. Some cells were stimulated with 10% FBS for 1 h or with drug plus serum for 1 h prior to harvesting cells. (C) Immunoblot for pS6, S6, actin and Flag-tagged caRheb from U20S cells stably expressing VCP-WT, VCP-E578Q or VCP-R155H and transfected with a plasmid expressing GFP or caRheb-Flag. The cells were starved of serum for 18 h and then restimulated with 10% FBS for 1 h. Some cells were also treated with 10 µm DBeQ at the time of serum stimulation. Samples were isolated and blotted under the same conditions. Gels are rearranged from the same experiment for presentation purposes.
Rheb expression increases the myofiber size and activates mTOR in VCP-IBM mice
Rheb expression can increase the myofiber size when transiently expressed in mouse muscle (22). To see whether Rheb expression was able to improve the mTOR defect in VCP-IBM mouse muscle, we electroporated caRheb-N153T-Flag or a GFP control plasmid into control, VCP-WT or VCP-RH mouse TA muscle. Seven days later, we harvested muscle, cryosectioned and immunostained using an anti-Flag antibody and anti-collagen IV to delineate the sarcolemmal membrane. The cross sectional area (CSA) of GFP expressing, GFP non-expressing, Flag expressing and Flag non-expressing fibers was quantitated from six animals per cohort. As expected, the CSAs of GFP expressing fibers were not different from GFP non-expressing fibers. Consistent with previous studies (22), Rheb increased the CSA of mature myofibers by 78% in control and 88% in VCP-WT mouse muscle (Fig. 7A–C). Rheb expression increased the CSA in VCP-RH mouse muscle by 47% (Fig. 7A–C). This increase correlated with an increase in the phosphorylation of an exogenously electroporated p70S6K-HA-tagged construct when caRheb-N153T was co-expressed in VCP-RH muscle (Fig. 7D).
Figure 7.

Rheb expression increases the myofiber size and activates mTOR in VCP-IBM mice. (A) Graph of the mean CSA of untransfected, caRheb-Flag or GFP transfected TA myofibers 7 days post-electroporation from 5-month-old control, VCP-WT or VCP-RH mice. **P < 0.001. Error bars denote standard error. (B) Immunofluorescence with anti-Flag (red) and anti-collagen IV (green) of TA myofibers 7 days post-electroporation from 5-month-old control, VCP-WT or VCP-RH mice image. Stars denote transfected fibers. Bar is 50 microns. (C) Histogram of the percent of myofibers with the indicated CSA for control, VCP-WT or VCP-RH mice electroporated with GFP or caRheb expressing plasmids. (D) Immunoblot for pp70S6k, p70SK1-HA, pS6 or S6 of VCP-WT or VCP-RH TA muscle following 7-day post-electroporation with GFP or caRheb-Flag with p70S6K1-HA expression vector. *denotes phosphorylated p70S6K1-HA.
DISCUSSION
Our study identified a defect in mTOR activity in VCP-IBM mice. Diminished mTOR function leads to a decrease in protein synthesis and an increase in autophagosome formation in these mice. When mTORC1 is further suppressed with rapamycin in presymptomatic VCP-IBM mice, they become weaker and have worsened pathology. Since VCP is required for autophagosome maturation and IBMPFD/ALS mutations in VCP impair this function resulting in the accumulation of non-degradative autophagosomes (6,8), an increase in mTOR-mediated autophagic initiation may be detrimental (see the model in Fig. 8). Breaking this cycle through the activation rather than inhibition of mTOR may prove therapeutic in IBMPFD/ALS.
Figure 8.
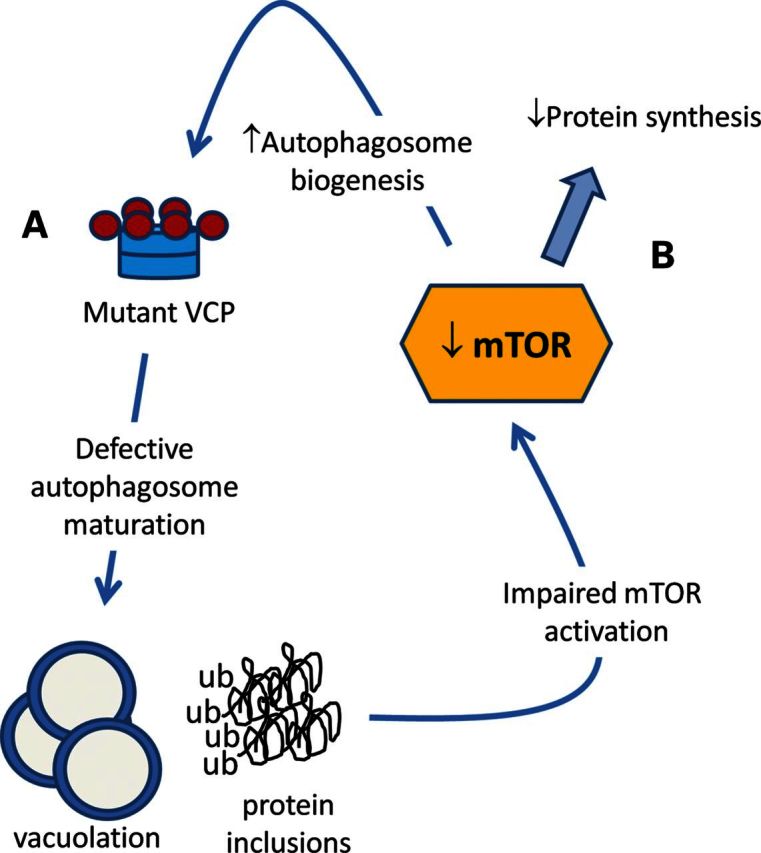
Model of mTOR function in IBMPFD/ALS. (A) Mutant VCP expression impairs autophagosome maturation leading to vacuolation and protein inclusions. (B) Diminished mTOR function leading to reduced protein synthesis and increased autophagosome biogenesis.
This mechanism of disease pathogenesis may not be unique to IBMPFD/ALS. Several muscular disorders are due to a defect in lysosome-mediated protein degradation including Pompe disease and X-linked myopathy with excessive autophagy (XMEA) (29,30). These vacuolar myopathies are characterized by the accumulation of autophagosome-lysosome membranes in the setting of failed autophagosome clearance. Fibroblasts from patients with XMEA fail to properly acidify their lysosomes resulting in the accumulation of non-degradative autophagosomes and also have a decrease in mTOR activity (30). Similarly, muscles from patients with sIBM, a myopathy with pathologic similarities to IBMPFD/ALS muscle disease, have diminished mTOR activity (31). Whether muscles from patients with Pompe disease, XMEA or sIBM have an increase in autophagosome biogenesis due to reduced mTOR activity remains to be established.
It is conceivable that other stimuli known to initiate autophagy in skeletal muscle may be detrimental to vacuolar myopathies such as IBMPFD/ALS. For example a patient's nutrient status or exercise regimen may result in enhanced autophagosome biogenesis similar to mTOR inhibition with rapamycin. In addition, autophagy is coordinately upregulated with proteasomal degradation during denervation and disuse atrophy (17,18). This may be especially relevant to IBMPFD/ALS in which denervation occurs in the setting of concomitant motor neuron disease and disuse atrophy occurs when disabling weakness is experienced later in the disease (1).
Why mTOR activity and its activation are diminished in VCP-IBM mouse muscle is unclear. Moreover, how inhibition of VCP activity disrupts mTOR function is not known. It is possible that mTOR is compensatorily downregulated in VCP-inhibited cells or VCP-IBM mouse muscle in response to diminished autophagic protein degradation. However, mice with a muscle-specific deletion of ATG7 that are deficient in skeletal muscle autophagy have unchanged mTOR function (16). Moreover, mice containing a BCL2 protein with mutations to multiple phosphorylation sites that render it unable to release from beclin1 and are thus deficient in stimulus-induced autophagy have normal AKT1-mTOR signaling (19). mTOR signaling requires translocation of mTOR to an acidic late endosomal compartment that contains Rheb (32). The integrity of the endolysosomal system is necessary for this process (33). Inhibition of VCP activity and IBMPFD/ALS mutant VCP expression disrupts endolysosomal trafficking resulting in enlarged non-acidic late endosomes (7). These aberrant endolysosomes may fail to localize Rheb or redistribute mTOR upon nutrient stimulation.
VCP may also be more directly involved in mTOR signaling. Proteasomal activity is required for mTOR activation (34). More specifically, the endogenous mTOR inhibitor DEPTOR is ubiquitinated by Skp, Cullin, F-box containing (SCF) (βTrCP) E3 ubiquitin ligase and subsequently degraded via the 26S proteasome (35,36). DEPTOR's degradation then leads to enhanced mTOR activity. VCP facilitates the degradation of ubiquitinated substrates via its interactions with selected ubiquitin adaptors and E3 ubiquitin ligases including SCF(βTrCP) (37). Whether DEPTOR is a VCP substrate remains to be established.
VCP may also associate with other members of the mTORC1 complex. The ubiquitin-binding protein, p62, also mutated in PDB and ALS (38–40) (both phenotypic variants of IBMPFD/ALS), has been shown to mediate mTOR activity via its direct association with raptor and mTOR (41). Upon nutrient stimulation, p62 is necessary for the proper localization of mTOR to lysosomal membranes. VCP through its association with ubiquitin adaptors segregates large macromolecular complexes at membrane interfaces such as the ER, endosome and mitochondrion (42). VCP could perform a similar role with mTORC1 components at the lysosome.
Mutations in VCP cause a spectrum of variably penetrant degenerative phenotypes in patients. These include IBM, PDB, FTD and ALS. IBM is the most common phenotypic feature associated with VCP mutations and our mouse model only expresses mutant VCP in skeletal muscle (15). This model has advantages and disadvantages. One clear advantage is the ability to isolate mouse behavioral studies to only changes in skeletal muscle strength and function since we would not have to be concerned about rapamycin's effect on bone, brain and spinal cord. In addition, we believe that muscle-specific VCP expression models both muscle pathology in IBMPFD/ALS but also other hereditary IBMs and vacuolar myopathies. It will be important to see whether the findings that we see in VCP-IBM mouse muscle are seen in other tissues affected in IBMPFD/ALS patients. However, it is conceivable that a similar pathogenic mechanism occurs in other tissues affected by VCP mutations. For example, treatment of an SOD1-G93A mouse model of ALS mice with rapamycin worsened pathology, increased protein inclusions and exacerbated weakness (43). In contrast treatment of a mouse model of Huntington's Disease with the rapamycin analog, CCI-779, improved behavior and reduced protein aggregate burden within the striatum (44).
The manipulation of autophagy as a potential therapy in skeletal muscle disease is particularly promising. Activation of reduced autophagy using mTOR inhibition or dietary modifications improves strength and muscle pathology in a mouse model of congenital muscular dystrophy associated with the absence of collagen VI (45). More recently, it was shown that rapamycin and its analog temsirolimus blocked the elevated mTOR activity that is present in mouse models of lamin A/C-associated myopathy and cardiomyopathy (24,25). mTOR inhibition normalized the diminished autophagy, improved muscle function and survival in laminopathy mouse models (24,25). In contrast, inhibition of autophagy with 3-methyladenine improved survival and muscle pathology in a mouse model of congenital muscular dystrophy associated with laminin-α2 chain deficiency (46). Whether it is better to enhance or inhibit autophagy in human muscle disease remains to be established.
In the present study, we utilized a caRheb construct to activate mTOR in VCP-IBM mouse muscle. caRheb effectively enhanced the phosphorylation of p70S6K and increased the myofiber size in VCP-IBM mouse muscle. Our study did not evaluate whether caRheb expression improved muscle strength, pathology or blocked autophagy due to the transient nature of caRheb expression. Rheb has been shown to inhibit ischemia-induced autophagy in Rheb transgenic mouse cardiac muscle (47). However, this inhibited autophagy resulted in enhanced cardiomyocyte death and larger infarct size (47). Future studies will need to be performed to see whether prolonged Rheb expression can abrogate weakness, vacuolation and inclusion bodies in VCP-IBM mice.
Using IBM-VCP mice, we identified a defect in mTOR signaling that results in enhanced autophagy and decreased protein synthesis. Since autophagosome maturation is defective in VCP-IBM muscle, the enhanced autophagy is detrimental to myofibers. Impaired mTOR signaling may directly relate to changes in VCP activity since VCP inhibition in cell culture also blocked mTOR signaling. We suggest that manipulation of the mTOR pathway may prove therapeutic in vacuolar myopathies such as VCP-IBM.
MATERIALS AND METHODS
Animals and experimental protocols
VCP-WT and VCP-RH (R155H) mice that express a human VCP complementary DNA (cDNA) under the control of a muscle creatine kinase promoter (15) were bred five generations to C57BL/6 and then to homozygosity. Control mice (C57BL/6) were purchased from Jackson Laboratories. All animal experimental protocols were approved by the Animal Studies Committee of Washington University School of Medicine. Mice were housed in a temperature-controlled environment with 12 h light–dark cycles where they received food and water ad libitum.
Depending on the experimental setting, mice were subjected to i.p. injection of either rapamycin (10 mg/kg body weight, LC Laboratories) every other day for 21 days, l-leucine (360 µMoles/100 g body weight, Sigma) for 1 h, or insulin (0.75 units/kg body weight, Sigma) for 5 min. Rapamycin was dissolved in 100% ethanol for a 62.5 mg/ml stock solution and stored at −20°C. Immediately prior to injection, rapamycin was diluted to 2.5 mg/ml in vehicle solution (5% PEG 400, 5% Tween 80 and 4% ethanol). Mice were weighed daily to confirm similar nutrition. Colchicine was dissolved in water and stored at −20°C as a stock solution at a concentration of 4 mg/ml. On the day of treatment, colchicine was diluted to 0.1 mg/ml in water prior to injection. Control mice received an equal volume of i.p. water.
Twenty-four hours after the last injection, mice were euthanized and skeletal muscle was dissected. For western blot analysis, muscle was flash frozen in liquid nitrogen and stored at −80°C. For immunohistochemistry, muscle was mounted on a cork with traganath gum and quickly cooled in liquid nitrogen-cooled isopentane for 5 min and then stored at −80°C. Total RNA was isolated immediately from freshly dissected skeletal muscle using an SV Total RNA isolation kit Z3100 (Promega) according to the manufacturer's instructions. Creatine phosphokinase was determined from serum of mice following direct cardiac puncture of isoflurane anesthesized mice prior to euthanizing by the Washington University Department of Comparative Anatomy.
Grip strength testing consisted of five separate measurements using a trapeze bar attached to a force transducer that recorded peak-generated force (Stoelting, WoodDale, IL, USA). Mice have the tendency to grab the bar with their forepaws and continue to hold while being pulled backwards by the tail, releasing only when unable to maintain grip. The resulting measurement was recorded and the average of the highest three measurements was determined to give the strength score. For every time point and strain, at least five animals were used. P-values were determined by a paired Student's t-test. To validate our results, another quantitative strength measurement was performed by quantifying the rotarod performance of mice. Mice tend to balance on a rotating rod and the time taken to fall off the rotating rod under constant acceleration is measured. Mice were trained at a constant speed of 2.5 rpm until they can remain on the rotating rod for 120 s. They were left undisturbed in the home cage for 1 h before being tested for rotarod performance on the rod accelerating at 3 rpm every 10 s.
For electroporation, mice were anesthetized using inhaled isoflurane. The skin overlying the TA muscle was shaved, and the animals were injected with a 30 μg endotoxin-free expression plasmid (see below for plasmid details) diluted in sterile phosphate-buffered saline (PBS) to a volume of 50 μl by using a 0.5 ml syringe fitted with a 29-gauge needle. Two-needle array electrodes (450121) (Harvard Apparatus, Holliston, MA, USA) were inserted into the muscle immediately after DNA delivery for electroporation. The distance between the electrodes was 5 mm, and the array was inserted longitudinally relative to the muscle fibers. In vivo electroporation parameters were the following: voltage, 75 V; pulse length, 50 ms; number of pulses, six pulses; pulse interval, 200 ms; desired field strength, 200 V/cm, given by a BTX ECM830 Electro Square Porator. Animals were allowed to recover for 7 days prior to muscle isolation.
Protein synthesis was determined using surface sensing of translation (SUnSET) (26,27). 0.04 µmol/gram body weight of puromycin is injected i.p. into mice 30 min before muscles were collected. The amount of puromycin incorporated into the nascent peptides is detected by western blot using a rabbit monoclonal antibody to puromycin.
Western blotting
Muscle tissues and cultured cells were homogenized using RIPA lysis buffer (50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 1% NP-40, 0.25% Na-deoxycholate and 1 mm EDTA) supplemented with protease inhibitor cocktail (Sigma-Aldrich), and lysates were centrifuged at 14000g for 10 min. Protein concentrations were determined using a BCA protein assay kit (Thermo Fisher Scientific). Aliquots of lysates were solubilized in Laemmli sample buffer and equal amounts of proteins were separated on 12% SDS-PAGE gels. Proteins were transferred to nitrocellulose membrane and the membrane was blocked with 5% nonfat dry milk in PBS for 1 h. The membrane was then incubated with primary antibodies, specific to the protein of interest, in 5% nonfat dry milk overnight at 4°C. After incubation with the appropriate secondary antibody conjugated with horseradish peroxidase, enhanced chemiluminescence (GH Healthcare, UK) was used for protein detection. If necessary, blots were stripped using Re-blot strong solution (Millipore) and reprobed with the appropriate antibodies. Autoradiographs of immunoblots were scanned using an Epson 636 Expression scanner and collected in Photoshop or immunoblots were obtained using the G:Box Chemi XT4, Genesys Version 1.1.2.0 (Syngene). Densitometry was measured with imageJ software (National Institute of Health). quantitative polymerase chain reaction was performed with primers to mouse Atrogin 1 and Murf1 as previously described (16). The values were normalized to GAPDH and represented as fold change. Antibodies used were the following: pAkt, Akt, pS6, S6, p4EBP1, 4EBP1, pp70S6K, p70S6K, pACC, pULK1, pAMPK, AMPK and Beclin 1 were obtained from Cell Signaling Technologies. Anti-LC3 (Sigma), anti-ULK1 (Sigma), anti-p62 (Proteintech), anti-BNIP3 (Abcam), anti-mono and polyubiquitinated conjugates (FK2) (Enzo life sciences, anti-FLAG (Sigma), anti-GFP (Sigma), anti-HA (Abcam), anti-actin (Sigma), anti-GAPDH (Abcam), anti-collagen IV (Abcam)). Secondary antibodies include anti-mouse HRP (Pierce), anti-rabbit HRP (cell signaling), anti-mouse AlexaFluor 543 and anti-rabbit AlexaFluro 488. Anti-puromycin was a gift from Dr Peter Walter.
Histochemistry and immunohistochemistry
Ten micron sections of mounted skeletal muscle were affixed to slides, treated for 10 min in ice cold acetone. Congo red staining was performed as previously described (15). For immunohistochemistry, the sections were blocked in PNB (PerkinElmer), incubated with primary antibody followed by the appropriate secondary antibody. The sections were mounted with Prolong Gold + DAPI (Invitrogen) (15). Slides were examined using a fluorescent microscope (Nikon 80i upright) and Roper Scientific EZ monochromeCCD camera with deconvolution software analysis (NIS Elements, Nikon). Nonfluorescent images were taken with a five megapixel color CCD (Nikon). Image processing and analyses were done with (NIS Elements 4.0) software and Adobe Photoshop CS3. The fiber CSA was quantified from at least 10 images of six sectioned muscles/condition. Hematoxylin and eosin stained sections were manually outlined and the CSA was then calculated using NIH elements. For electroporation studies, a minimum of 150 electroporated fibers/condition were counted from immunofluorescent images that were manually defined and then CSA calculated using NIH elements. Quantitation of vacuoles was performed using congo red staining of two sections from four treated or untreated mouse cohorts for a total of eight sectionsper cohort. The number of vacuolated fibers was expressed as a percentage of total fibers obtained from each section.
Cell culture
U20S cells and stable tetracycline inducible U20S cells expressing pcDNA4.0 VCP-WT-myc, VCP-E578Q-myc or VCP-R155H myc were cultured in DMEM containing 4 mm l-glutamine (Invitrogen; 11965–084), 10% FBS (Atlanta Biologicals; S10350) and penicillin (50 IU)/streptomycin (50 µg/ml) (Invitrogen; 15140). Stably expressing U20S cells were also incubated with 50 µg/ml hygromycin B (Invitrogen; 10687–010), 50 µg/ml zeocin (Invitrogen; R25001) and induced with 1 µg/ml tetracycline hydrochloride (Sigma; T76600) 24 h prior to experimentation. Cells were maintained in 5% CO2 at 37°C in six-well tissue culture-treated plates until the cells were 80–85% confluent.
For cell starvation and nutrient stimulation, growth medium was removed and the cells were rinsed twice with PBS (Invitrogen; 14190–136) and the cells were serum starved (no serum in the growth media) for 15 h. In some cases, the cells were treated with 0.1% dimethyl sulfoxide (DMSO), 10 µm rapamycin (LC laboratories; R-5000) and 10 µm of the VCP inhibitor, N2,N4-dibenzylquinazoline-2,4-diamine (DBeQ; a gift from Dr Frank Schoenen and Raymond Deshaies) for 30 min and then the cells were restimulated with the growth media containing 10% FBS for 1 h along with the drugs.
Cell transfection were performed using Fugene6 (Roche; 11988484001) according to the manufacturer's instructions with 1 µg each of pcDNA3-FLAG-Rheb-N153T (Addgene, plasmid 19997) and a GFP control plasmid (pEGFPN1). The transfection complex was removed and the cells were replaced with new media after 24 h.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (NIH) AG031867 and AG042095 (C.C.W.), the Neuroscience Blueprint Core Grant NS057105 to Washington University, the Hope Center for Neurological Disorders, the Muscular Dystrophy Association (C.C.W.) and the Washington University Center for Musculoskeletal Research P30 AR057235.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Johnson J.O., Mandrioli J., Benatar M., Abramzon Y., Van Deerlin V.M., Trojanowski J.Q., Gibbs J.R., Brunetti M., Gronka S., Wuu J., et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. doi:10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watts G.D., Wymer J., Kovach M.J., Mehta S.G., Mumm S., Darvish D., Pestronk A., Whyte M.P., Kimonis V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004;36:377–381. doi: 10.1038/ng1332. doi:10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 3.Weihl C.C., Pestronk A., Kimonis V.E. Valosin-containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto-temporal dementia. Neuromuscul. Disord. 2009;19:308–315. doi: 10.1016/j.nmd.2009.01.009. doi:10.1016/j.nmd.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weihl C.C., Temiz P., Miller S.E., Watts G., Smith C., Forman M., Hanson P.I., Kimonis V., Pestronk A. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry. 2008;79:1186–1189. doi: 10.1136/jnnp.2007.131334. doi:10.1136/jnnp.2007.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubbers C.U., Clemen C.S., Kesper K., Boddrich A., Hofmann A., Kamarainen O., Tolksdorf K., Stumpf M., Reichelt J., Roth U., et al. Pathological consequences of VCP mutations on human striated muscle. Brain. 2007;130:381–393. doi: 10.1093/brain/awl238. doi:10.1093/brain/awl238. [DOI] [PubMed] [Google Scholar]
- 6.Tresse E., Salomons F.A., Vesa J., Bott L.C., Kimonis V., Yao T.P., Dantuma N.P., Taylor J.P. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010;6:1–11. doi: 10.4161/auto.6.2.11014. doi:10.4161/auto.6.1.10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritz D., Vuk M., Kirchner P., Bug M., Schutz S., Hayer A., Bremer S., Lusk C., Baloh R.H., Lee H., et al. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat. Cell. Biol. 2011;13:1116–1123. doi: 10.1038/ncb2301. doi:10.1038/ncb2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ju J.S., Fuentealba R.A., Miller S.E., Jackson E., Piwnica-Worms D., Baloh R.H., Weihl C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 2009;187:875–888. doi: 10.1083/jcb.200908115. doi:10.1083/jcb.200908115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zehner M., Chasan A.I., Schuette V., Embgenbroich M., Quast T., Kolanus W., Burgdorf S. Mannose receptor polyubiquitination regulates endosomal recruitment of p97 and cytosolic antigen translocation for cross-presentation. Proc. Natl Acad. Sci. U.S.A. 2011;108:9933–9938. doi: 10.1073/pnas.1102397108. doi:10.1073/pnas.1102397108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramanathan H.N., Ye Y. The p97 ATPase associates with EEA1 to regulate the size of early endosomes. Cell Res. 2011;22:346–359. doi: 10.1038/cr.2011.80. doi:10.1038/cr.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chou T.F., Brown S.J., Minond D., Nordin B.E., Li K., Jones A.C., Chase P., Porubsky P.R., Stoltz B.M., Schoenen F.J., et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl Acad. Sci. U.S.A. 2011;108:4834–4839. doi: 10.1073/pnas.1015312108. doi:10.1073/pnas.1015312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Badadani M., Nalbandian A., Watts G.D., Vesa J., Kitazawa M., Su H., Tanaja J., Dec E., Wallace D.C., Mukherjee J., et al. VCP associated inclusion body myopathy and Paget disease of bone knock-in mouse model exhibits tissue pathology typical of human disease. PLoS One. 2010;5:e13183. doi: 10.1371/journal.pone.0013183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malicdan M.C., Noguchi S., Nonaka I., Saftig P., Nishino I. Lysosomal myopathies: an excessive build-up in autophagosomes is too much to handle. Neuromuscul. Disord. 2008;18:521–529. doi: 10.1016/j.nmd.2008.04.010. doi:10.1016/j.nmd.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 14.Custer S.K., Neumann M., Lu H., Wright A.C., Taylor J.P. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet. 2010;19:1741–1755. doi: 10.1093/hmg/ddq050. doi:10.1093/hmg/ddq050. [DOI] [PubMed] [Google Scholar]
- 15.Weihl C.C., Miller S.E., Hanson P.I., Pestronk A. Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum Mol. Genet. 2007;16:919–928. doi: 10.1093/hmg/ddm037. doi:10.1093/hmg/ddm037. [DOI] [PubMed] [Google Scholar]
- 16.Masiero E., Agatea L., Mammucari C., Blaauw B., Loro E., Komatsu M., Metzger D., Reggiani C., Schiaffino S., Sandri M. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. doi:10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 17.Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S.J., Di Lisi R., Sandri C., Zhao J., et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. doi:10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Zhao J., Brault J.J., Schild A., Cao P., Sandri M., Schiaffino S., Lecker S.H., Goldberg A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. doi:10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 19.He C., Bassik M.C., Moresi V., Sun K., Wei Y., Zou Z., An Z., Loh J., Fisher J., Sun Q., et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. doi:10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ju J.S., Varadhachary A.S., Miller S.E., Weihl C.C. Quantitation of ‘autophagic flux’ in mature skeletal muscle. Autophagy. 2010;6:929–935. doi: 10.4161/auto.6.7.12785. doi:10.4161/auto.6.7.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sengupta S., Peterson T.R., Sabatini D.M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. doi:10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodman C.A., Miu M.H., Frey J.W., Mabrey D.M., Lincoln H.C., Ge Y., Chen J., Hornberger T.A. A phosphatidylinositol 3-kinase/protein kinase B-independent activation of mammalian target of rapamycin signaling is sufficient to induce skeletal muscle hypertrophy. Mol. Biol. Cell. 2010;21:3258–3268. doi: 10.1091/mbc.E10-05-0454. doi:10.1091/mbc.E10-05-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Risson V., Mazelin L., Roceri M., Sanchez H., Moncollin V., Corneloup C., Richard-Bulteau H., Vignaud A., Baas D., Defour A., et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J. Cell Biol. 2009;187:859–874. doi: 10.1083/jcb.200903131. doi:10.1083/jcb.200903131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi J.C., Muchir A., Wu W., Iwata S., Homma S., Morrow J.P., Worman H.J. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin a/c gene mutation. Sci. Transl. Med. 2012;4:144ra102. doi: 10.1126/scitranslmed.3003875. doi:10.1126/scitranslmed.3003875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramos F.J., Chen S.C., Garelick M.G., Dai D.F., Liao C.Y., Schreiber K.H., Mackay V.L., An E.H., Strong R., Ladiges W.C., et al. Rapamycin reverses elevated mTORC1 signaling in Lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012;4:144ra103. doi: 10.1126/scitranslmed.3003802. doi:10.1126/scitranslmed.3003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt E.K., Clavarino G., Ceppi M., Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. doi:10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 27.Goodman C.A., Mabrey D.M., Frey J.W., Miu M.H., Schmidt E.K., Pierre P., Hornberger T.A. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 2011;25:1028–1039. doi: 10.1096/fj.10-168799. doi:10.1096/fj.10-168799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung C.H., Jun C.B., Ro S.H., Kim Y.M., Otto N.M., Cao J., Kundu M., Kim D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. doi:10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukuda T., Ewan L., Bauer M., Mattaliano R.J., Zaal K., Ralston E., Plotz P.H., Raben N. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann. Neurol. 2006;59:700–708. doi: 10.1002/ana.20807. doi:10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- 30.Ramachandran N., Munteanu I., Wang P., Aubourg P., Rilstone J.J., Israelian N., Naranian T., Paroutis P., Guo R., Ren Z.P., et al. VMA21 deficiency causes an autophagic myopathy by compromising V-ATPase activity and lysosomal acidification. Cell. 2009;137:235–246. doi: 10.1016/j.cell.2009.01.054. doi:10.1016/j.cell.2009.01.054. [DOI] [PubMed] [Google Scholar]
- 31.Nogalska A., D'Agostino C., Terracciano C., Engel W.K., Askanas V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am. J. Pathol. 2010;177:1377–1387. doi: 10.2353/ajpath.2010.100050. doi:10.2353/ajpath.2010.100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sancak Y., Peterson T.R., Shaul Y.D., Lindquist R.A., Thoreen C.C., Bar-Peled L., Sabatini D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. doi:10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flinn R.J., Yan Y., Goswami S., Parker P.J., Backer J.M. The late endosome is essential for mTORC1 signaling. Mol. Biol. Cell. 2010;21:833–841. doi: 10.1091/mbc.E09-09-0756. doi:10.1091/mbc.E09-09-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh P., Wu M., Zhang H., Sun H. mTORC1 signaling requires proteasomal function and the involvement of CUL4-DDB1 ubiquitin E3 ligase. Cell Cycle. 2008;7:373–381. doi: 10.4161/cc.7.3.5267. doi:10.4161/cc.7.3.5267. [DOI] [PubMed] [Google Scholar]
- 35.Zhao Y., Xiong X., Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell. 2011;44:304–316. doi: 10.1016/j.molcel.2011.08.029. doi:10.1016/j.molcel.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao D., Inuzuka H., Tan M.K., Fukushima H., Locasale J.W., Liu P., Wan L., Zhai B., Chin Y.R., Shaik S., et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell. 2011;44:290–303. doi: 10.1016/j.molcel.2011.08.030. doi:10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alexandru G., Graumann J., Smith G.T., Kolawa N.J., Fang R., Deshaies R.J. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell. 2008;134:804–816. doi: 10.1016/j.cell.2008.06.048. doi:10.1016/j.cell.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fecto F., Yan J., Vemula S.P., Liu E., Yang Y., Chen W., Zheng J.G., Shi Y., Siddique N., Arrat H., et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. doi:10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 39.Laurin N., Brown J.P., Morissette J., Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 2002;70:1582–1588. doi: 10.1086/340731. doi:10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hocking L.J., Lucas G.J., Daroszewska A., Mangion J., Olavesen M., Cundy T., Nicholson G.C., Ward L., Bennett S.T., Wuyts W., et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget's disease. Hum. Mol. Genet. 2002;11:2735–2739. doi: 10.1093/hmg/11.22.2735. doi:10.1093/hmg/11.22.2735. [DOI] [PubMed] [Google Scholar]
- 41.Duran A., Amanchy R., Linares J.F., Joshi J., Abu-Baker S., Porollo A., Hansen M., Moscat J., Diaz-Meco M.T. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. doi:10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyer H., Bug M., Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell. Biol. 2012;14:117–123. doi: 10.1038/ncb2407. doi:10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X., Li L., Chen S., Yang D., Wang Y., Wang Z., Le W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7:412–425. doi: 10.4161/auto.7.4.14541. doi:10.4161/auto.7.4.14541. [DOI] [PubMed] [Google Scholar]
- 44.Ravikumar B., Vacher C., Berger Z., Davies J.E., Luo S., Oroz L.G., Scaravilli F., Easton D.F., Duden R., O'Kane C.J., et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004;36:585–595. doi: 10.1038/ng1362. doi:10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 45.Grumati P., Coletto L., Sabatelli P., Cescon M., Angelin A., Bertaggia E., Blaauw B., Urciuolo A., Tiepolo T., Merlini L., et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010;16:1313–1320. doi: 10.1038/nm.2247. doi:10.1038/nm.2247. [DOI] [PubMed] [Google Scholar]
- 46.Carmignac V., Svensson M., Korner Z., Elowsson L., Matsumura C., Gawlik K.I., Allamand V., Durbeej M. Autophagy is increased in laminin alpha2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet. 2011;20:4891–4902. doi: 10.1093/hmg/ddr427. doi:10.1093/hmg/ddr427. [DOI] [PubMed] [Google Scholar]
- 47.Sciarretta S., Zhai P., Shao D., Maejima Y., Robbins J., Volpe M., Condorelli G., Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. doi:10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]