

Abstract
Blood pressure (BP) is a heritable determinant of risk for cardiovascular disease (CVD). To investigate genetic associations with systolic BP (SBP), diastolic BP (DBP), mean arterial pressure (MAP) and pulse pressure (PP), we genotyped ∼50 000 single-nucleotide polymorphisms (SNPs) that capture variation in ∼2100 candidate genes for cardiovascular phenotypes in 61 619 individuals of European ancestry from cohort studies in the USA and Europe. We identified novel associations between rs347591 and SBP (chromosome 3p25.3, in an intron of HRH1) and between rs2169137 and DBP (chromosome1q32.1 in an intron of MDM4) and between rs2014408 and SBP (chromosome 11p15 in an intron of SOX6), previously reported to be associated with MAP. We also confirmed 10 previously known loci associated with SBP, DBP, MAP or PP (ADRB1, ATP2B1, SH2B3/ATXN2, CSK, CYP17A1, FURIN, HFE, LSP1, MTHFR, SOX6) at array-wide significance (P < 2.4 × 10−6). We then replicated these associations in an independent set of 65 886 individuals of European ancestry. The findings from expression QTL (eQTL) analysis showed associations of SNPs in the MDM4 region with MDM4 expression. We did not find any evidence of association of the two novel SNPs in MDM4 and HRH1 with sequelae of high BP including coronary artery disease (CAD), left ventricular hypertrophy (LVH) or stroke. In summary, we identified two novel loci associated with BP and confirmed multiple previously reported associations. Our findings extend our understanding of genes involved in BP regulation, some of which may eventually provide new targets for therapeutic intervention.
INTRODUCTION
Blood pressure (BP) is a cardinal risk factor for cardiovascular disease (CVD). Systolic BP (SBP) and diastolic (DBP) levels are associated with increased risk of atherosclerotic vascular disease and other cardiovascular causes of death (1). Much of the excess CVD risk imparted by BP elevation can be ameliorated through interventions to decrease BP (2). The identification of novel genes and pathways involved in BP regulation may highlight new ways of reducing BP and CVD risk associated with hypertension. The mean arterial pressure (MAP) and pulse pressure (PP, the difference between SBP and DBP) are single BP components associated with CVD risk (3–5). The latter is an indicator of conduit artery stiffness and is known to increase with age, as aortic elasticity decreases. To date, ∼50 common genetic variants associated with BP and hypertension have been reported, largely through genome-wide association studies (GWAS), meta-analyses and admixture mapping approaches (6–16). The identification of common variants associated with BP may be enriched through further gene-centric approaches (17–19). Accordingly, we tested the hypothesis that candidate gene analysis would identify known and novel associations with SBP, DBP, MAP and PP and would confirm previously reported associations. To further investigate and discover associations with BP, we genotyped approximately 50 000 single-nucleotide polymorphisms (SNPs) on a gene-centric array (ITMAT-Broad_CARe [IBC] array, Illumina San Diego, CA, USA) that captures variation in ∼2100 candidate genes for cardiovascular traits including BP (20) in 61 619 individuals of European ancestry. We identified two novel BP-associated loci, at the candidate genes MDM4 and HRH1 and have validated these associations through in silico replication analysis in a large set of independent samples.
RESULTS
Discovery association analyses
In the primary discovery meta-analysis, four BP traits were analyzed in 61 619 individuals from 27 cohorts, as described in Table 1. We analyzed SBP, DBP, MAP and PP as continuous traits. Cohort characteristics, including age, sex, BP values and the proportion of individuals treated with BP lowering medications, are provided in Table 1. The details of the cohorts are provided in the Supplementary Material, Table S1 and Supplementary Materials.
Table 1.
Clinical characteristics of discovery and replication cohorts
| Age | Female/ male | SBP | DBP | MAP | PP | BMI | Taking anti- hypertensive medication | |
|---|---|---|---|---|---|---|---|---|
| Discovery cohorts | ||||||||
| AMC-PAS | 42.9 ± 5.3 | 180/563 | 128.6 ± 17.9 | 79.9 ± 10.7 | 96.1 ± 12.1 | 48.8 ± 13.1 | 26.9 ± 4.1 | 33.2% |
| Amish | 47.6 ± 15.0 | 713/691 | 121.9 ± 16.5 | 75.3 ± 9.4 | 90.8 ± 10.8 | 46.6 ± 12.2 | 27.3 ± 5.0 | 16.4% |
| ARIC | 54.2 ± 5.7 | 5124/4453 | 118.3 ± 17 | 71.5 ± 10 | 87.1 ± 11.3 | 46.7 ± 12.7 | 26.9 ± 4.9 | 25.1% |
| BHS | 22.5 ± 4.4 | 291/228 | 111.5 ± 10.2 | 71.8 ± 8.5 | 85 ± 8.3 | 39.7 ± 8 | 24.7 ± 6.1 | 2.7% |
| CARDIA | 40.6 ± 4.1 | 703/623 | 102.2 ± 30.5 | 71.9 ± 11.2 | 84.5 ± 11.4 | 37.7 ± 8.8 | 26.9 ± 6.4 | 3.7% |
| CCCS | 64.2 ± 9.7 | 555/1402 | 136.94 ± 19.04 | 78.33 ± 10.61 | 156.48 ± 23.57 | 58.61 ± 15.69 | 29.2 (4.8)/ 29.8 (6.5) | 88.7% |
| CFS | 40.9 ± 19.9 | 302/252 | 121.1 ± 16.6 | 71.8 ± 11.7 | 88.2 ± 12.2 | 49.3 ± 12.4 | 30.1 ± 8.8 | 8.8% |
| CHS | 72.6 ± 6.3 | 2208/1722 | 135.3 ± 21.5 | 69.9 ± 11.6 | 91.7 ± 12.9 | 65.4 ± 18.6 | 26.3 ± 4.8 | 39.7% |
| CLEAR | 67.8 ± 9.6 | 0/1365 | 151.4 ± 22.4 | 82.2 ± 12.3 | 105.3 ± 13.8 | 69.2 ± 18.4 | 28.1 ± 5.0 | 67.0% |
| EPIC_NL | 54.06 ± 10.11 | 4057/1137 | 133.13 ± 21.22 | 80.46 ± 10.93 | 97.98 ± 13.20 | 52.65 ± 15.68 | 26.77 ± 4.45 | N/A |
| FHS | 40.9 ± 9.1 | 3775/3134 | 118.6 ± 14.3 | 76.4 ± 9.8 | 90.5 ± 10.6 | 42.2 ± 9.4 | 26.1 ± 5 | 5.5% |
| GIRaFH | 44.5 ± 11.7 | 882/812 | 134.9 ± 19.2 | 82.0 ± 10.5 | 99.7 ± 12.3 | 52.9 ± 14.4 | 25.1 ± 3.5 | 9.3% |
| GQ2 | 65.5 ± 10.5 | 385/93 | 130.51 ± 22.40 | 72.71 ± 12.89 | 149.78 ± 27.66 | 57.81 ± 18.47 | 29.7 (7.7)/ 29.5 (6.3) | 76.2% |
| INVEST | 69.4 ± 9.5 | 467/580 | 160.8 ± 17.4 | 90.6 ± 10.1 | 114.0 ± 10.6 | 70.1 ± 15.9 | 29.0 (4.7)/ 28.4 (6.2) | 82.6% |
| LURIC | 58.1 ± 8.6 | 558/1480 | 151.2 ± 24.4 | 89.8 ± 12.1 | 110.3 ± 15.0 | 61.4 ± 17.6 | 27.7 ± 4.2 | 85.6% |
| MEDAL | 62.9 ± 9.0 | 1178/2820 | 136.8 ± 16.0 | 81.7 ± 9.6 | 94.9 ± 8.6 | 52.9 ± 11.5 | 30.5 ± 6.3 | 1.4% |
| MESA | 62.7 ± 10.3 | 1199/1097 | 123.5 ± 20.8 | 70.1 ± 10.2 | 87.9 ± 12.3 | 53.4 ± 16.7 | 27.8 ± 5.1 | 33.3% |
| MONICA/ KORA F3 | 57.6 ± 8.1 | 755/649 | 131.8 ± 19.4 | 83.3 ± 10.3 | 99.5 ± 12.6 | 48.5 ± 13.2 | 27.8 ± 4.5 | 30.1% |
| MONICA/ KORA S12 | 51.8 ± 9.9 | 431/549 | 133.6 ± 19.1 | 81.5 ± 11.1 | 98.8 ± 12.5 | 52.1 ± 14.6 | 27.2 ± 4.0 | 16.6% |
| NSHS95 | 49.4 ± 18.4 | 857/899 | 126.6 ± 17.7 | 76.7 ± 11.6 | 93.3 ± 11.8 | 49.9 ± 15.8 | 27.1 ± 5.5 | N/A |
| PEAR | 50.1 ± 9.4 | 194/244 | 151.8 ± 12.4 | 98.0 ± 5.7 | 115.9 ± 6.9 | 53.7 ± 10.8 | [30.3 (4.4)/ 30.4 (6.1)] | 0% |
| PennCAC | 56.0 ± 8.0 | 631/1145 | 132 ± 23.2 | 72.4 ± 11.2 | 52.6 ± 12.6 | 59.5 ± 19.7 | 29.8 ± 5.9 | N/A |
| PennCath | 52.0 ± 9.0 | 739/1386 | 127 ± 15.1 | 76.7 ± 9.5 | 62.9 ± 10.5 | 51.9 ± 12.3 | 30.1 ± 5.9 | 32.8% |
| SMART | 59.36 ± 12.25 | 206/299 | 158.64 ± 18.57 | 94.76 ± 11.80 | 116.06 ± 12.87 | 63.88 ± 13.78 | 27.35 ± 4.62 | 39.4% |
| WHI | 68.0 ± 6.6 | 7606/0 | 133.0 ± 18.8 | 75.0 ± 9.7 | 94.3 ± 11.0 | 58.0 ± 16.2 | 28.3 (6.2) | 33.3% |
| Replication cohorts | ||||||||
| AIBIII | 52.8 (9.2) | 249/209 | 119.9 (13.7) | 75.4 (7.6) | 90.2 (8.8) | 44.5 (10.3) | 25.7 (3.6) | 100.0% |
| ASCOT | 63 (8.1) | 224/1015 | 161.4 (17.8) | 92.9 (9.9) | 115.7 (10.6) | 68.5 (16.1) | 29.1 (4.6) | 52.9% |
| BRIGHT (controls) | 58.7 (8.9) | 1088/647 | 123 (10.5) | 76.4 (7.2) | 91.9 (7.5) | 46.7 (8.3) | 25.3 (3.3) | 100.0% |
| BRIGHT (cases) | 58 (10.3) | 1144/775 | 154.3 (21.1) | 93.9 (11.3) | 114 (13.3) | 60.4 (15.7) | 58 (10.3) | 51.8% |
| BWHHS | 68.85 (5.51) | 3373/0 | 146.53 (26.59) | 79.16 (12.85) | 102 (15) | 67.6 (19) | 27.25 (5.95) | 76.8% |
| GRAPHIC | 39.30 (14.50) | 1004/1020 | 127.09 (17.84) | 79.12 (10.96) | 95.1 (12.5) | 48 (11.9) | 26.11 (4.61) | 93.7% |
| LIFELINES | 47.3 ± 11.2 | 4640/3483 | 127.9 ± 15.7 | 75.1 ± 9.1 | 52.7 ± 11.8 | 92.7 ± 10.3 | 26.3 ± 4.3 | 15.5% |
| MDC | 57.8 (5.9) | 1074/772 | 115.6 (5.8) | 73.6 (5.3) | 87.6 (4.7) | 42 (6) | 24.3 (3.3) | 100.0% |
| NBS | 41.36 (12.37) | 1183/1169 | N/A | N/A | N/A | N/A | N/A | N/A |
| NESDA | 41.5 ± 12.7 | 1166/551 | 135.3 ± 20.2 | 81.7 ± 11.8 | 99.6 ± 13.9 | 53.6 ± 12.7 | 25.5 ± 4.9 | 12.8% |
| NORDIL | 56 (4) | 979/940 | 177.3 (14.6) | 105.9 (5.5) | 129.7 (7.1) | 71.5 (13.9) | 28.3 (4.6) | 100.0% |
| PREVEND | 49.6 ± 12.5 | 1752/1869 | 129.1 ± 19.9 | 74.1 ± 9.9 | 54.9 ± 13.9 | 92.4 ± 12.5 | 26.1 ± 4.3 | 14.2% |
| Procardis | 59.34 (9.93) | 1634/1564 | 130.75 (17.11) | 79.63 (10.03) | 96.7 (11.2) | 51.1 (13.4) | 26.81 (4.37) | 83.8% |
| Rotterdam Study | 69.4 (9.1) | 3327/2327 | 144.1 (24.2) | 76.9 (12.6) | 99.3 (15.0) | 67.1 (18.5) | 26.3 (3.7) | 32.5% |
| TRAILS clinical cohort | 15.8 ± 0.6 | 97/217 | 119.1 ± 12.6 | 61.0 ± 6.6 | 80.4 ± 7.3 | 58.1 ± 11.4 | 21.5 ± 3.6 | N/A |
| TRAILS population cohort | 16.2 ± 0.7 | 693/642 | 118.1 ± 12.4 | 61.1 ± 6.9 | 80.1 ± 7.4 | 57.7 ± 10.6 | 21.2 ± 3.2 | N/A |
| WGHS | 54.2 ± 7.1 | 22 625/0 | 125.5 ± 16.4 | 78.0 ± 10.7 | 93.8 ± 11.9 | 47.6 ± 10.4 | 25.9 ± 5.0 | 12.9% |
| WHII | 60.83 (6.0) | 1845/3210 | 128.1 (16.7) | 74.6 (10.5) | 92.4 (11.8) | 53.5 (11.2) | 26.7 (4.3) | 81.4% |
Mean + standard deviation is given for each phenotype, except % were indicated. BP values shown are actual values without modification for medication treatment.
Association analyses were successfully carried out for up to 48 372 SNPs, and summaries of the quality-control (QC) steps and numbers of SNPs removed at each step are provided in Supplementary Material, Table S2A. Cohort-specific genomic control inflation factors, λGC, did not suggest the presence of inflation (Supplementary Material, Table S3). Meta-analysis quantile–quantile plots are shown in Supplementary Material, Fig. S1 and P-values for association for all SNPs are provided in Supplementary Material, Table S4. We identified 22 significant SNP-trait associations with SBP, DBP, MAP and PP at 12 different loci (P < 2.4 × 10−6), including two novel loci near HRH1 and MDM4 for BP traits and one novel SNP-trait association in the SOX6 locus, a region previously described in association with MAP.
Replication analyses
Replication testing was performed in 65 866 additional individuals, including 43 266 individuals in seven cohorts with genome-wide SNP genotypes imputed to HapMap (Supplementary Material, Table S2B) and 22 600 individuals genotyped on the same IBC chip used for the discovery analyses. Through the joint analysis of SNPs considered relevant during the discovery phase combined with replication data, we identified robust association of 22 SNP-trait associations at 12 independent loci meeting our array-wide significance threshold of P < 2.4 × 10−6: six loci were associated with DBP (MTHFR, MDM4, HFE, SH2B3/ATXN2, CSK, FURIN), nine loci were associated with SBP (MTHFR, HRH1, CYP17A1, LSP1, SOX6, ATP2B1, SH2B3/ATXN2, CSK, FURIN), six loci were associated with MAP (MTHFR, ADRB1, ATP2B1, ATXN2, CSK, FURIN) and one locus associated with PP (CYP17A1). The association findings are summarized in Table 2.
Table 2.
Loci associated with hypertension traits using discovery and replication data (Betas and SEs corresponding to the trait are highlighted in bold)
| Discovery results |
Replication results |
Discovery + replication results | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | SNP | CHR | BP | A1 | A2 | CAF | Beta | SE | DBP | MAP | PP | SBP | DBP | MAP | PP | SBP | DBP | MAP | PP | SBP |
| DBP | ||||||||||||||||||||
| MTHFR | rs13306561 | 1 | 11788391 | G | A | 0.154 | −0.5224 | 0.0852 | 8.641E−10 | 9.93E−12 | 1.33E−09 | 2.6E−13 | 6.91E−14 | 8.51E−04 | 3.82E−11 | 1.75E−21 | 4.49E−24 | 3E−19 | ||
| MDM4 | rs2169137 | 1 | 202764536 | G | C | 0.271 | −0.3552 | 0.0702 | 4.201E−07 | 0.007492 | 5.86E−08 | |||||||||
| HFE | rs1799945 | 6 | 26199158 | G | C | 0.146 | 0.406 | 0.0864 | 2.615E−06 | 6.34E−12 | 2.78E−16 | |||||||||
| ATXN2 | rs10774625 | 12 | 110394602 | A | G | 0.505 | 0.4929 | 0.0764 | 1.103E−10 | 4.61E−10 | 1.56E−07 | 5.98E−09 | 8.88E−11 | 9.37E−10 | 4.94E−18 | 2.4E−19 | 8.12E−16 | |||
| CSK | rs7085 | 15 | 72882536 | T | C | 0.281 | 0.4403 | 0.0677 | 7.936E−11 | 5.01E−11 | 6.68E−11 | 1.58E−05 | 3.54E−08 | 1.33E−05 | 2.5E−14 | 2.14E−17 | 3.48E−14 | |||
| FURIN | rs2071410 | 15 | 89221944 | G | C | 0.371 | 0.3661 | 0.066 | 2.882E−08 | 4.30E−08 | 0.00013 | 1.19E−08 | 2.27E−09 | 2.36E−09 | 1.78E−15 | 5.31E−16 | 2.92E−12 | |||
| MAP | ||||||||||||||||||||
| MTHFR | rs13306561 | 1 | 11788391 | G | A | 0.154 | −0.6665 | 0.0979 | 8.641E−10 | 9.93E−12 | 1.33E−09 | 2.6E−13 | 6.91E−14 | 8.51E−04 | 3.82E−11 | 1.75E−21 | 4.49E−24 | 3E−19 | ||
| ADRB1 | rs7076938 | 10 | 115779365 | C | T | 0.282 | −0.3869 | 0.0776 | 6.05E−07 | 9.72E−09 | 3.14E−14 | |||||||||
| ATP2B1 | rs2681472 | 12 | 88533090 | G | A | 0.172 | −0.612 | 0.1129 | 5.87E−08 | 3.99E−09 | 6.95E−10 | 3.3E−11 | 2.24E−16 | 7.68E−19 | ||||||
| ATXN2 | rs10774625 | 12 | 110394602 | A | G | 0.505 | 0.5343 | 0.0857 | 1.103E−10 | 4.61E−10 | 1.56E−07 | 5.98E−09 | 8.88E−11 | 9.37E−10 | 4.94E−18 | 2.4E−19 | 8.12E−16 | |||
| CSK | rs7085 | 15 | 72882536 | T | C | 0.281 | 0.5109 | 0.0778 | 7.936E−11 | 5.01E−11 | 6.68E−11 | 1.58E−05 | 3.54E−08 | 1.33E−05 | 2.5E−14 | 2.14E−17 | 3.48E−14 | |||
| FURIN | rs6227 | 15 | 89226236 | T | C | 0.316 | 0.764 | 0.1506 | 4.081E−08 | 3.65E−09 | 3.88E−07 | 1.86E−06 | 1.02E−10 | 3.77E−09 | 9.83E−13 | 3.35E−18 | 8.01E−15 | |||
| PP | ||||||||||||||||||||
| CYP17A1 | rs3824755 | 10 | 104585839 | C | G | 0.096 | −0.6438 | 0.1289 | 5.93E−07 | 2.46E−07 | 6.06E−04 | 0.000384 | 1.52E−09 | 3.77E−10 | ||||||
| SBP | ||||||||||||||||||||
| MTHFR | rs13306561 | 1 | 11788391 | G | A | 0.154 | −0.8657 | 0.1428 | 8.641E−10 | 9.93E−12 | 1.33E−09 | 2.6E−13 | 6.91E−14 | 8.51E−04 | 3.82E−11 | 1.75E−21 | 4.49E−24 | 3E−19 | ||
| HRH1 | rs347591 | 3 | 11265122 | G | T | 0.345 | −0.5284 | 0.1071 | 8.14E−07 | 0.001192 | 1.57E−08 | |||||||||
| CYP17A1 | rs3824755 | 10 | 104585839 | C | G | 0.096 | −0.6438 | 0.1289 | 5.93E−07 | 2.46E−07 | 6.06E−04 | 0.000384 | 1.52E−09 | 3.77E−10 | ||||||
| LSP1 | rs661348 | 11 | 1861868 | C | T | 0.437 | 0.472 | 0.1038 | 5.43E−06 | 7.55E−11 | 3.39E−15 | |||||||||
| SOX6 | rs2014408 | 11 | 16321858 | T | C | 0.21 | 0.5571 | 0.1246 | 7.74E−06 | 1.03E−05 | 5.71E−10 | |||||||||
| ATP2B1 | rs2681472 | 12 | 88533090 | G | A | 0.172 | −0.9733 | 0.1654 | 5.87E−08 | 3.99E−09 | 6.95E−10 | 3.3E−11 | 2.24E−16 | 7.68E−19 | ||||||
| ATXN2 | rs10774625 | 12 | 110394602 | A | G | 0.505 | 0.6614 | 0.1261 | 1.103E−10 | 4.61E−10 | 1.56E−07 | 5.98E−09 | 8.88E−11 | 9.37E−10 | 4.94E−18 | 2.4E−19 | 8.12E−16 | |||
| CSK | rs7085 | 15 | 72882536 | T | C | 0.281 | 0.7412 | 0.1135 | 7.936E−11 | 5.01E−11 | 6.68E−11 | 1.58E−05 | 3.54E−08 | 1.33E−05 | 2.5E−14 | 2.14E−17 | 3.48E−14 | |||
| FURIN | rs6227 | 15 | 89226236 | T | C | 0.316 | 0.764 | 0.1506 | 4.081E−08 | 3.65E−09 | 3.88E−07 | 1.86E−06 | 1.02E−10 | 3.77E−09 | 9.83E−13 | 3.35E−18 | 8.01E−15 | |||
We confirmed previously reported BP associations at 10 loci, and identified two novel loci: rs347591 associated with SBP (chromosome 3p25, in an intron of HRH1, P = 1.57 × 10−8) (Fig. 1A); rs2169137 associated with DBP (chromosome 1q32, in an intron of MDM4, P = 5.9×10−8) (Fig. 1B). We additionally found evidence of association of rs281413 (chromosome 19p13 in an intron of ICAM3) with DBP, in our discovery analysis, although it was not confirmed in the replication analysis (P = 3.08 × 10−6 in discovery, P = 1.4 × 10−5 in the joint analysis). Finally, one of the SNP-trait associations we identified was novel in our analysis, with the association of rs2014408 with SBP (chromosome 11p15, in an intron of SOX6, P = 5.71 × 10−10), whereas previously only association with MAP had been reported. A second Bonferroni correction of our results for testing four traits did not result in a change in the overall results, so we present the original results here, as the four traits are highly correlated. Full association results for SNPs in our discovery analysis with association P < 1 × 10−5 are reported in Supplementary Material, Table S5. Despite ascertainment biases in some of the discovery cohorts, due to inclusion or exclusion based upon BP or hypertension status (as noted in Supplementary Material, Table S1), we show replication of the key findings. We comprehensively compared the results of our analysis with all published associations at the time of this report (6–13,19) (Supplementary Material, Table S6). We reviewed 77 previously reported loci for our BP traits of interest and found that 43 were represented on our genotyping array, with one region containing a proxy SNP (r2 = 0.66) rather than the index SNP previously reported (Supplementary Material, Table S4). At a nominal association threshold (P = 0.05), 32 SNPs were associated with one or more BP traits in our study, and with a multiple testing correction (P < 0.00116), we observed 21 SNPs with BP associations.
Figure 1.
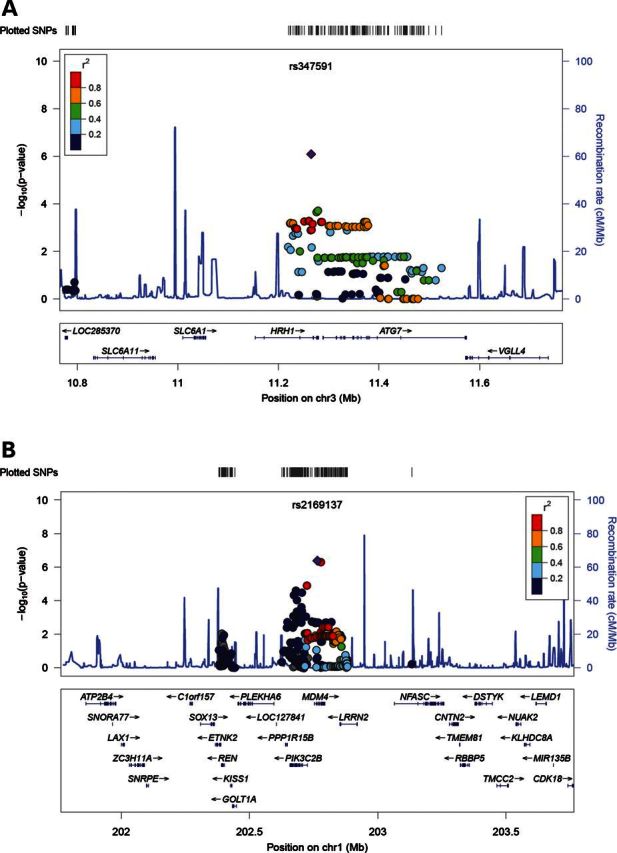
Regional association plots of the (A) HRH1 and (B) MDM4 loci are shown with negative log10 (P-value) on the y-axis and chromosomal position on the x-axis.
Sex interaction
In a secondary sex-specific analysis of our discovery sample (Supplementary Material, Table S7), we had no new significant associations. To follow-up possible sex differences in the two novel associations identified in our discovery efforts, we tested for interactions of rs347591 (HRH1) and rs2169137 (MDM4) with sex and identified a modest sex-specific effect for all four continuous BP traits at rs2169137 (P-value for interaction was 0.0058 for SBP; 0.055 for DBP; 0.014 for MAP and 0.033 for PP) (Supplementary Material, Table S8), with the association observed in women but not in men (in females only SBP beta was 0.041, se 0.322, and in males only SBP beta was 0.82, se 0.307, P-value for interaction of rs2169137 with sex was 0.0058).
Conditional analyses
For the loci described in Table 2, except those containing fewer than three genome-wide significant SNPs, conditional analyses were conducted using the allele dosage of the SNPs within a 500 kb (±250 kb) window around the most significant SNP per locus as a covariate in a subset of discovery cohorts (ARIC, CARDIA, CHS and MESA). Statistical models were identical to those used in the discovery analyses except for the additional SNP covariates. No SNPs remained significant in these conditional models after correcting the total number of tests (SNPs) in the 500 kb window, suggesting that only the strongest signal of association at each locus explained our findings. The complete results for these analyses are shown in Supplementary Material, Table S9.
Annotations of the identified loci
Annotations for the 12 loci associated with BP traits in our study showed that the majority of the variants we identified were located within an intron of the corresponding gene (rs13306561 in MTHFR; rs2169137 in MDM4; rs3824755 in CYP17A1; rs661348 in LSP1; rs2014408 in SOX6; rs2681472 in ATP2B1; rs4766578 and rs10774625 in ATXN2; rs8032315 and rs2071410 in FURIN; rs347591in HRH1). In aggregate, the associated SNPs accounted for 0.4–0.6% of the phenotypic variance in SBP, DBP, MAP and PP (Supplementary Material, Table S10). These estimates are in line with other association studies in which <1% of the overall phenotypic variance was explained by the common variants identified (6).
Expression QTL (eQTL) analysis showed that several of our associated SNPs (or proxy SNPs that were highly correlated with our lead SNP of interest [r2 > 0.8]) were associated with the expression levels of nearby genes, as assessed by the microarray analysis of RNA expression in various tissues, including blood, liver, lymph tissues, lymphoblastoid cell lines (LCLs) and peripheral blood T cells and monocytes, brain, adipose tissue and liver. These results are summarized in Supplementary Material, Table S11. Testing for eQTL associations at our novel loci showed no eQTL associations in the HRH1 region, but we did observe significant eQTL associations in the MDM4 region. Expression of MDM4 transcripts in lymph tissues, LCLs and monocytes was associated with rs4951401 (P = 1.59 × 10−7 in lymph tissue) and rs4245739 (P = 9.9 × 10−11 in LCLs and P = 6.68 × 10−12 in monocytes). Both rs4951401 and rs4245739 are highly correlated with the lead SNP we identified in our association analyses, rs2169137 in MDM4 (r2 0.963 and 0.927, respectively). Additional eQTL associations were identified for rs129128 with the HFE transcript (chromosome 6p22, P = 3.03 × 10−8), rs3184504 and rs653178 with SH2B3 and ATXN2 transcripts (12q24, P = 3.5 × 10−7); rs2470893 with the CSK transcript (15q24, P = 4.6 × 10−9).
Additionally, we queried the ENCODE database (http://www.regulomedb.org/). http://www.regulomedb.org/. We examined the annotations in the region of MDM4 SNPs (these SNPs include rs4245739, rs4951401, rs2169137). These SNPs show no new ENCODE regulatory annotations. We also queried HRH1 (rs347591) which did not have an eQTL association. This SNP does show overlap via position weight matrix for the HTF (HER2) transcription factor and also via DNA footprinting. This is potentially encouraging but no ChIP-seq data are available for HTF, so this is not necessarily strongly confirmed. The SNPs which do overlap with features are those with eQTL associations in HFE (rs129128), SH2B3 (rs3184504) and CSK (rs2470893).
Pleiotropy evaluations
We tested association of rs347591 (chromosome 3p25, HRH1) and rs2169137 (chromosome 1q32, MDM4) with traits known to be associated with hypertension, including coronary artery disease (CAD), left ventricular hypertrophy (LVH) and stroke. In these analyses, no additional trait associations with these SNPs were identified (Supplementary Material, Table S12).
DISCUSSION
Using a genotyping array that covers common genetic variation in ∼2100 candidate genes for several cardiovascular traits including BP, we identified robust associations at 11 known and two novel loci associated with continuous BP traits. In our primary discovery experiment, we identified 21 loci associated with our traits of interest in an analysis of 61 619 individuals (P < 2.4 × 10−6). Through the joint analysis of SNPs considered relevant during the discovery phase combined with replication data, in a total of 127 485 individuals, we identified robust association of 22 SNP-trait associations at 12 independent loci meeting our significance threshold of P < 2.4 × 10−6. Associations at two of these loci were not previously known to be associated with BP. The association of SBP with variants in the HRH1 locus, and the association of DBP with variants in the MDM4 locus were novel and replicated in silico in an additional 65 866 individuals. Additionally, the SOX6 locus contains a novel SNP-trait association for SBP with rs2014408; previously an association with MAP was shown for this locus (19). Finally, one additional region, chromosome 19p13.2 containing ICAM3, was significant in our discovery experiment but was not replicated in the joint analysis of discovery and replication samples. Only one of the replication cohorts had data available for this SNP (UK, P = 0.466). Although still meeting the array-wide significance criterion in the combined analysis, we do not consider this association to be robust in the replication experiment.
The histamine receptor H1 (HRH1) gene product is expressed in numerous tissues, such as smooth muscle and neurons. HRH1 is expressed in the nucleus tractus solitarii, where it has a role in regulating arterial pressure in rats (21). Congenic mapping in a rodent model linked HRH1 to autoimmune T-cell responses and vascular responses regulated by histamine after Bordetella pertussis toxin sensitization (22), and this locus appears to have a role in regulating blood brain barrier permeability (23). In a mouse model of atherosclerosis, apolipoprotein E-null mice treated with a histamine H1 receptor selective antagonist developed 40% fewer aortic plaques compared with mice treated with a H2 receptor antagonist and higher levels of inflammatory markers within the plaques and higher numbers of inflammatory cells, despite equivalent plasma lipoprotein levels, suggesting that the H1 receptor enhances low density lipoprotein cholesterol permeability into the intimal space of the artery (24). HRH1 was selected for the IBC array as a lower priority gene based on the presence in a Protein ANalysis Through Evolutionary Relationships (PANTHER) inflammatory/immune response pathway. The HRH1 gene is located ∼249 kb from the ATP2B2 gene with no linkage disequilibrium (LD) detected in HapMap CEU between common variants in these genes. Although ATP2B2 has not been directly implicated in BP or hypertension, it is one of the Ca(2+)-ATPases, a family of plasma membrane pumps encoded by at least three additional genes: ATP2B1 on chromosome 12q21; ATP2B3 on Xq28 and ATP2B4 on 1q25. ATP2B1 contains a robust SNP association for SBP, DBP and hypertension (6). Mice null for Atp2b1 are embryolethal (25), but recently a mouse with conditional knockout for the Atp2b1 in vascular smooth muscle cells was generated and found to have significantly elevated BP (26). In humans, no associations have been found between variants in the ATP2B2 gene and BP or other vascular traits. Additionally, within the recombination interval containing HRH1 and the peak association signal, SNPs within the ATG7 gene were associated but well below the statistical significance threshold for association, decreasing the likelihood that variants in ATG7 are driving the signal in this region.
Mouse double minute 4 homolog (MDM4) encodes a nuclear protein that is a critical regulator of p53 tumor suppressor protein by binding to this protein and inhibiting its activity, promoting cell viability and growth. MDM4 does not have a described vascular function, but it has a critical role in regulating p53, a transcription factor which plays an important role in regulating target genes that induce cell cycle arrest, apoptosis and cell senescence (27). MDM4 was also selected for the IBC array as a lower priority gene, based on the presence in a PANTHER apoptosis pathway. Mice null for p53 shows a variety of aging-related phenotypes (28). MDM2, another regulator of p53, has been shown to have a functional role in aldosterone-induced vascular remodeling (29), and MDM2 and MDM4 have been shown to have non-overlapping but a similar regulation of p53 pathways (30–32). A genome-wide linkage analysis for loci associated with PP has shown suggestive linkage on chromosome 1 (BP QTL 77, LOD = 2.7) for blacks in the HyperGEN Network (33), with MDM4 overlapping with this linkage peak. An overlap with the MDM4 region has also been shown for 10 quantitative trait loci (QTL) related to BP in rats (Human MDM4: http://rgd.mcw.edu/rgdweb/report/gene/main.html?id=1319584; Rats MDM4: http://rgd.mcw.edu/rgdweb/search/qtls.html?term=Mdm4[gene]&speciesType=3). The sex interaction we observed is of unclear significance since it was not confirmed in independent samples. MDM4 has also been associated with measures of cognitive performance in a GWAS (34). Within the recombination interval containing MDM4, correlated SNPs in PIK3C2B were associated, so we cannot exclude that the association signal may in part be due to variants in this gene. PIK3C2B has no known function related to BP or vascular disease.
The association of the SOX6 locus with SBP expands our prior knowledge of associations in this region with associated BP traits, with this locus previously reported as associated with MAP (11). MAP is derived from a calculation incorporating SBP and therefore these traits are highly correlated. Additionally, we replicated previous reports of 32 SNP-trait associations with BP, with acknowledgement that some of the cohorts in the published literature were genotyped on multiple platforms and have therefore been included in not only our IBC analysis but also previous reports.
The strengths of this study include the large size (n = 61 619) of the discovery meta-analysis with replication in an additional 65 866 individuals and access to gene expression data from humans subjects. This meta-analysis has led to the identification of two novel signals not previously detected by prior association studies. Another unreported locus was found in the discovery phase, e.g. ICAM3, which could not be replicated due to the inadequate sample size in the replication cohorts with high-quality genotypes for this SNP. The non-replication may also be explained by GWAS with less dense coverage used for the replication phase. In addition, future functional studies are needed to fully comprehend the underlying mechanisms responsible for the detected associations. However, we did find that the MDM4 locus was related to expression of MDM4 transcripts in several tissues, and the lead SNP identified in our study is in tight LD with the expression-associated SNPs, suggesting a transcriptional effect of the associated SNP we identified. Finally, it needs to be emphasized that the results were generated in European ancestry populations and that additional studies are needed in other ethnic groups.
In summary, our study has identified two novel loci containing the HRH1 and MDM4 genes associated with BP traits of clinical significance. The identification of these loci expands our understanding of the genetic determinants of BP.
MATERIALS AND METHODS
Study subjects
The phenotype and genotype data of 61 619 individuals of European ancestry, belonging to 27 participating studies (Supplementary Material, Table S1), were analyzed in the discovery phase, and additional 65 866 individuals of European ancestry from 18 additional studies were used in the replication phase. Individuals of European ancestry, as confirmed by principal component analysis of genetic ancestry, were analyzed in this study. All individuals in these studies provided informed consent, and each study was approved by its own local ethics committee. More detailed information about each participating cohort is provided in the Supplementary Material.
Phenotype
BP ascertainment in each study was performed according to the protocols described in the Supplementary Material. PP was defined as SBP minus DBP, and MAP was defined as 2/3 DBP plus 1/3 SBP. In the discovery analyses, each cohort provided regression models for its data, adjusted for age, age-squared, body mass index (BMI) and study-specific corrections for population substructure (based on principal component analysis). For individuals taking BP lowering medications, the BP values were adjusted by adding 15mmHg to the SBP and 10mmHg to the DBP in the discovery and replication cohorts. These adjustments were also implemented prior to the calculation of estimated off-treatment MAP and PP.
Genotyping and quality control
A total of 51 859 SNPs were genotyped and after filtering for an mismatches >30% with HapMap, removing SNPs without an rsID in dbSNP129 and SNPs with more than two possible bases for a single SNP, 48 372 SNPs included in the discovery meta-analyses, all present in at least one of the three versions of the Illumina HumanCVD BeadChip (‘Cardiochip’, ITMAT-Broad_CARe [IBC] array, Illumina San Diego, CA, USA) (20), which was used by all cohorts participating in the discovery analysis.
For the IBC array, gene and specific SNP information was assimilated from 2400 published studies systematically analyzed up until May 2007. An emphasis was placed on the sample size, data quality and strength of the described associations. Genes with known or putative association with phenotypes for sleep, lung and blood diseases were also nominated. Several pathway-based tools were used to identify additional biologically plausible candidate genes: Kyoto Encyclopedia of Genes and Genomes; PANTHER and BioCarta. These tools were employed to collate additional genes from key pathways including lipid metabolism, thrombogenesis, circulation and gas exchange, insulin resistance, metabolism, and inflammation, oxidative stress and apoptosis.
Early access was provided to a number of unpublished mouse atherosclerosis expression QTL (eQTL) datasets. Genes predicted to be causal for the atherosclerotic lesion size in genetic crosses of mice with differing susceptibility to atherosclerosis were identified. Early access was provided to a number of key findings from a number of CVD-related GWASs.
Genotypes were called using Beadstudio (Illumina) and the data were processed using stringent QC filters, as summarized in Supplementary Material, Tables S2 and S3. Variants with minor allele frequencies <1% were excluded from the analysis. Further details on genotyping methods are provided in the Supplementary Material, Tables S2 and S3.
QC measures were taken during the various steps of this work. Individuals with <90% call rate (completeness) across all SNPs were removed. SNPs with <95% call rate (completeness) or SNPs causing heterozygous haploid genotype calls were removed across all remaining individuals. SNPs with P < 1 × 10−7 for the Hardy–Weinberg Equilibrium test were also removed. In the NHLBI Candidate Gene Association Resource (CARe) samples (35), SNPs associated with chemistry plate effects were also removed.
Statistical association and meta-analysis
Initial association analyses were calculated within each cohort for males and females separately, adjusting for age, age-squared, BMI and study center when appropriate. In each cohort, except FHS, CFS and Amish, association analysis was performed using PLINK (36) using linear regression under an additive genetic model. The family structure was modeled using a linear mixed effects model implemented in R (37) in FHS and CFS, and the Mixed Model Analysis for Pedigrees software program in the Amish (38).
Meta-analysis was conducted using the summary statistics contributed by each discovery study, using an inverse variance weighted, fixed-effects method. At the meta-analysis stage of analysis, SNPs with frequencies incompatible with HapMap frequencies were removed (defined as >30% difference in the allele frequencies). Two analysis groups independently performed the meta-analysis using different software packages: METAL (39) and MANTEL (40); both applied a fixed-effects model weighted by inverse variance. The results from both the groups were compared and a concordance check was performed as a validation of the results (data not shown). Genomic control (41) was applied to each study result and then to the meta-analysis summary data to control effects possibly due to population stratification or cryptic relatedness. Quantile–quantile plots are shown for each trait in Supplementary Material, Fig. S1. Previous studies using the IBC array have used different significance thresholds from P < 1 × 10−5 to 3 × 10−6 [(42) and (43), respectively]. The CARe IBC array studies (35), which are included in this meta-analysis, determined that after accounting for LD, the effective number of independent tests was ∼20 500 for Europeans producing an experimental or ‘array-wide’ statistical threshold of P = 2.4 × 10−6, respectively, to maintain an false-positive rate of 5% (44) and thus, we have adopted these thresholds for this study. Since we have analyzed four traits, although highly correlated, we examined the effect of further Bonferroni correction for four tests. For each associated locus, the LD patterns were examined and independence between the loci identified in this study was verified using SNAP (45) (r2 < 0.3).
For loci with multiple SNPs showing association with the traits, we also conducted conditional analyses to evaluate independent signals. At each locus with variants associated with BP traits, we added the most significant SNP within the locus as a covariate in the association tests in each cohort in the NHLBI CARe consortium. Then, we performed meta-analysis of cohort-specific conditional analysis results. This conditional analysis was performed for the SNPs within a 500 kb region around the most significant SNP. The P-values for SNP association testing were then recorded, respectively, for associated locus for each trait.
Replication analysis
Independent SNPs with P < 1.0 × 10−5 in the discovery analysis were carried forward for replication analysis using independent samples for each trait. The significance threshold for association in the replication phase was a Bonferroni-corrected P-value based on an α = 0.05 and the final number of independent (r2 < 0.3) SNPs tested, and we also combined the discovery and replication data in a meta-analysis, in which evidence of positive replication was defined by P < 2.4 × 10−6 in the meta-analysis of combined discovery and replication samples. Associated loci were tested for replication by carrying forward to replication testing the lead SNP (minimum P-value) at each locus.
Interaction testing
For the lead SNP identified in novel genes, we tested for interactions with sex by first calculating the residual after adjusting for age, age-squared, BMI, sex and 10 principle components, and then performed sex-specific linear regression on a SNP for each cohort separately. This analysis was done using the PLINK G × E function. The sex-specific estimates were further combined by meta-analysis for men and women separately, using METAL (39). The interaction between gene and sex was tested by comparing the regression coefficients in men and women. That is, we calculated a T statistic: 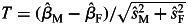 , where
, where 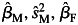 and
and 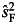 are the estimated regression coefficients and their standard errors from male and female meta-analysis, respectively. T follows the standard normal distribution.
are the estimated regression coefficients and their standard errors from male and female meta-analysis, respectively. T follows the standard normal distribution.
Variance explained
The proportion of the trait variance explained by the discovered associations was calculated by first obtaining the residuals after adjusting for age, age-squared, BMI and 10 PCs and then performing linear regression on all 21 identified associated variants together in a subset of the cohorts comprising the NHLBI CARe consortium cohorts (ARIC, CHS, MESA, CARDIA, CFS). The variance explained was calculated by a standard analysis of variance.
Annotation
eQTL analysis
We identified alias rsIDs for significant index SNPs using SNAP (45). Further proxy SNPs in high LD (r2 = 1.0) were identified with SNAP using multiple HapMap CEU builds. Current and alias rsIDs were searched for primary SNPs and LD proxies against a collected database of expression SNP (eSNP) results including the following tissues: fresh lymphocytes (46), fresh leukocytes (47), leukocyte samples in individuals with celiac disease (48)), LCLs derived from asthmatic children (49), HapMap LCL from three populations (50), a separate study on HapMap CEU LCL (51), peripheral blood monocytes (52,53), omental and subcutaneous adipose (54,55), stomach (55) and whole blood samples (54,56), endometrial carcinomas (57), brain cortex (52,58), three large studies of brain regions including prefrontal cortex, visual cortex and cerebellum, respectively (Emilsson, personal communication), liver (55,59,60), osteoblasts (61), skin (62) and additional fibroblast, T-cell and LCL sample datasets (63). The collected eSNP results met the criteria for statistical thresholds for association with gene transcript levels as described in the original papers. In each case, where an index or proxy SNP was associated with a transcript, we further examined the strongest eSNP for that transcript within that dataset (best eSNP), and the LD between the best eSNP and BP-selected eSNPs to assess the concordance of the BP association and expression signals. Annotation of SNPs in the region was done with the SNAP web-based tool (45).
Evaluation of pleiotropy of BP variants with cardiovascular disease
We evaluated the effect of the novel loci identified in our study with traits known to be related to elevated hypertension including CAD, LVH and stroke. The definitions of the CAD, LVH and stroke phenotypes and association tests were carried out as described in the Supplementary Material. Evidence of association for the additional traits was alpha of 0.05 adjusted by the two SNPs tested for association (P = 0.025).
SUPPLEMENTARY MATERIAL
Discovery cohort Acknowledgements
AMC-PAS: Funding for PAS was provided by Ipse Movet; Bloodomics (LSHM-CT-2004-503485).
Amish: The Amish studies were funded by the National Institutes of Health (R01 HL088119, R01 AG18728, U01 HL72515, U01 GM074518 with additional funding for cardiochip analysis provided by an American Heart Association Scientist Development grant (0830146N to H.S.). Genotyping of cardiochip was carried out in the Genomics Core at the University of Maryland, Baltimore with support from the Mid-Atlantic Nutrition and Obesity Research Center (NIH P30 DK072488).
ARIC: The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HSN268201100009C, HHSN268201100010C, HHSN268201100011C and HHSN268201100012C), R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402 and National Institutes of Health contract HHSN268200625226C. Atherosclerotic Risk in Communities: University of North Carolina at Chapel Hill (N01-HC-55015, N01-HC-55018), Baylor Medical College (N01-HC-55016), University of Mississippi Medical Center(N01-HC-55021), University of Minnesota (N01-HC-55019), Johns Hopkins University (N01-HC-55020), University of Texas, Houston (N01-HC-55022). The authors thank the staff and participants of the ARIC study for their important contributions. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research.
BHS: ENS, SSM and NJS are supported in part by NIH/NCRR Grant Number UL1 RR025774. The BHS was supported by grants HD-061437 and HD-062783 from the National Institute of Child Health and Human Development, and AG-16592 from the National Institute on Aging.
CARDIA: Coronary Artery Risk in Young Adults: University of Alabama at Birmingham (N01-HC-48047, N01-HC-95095), University of Minnesota (N01-HC-48048), Northwestern University (N01-HC-48049), Kaiser Foundation Research Institute (N01-HC-48050), Tufts-New England Medical Center (N01-HC-45204), Wake Forest University (N01-HC-45205), Harbor-UCLA Research and Education Institute (N01-HC-05187), University of California, Irvine (N01-HC-45134, N01-HC-95100).
CCCS: CCCS was supported by grant P50HL81011 from the National Heart, Lung and Blood Institute, National Institutes of Health, US Department of Health and Human Services. The authors thank the CHARISMA investigators for phenotype and sample collection.
CFS: Cleveland Family Study was supported by funding from the National Heart, Lung and Blood Institute, National Institutes of Health, US Department of Health and Human Services and Case Western Reserve University (R01-HL-46380, M01-RR-00080).
CHS: Cardiovascular Health Study: University of Washington (N01-HC-85079, N01-HC-55222, U01-HL-080295),Wake Forest University (N01-HC-85080), Johns Hopkins University (N01-HC-85081, N01-HC-15103), University of Pittsburgh (N01-HC-85082), University of California, Davis (N01-HC-85083), University of California, Irvine (N01-HC-85084), New England Medical Center (N01-HC-85085), University of Vermont (N01-HC-85086), Georgetown University (N01-HC-35129), University of Wisconsin (N01-HC-75150).
CLEAR: CLEAR support (GPJ) came from R01 HL67406, the Northwest Institute of Genetic Medicine and the State of Washington Life Sciences Discovery Fund. The CLEAR investigators sincerely thank the participants for their efforts.
EPIC-NL: The EPIC-NL study was funded by ‘Europe against Cancer’ Program of the European Commission (SANCO), Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch Cancer Society; ZonMW the Netherlands Organization for Health Research and Development, World Cancer Research Fund (The Netherlands). Genotyping of the IBC-chip was funded by IOP Genomics grant IGE05012 from NL Agency.
FHS: The Framingham Heart Study research included in this study is funded by NIH grant/contract N01-HC-25195, R01-HL-092577, R01-HL-076784, R01-AG-028321 and by the NIH Intramural Research Program.
INVEST: The INternational VErapamil SR Trandolapril (INVEST) genetic substudy was funded by NIH grants HL074730, HL69758, HL077113, GM074492 and RR017568, a grant from Abbott Pharmaceuticals and the Florida Opportunity Fund and NIH/NCRR clinical and Translational Science Award (CTSA) to University of Florida UL1 TR000064. INVEST and Pharmacogenomics Evaluation of Antihypertensive Responses (PEAR) studies thank the participants and the investigators who made this collection possible.
LURIC: LURIC thanks their participants and researchers and acknowledges that it has received funding through the sixth Framework Program (integrated project Bloodomics, grant LSHM-CT-2004-503485) and seventh Framework Program (integrated project Atheroremo, Grant Agreement number 201668) of the European Union.
MEDAL: MEDAL was supported by Merck & Co (Whitehouse Station, NJ, USA). The authors thank Amarjot Kaur and the current or former employees of Merck Research Laboratories who contributed to the conduct and analysis of the MEDAL data.
MESA: Multi-Ethnic Study of Atherosclerosis: support for MESA is provided by contracts N01-HC-95159 through N01-HC-95169 and CTSA UL1-RR-024156. Funding for genotyping was provided by NHLBI Contract N02-HL-6-4278 and N01-HC-65226.
MONICA/KORA: The MONICA/KORA Augsburg studies were financed by the Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany and supported by grants from the German Federal Ministry of Education and Research (BMBF). Part of this work was financed by the German National Genome Research Network (NGFNplus, project number 01GS0834), by the German Research Foundation (TH-784/2-1 and TH-784/2-2), by the European Foundation for the Study of Diabetes and through additional funds from the Helmholtz Zentrum München, the German Diabetes Center and the University of Ulm. Furthermore, the research was supported within the Munich Center of Health Sciences (MC Health) as part of the Ludwig Maximilians University innovative.
NSHS95: Nova Scotia Health Survey 1995 (NSHS95) was supported by grants HL-091099, HL-080665, HL-076857, HL-084034, HL-088117, HL-07854 and HL-072866 from the NHLBI; by the National Health and Welfare of Canada, Ottawa, Ontario; by the Nova Scotia Department of Health, Halifax and by the Heart and Stroke Foundation of New Brunswick, Saint John.
PEAR: PEAR was supported by the NIH Pharmacogenetics Research Network grant U01-GM074492; and CTSA grants UL1-TR000064 (University of Florida), UL1-TR000454 (Emory University) and UL1-TR000135 (Mayo Clinic); and funds from the Mayo Foundation.
PennCAC: The University of Pennsylvania Coronary Artery Calcification Study (PennCAC) gratefully acknowledges internal funding from the University of Pennsylvania and the participation of the study subject and is indebted to the investigators on these teams.
PennCath: The University of Pennsylvania Catheterization study program (PennCATH) gratefully acknowledges internal funding from the University of Pennsylvania and the participation of the study subject and is indebted to the investigators on these teams.
PROCARDIS: This work was supported by the British Heart Foundation; by the European Community Sixth Framework Program (grant number LSHM-CT-2007-037273) and by AstraZeneca AB. R.C., M.F. is supported by the British Heart Foundation Center for Research Excellence.
SMART: Folkert W. Asselbergs is supported by a clinical fellowship from the Netherlands Organization for Health Research and Development (ZonMw grant 90700342). M.F.L.M. was financially supported by EUGeneHeart, grant number LSHM-CT-2005-018833. The authors acknowledge all MR technicians, research nurses and medical students involved in SMART Heart for valuable support.
WHI: The Women's Health Initiative (WHI) program is funded by the National Heart, Lung and Blood Institute, National Institutes of Health, US Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C and HHSN271201100004C.
Replication cohort acknowledgements
This work was supported by the British Heart Foundation (grant number PG/07/131/24254 to P.B.M.) for HumanCVD BeadChip genotyping for the AIBIII, ASCOT, BRIGHT, MDC and NORDIL cohorts; by the Wellcome Trust (grant number 093078/Z/10/Z to T.J.) and in part by a VIP award from the Wellcome Trust to Queen Mary University of London in the 2009/2010 academic year.
AIBIII: The Allied Irish Bank workers III study was supported by the Higher Education Authority (Ireland), Program for research in Third-Level Institutions Cycle 3, Program for Human Genomics. We thank the Allied Irish Bank and their employees for facilitating the study.
ASCOT: This work was supported by Pfizer, New York, NY, USA, for the ASCOT study and the collection of the ASCOT DNA repository; by Servier Research Group, Paris, France; and by Leo Laboratories, Copenhagen, Denmark. We thank all ASCOT trial participants, physicians, nurses and practices in the participating countries for their important contribution to the study. In particular, we thank Clare Muckian and David Toomey for their help in DNA extraction, storage and handling.
BRIGHT: "This work was supported by the Medical Research Council of Great Britain (grant number G9521010D) and by the British Heart Foundation (grant number PG/02/128). A.F.D. was supported by the British Heart Foundation (grant numbers RG/07/005/23633, SP/08/005/25115) and by the European Union Ingenious HyperCare Consortium: Integrated Genomics, Clinical Research, and Care in Hypertension (grant number LSHM-C7-2006-037093). The BRIGHT study is extremely grateful to all the patients who participated in the study and the BRIGHT nursing team. We would also like to thank the Barts Genome Center staff for their assistance with this project. This work forms part of the research themes contributing to the translational research portfolio for Barts and the London Cardiovascular Biomedical Research Unit, which is supported and funded by the National Institute for Health Research.
BWHHS: The British Women's Heart and Health Study is supported by funding from the British Heart Foundation and the Department of Health Policy Research Program (England). We thank the BWHHS data collection team, general practitioners who helped with recruitment of participants and the participants. We thank all of the participants and the general practitioners, research nurses and data management staff who supported data collection and preparation. The BWHHS is coordinated by Shah Ebrahim (PI), Debbie Lawlor and Juan-Pablo Casas, with genotyping funded by the BHF (PG/07/131/24254, PI Tom Gaunt).The BRIGHT study was supported by the Medical Research Council of Great Britain (G9521010D) and the British Heart Foundation. (PG/02/128).
GRAPHIC: This work was supported by the British Heart Foundation (grant numbers RG/2001004, PG/07/132/24256) for recruitment and genotyping of the GRAPHIC cohort. N.J.S. was supported by a British Heart Foundation Chair of Cardiology (grant number CH/03/001. This study is part of the research portfolio supported by the Leicester National Institute for Health Research Biomedical Research Unit in Cardiovascular Disease.
Lifelines: The LifeLines Cohort Study, and generation and management of GWAS genotype data for the LifeLines Cohort Study, is supported by the Netherlands Organization of Scientific Research NWO (grant 175.010.2007.006), the Economic Structure Enhancing Fund (FES) of the Dutch government, the Ministry of Economic Affairs, the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the Northern Netherlands Collaboration of Provinces (SNN), the Province of Groningen, University Medical Center Groningen, the University of Groningen, Dutch Kidney Foundation and Dutch Diabetes Research Foundation.
We thank Behrooz Alizadeh, Annemieke Boesjes, Marcel Bruinenberg, Noortje Festen, Ilja Nolte, Lude Franke, Mitra Valimohammadi for their help in creating the GWAS database, and Rob Bieringa, Joost Keers, René Oostergo, Rosalie Visser and Judith Vonk for their work related to data collection and validation. The authors are grateful to the study participants, the staff from the LifeLines Cohort Study and Medical Biobank Northern Netherlands, and the participating general practitioners and pharmacists. LifeLines Scientific Protocol Preparation: Rudolf de Boer, Hans Hillege, Melanie van der Klauw, Gerjan Navis, Hans Ormel, Dirkje Postma, Judith Rosmalen, Joris Slaets, Ronald Stolk, Bruce Wolffenbuttel; LifeLines GWAS Working Group: Behrooz Alizadeh, Marike Boezen, Marcel Bruinenberg, Noortje Festen, Lude Franke, Pim van der Harst, Gerjan Navis, Dirkje Postma, Harold Snieder, Cisca Wijmenga, Bruce Wolffenbuttel.
MDC: This work was supported by the Swedish Medical Research Council; by the Swedish Heart and Lung Foundation; by the Medical Faculty of Lund University, Malmo University Hospital; by the Albert P_ahlsson Research Foundation; by the Crafoord foundation; by the Ernhold Lundstroms Research Foundation, the Region Skane; by the Hulda and Conrad Mossfelt Foundation; by the King Gustaf V and Queen Victoria Foundation; by the Lennart Hanssons Memorial Fund and by the Marianne and Marcus Wallenberg Foundation.
NBS: This work was supported by the Wellcome Trust (grant number 076113/C/04/Z); by the National Institute for Health Research (grant number RP-PG-0310-1002) program grant to NHSBT, and a National Institute for Health Research grant to the Cambridge Comprehensive Biomedical Research Center; and by the British Heart Foundation (grant number PG/07/132/24256) for HumanCVD BeadChip genotyping for the UKBS-CC cohort. We acknowledge use of DNA from the UK Blood Services collection of Common Controls (UKBS-CC collection), funded by the Wellcome Trust and by the National Institute for Health Research. The collection was established as part of the Wellcome Trust Case Control Consortium (WTCCC 2007).
NESDA: The infrastructure for the NESDA study is funded through the Geestkracht program of the Dutch Scientific Organization (ZON-MW, grant number 10-000-1002) and matching funds from participating universities and mental health care organizations. Genotyping in NESDA was funded by the Genetic Association Information Network of the Foundation for the US National Institutes of Health. Statistical analyses were carried out on the Genetic Cluster Computer (http://www.geneticcluster.org), which is financially supported by the Netherlands Scientific Organization (NWO 480-05-003) along with a supplement from the Dutch Brain Foundation.
NORDIL: This work was supported by the British Heart Foundation (grant number CH/98001 to A.F.D., RG/07/005/23633 to A.F.D., S.P. and C.D.) and a Special Project, for genotyping of the Swedish extremes from the NORDIL and MDC cohorts; and by Pharmacia. We thank Professor Thomas Hedner (Department of Clinical Pharmacology, Sahlgrenska Academy, Gotheburg, Sweden) and Professor Sverre Kjeldsen (Ullevaal University Hospital, University of Oslo, Oslo, Norway), who are investigators of the NORDIL study. Professor Kjeldsen is also an investigator of the ASCOT trial.
PREVEND: PREVEND genetics is supported by the Dutch Kidney Foundation (Grant E033), the National Institutes of Health (grant LM010098), The Netherlands organization for health research and development (NWO VENI grant 916.761.70) and the Dutch Inter University Cardiology Institute Netherlands.
Procardis: This work was supported by the British Heart Foundation; by the European Community Sixth Framework Program (grant number LSHM-CT-2007-037273) and by AstraZeneca AB. R.C., M.F. is supported by the British Heart Foundation Center for Research Excellence.
Rotterdam Study: The contributions of inhabitants, general practitioners and pharmacists of the Ommoord district to the Rotterdam Study are gratefully acknowledged. The Rotterdam Study is supported by the Erasmus Medical Center Rotterdam, the Erasmus University Rotterdam, the Netherlands Organization for Scientific Research (NWO), the Netherlands Organization for Health Research and Development (ZonMW), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry of Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam.
TRAILS: TRAILS (Tracking Adolescents’ Individual Lives Survey) is a collaborative project involving various departments of the University Medical Center and University of Groningen, the Erasmus University Medical Center Rotterdam, the University of Utrecht, the Radboud Medical Center Nijmegen, and the Parnassia Bavo group, all in the Netherlands. TRAILS has been financially supported by grants from the Netherlands Organization for Scientific Research NWO (Medical Research Council program grant GB-MW 940-38-011; ZonMW Brainpower grant 100-001-004; ZonMw Risk Behavior and Dependence grants 60-60600-98-018 and 60-60600-97-118; ZonMw Culture and Health grant 261-98-710; Social Sciences Council medium-sized investment grants GB-MaGW 480-01-006 and GB-MaGW 480-07-001; Social Sciences Council project grants GB-MaGW 457-03-018, GB-MaGW 452-04-314, and GB-MaGW 452-06-004; NWO large-sized investment grant 175.010.2003.005; NWO Longitudinal Survey and Panel Funding 481-08-013); the Sophia Foundation for Medical Research (projects 301 and 393), the Dutch Ministry of Justice (WODC), the European Science Foundation (EuroSTRESS project FP-006), the Dutch Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL CP32) and the participating universities. We are grateful to all adolescents, their parents and teachers who participated in this research and to everyone who worked on this project and made it possible. Statistical analyses were carried out on the Genetic Cluster Computer (http://www.geneticcluster.org), which is financially supported by the Netherlands Scientific Organization (NWO 480-05-003) along with a supplement from the Dutch Brain Foundation.
WGHS: Women's genome health study: The WGHS is funded by the Donald W. Reynolds Foundation (Las Vegas, NV), the Fondation LeDucq (Paris, France), the National Heart, Lung and Blood Institute (NHLBI; HL043851) and the National Cancer Institute (NCI; CA047988). Funding for genotyping and collaborative scientific support was provided by Amgen.
WHII: This work was supported by the British Heart Foundation (grant numbers PG/07/133/24260, RG/08/008, SP/07/007/23671), Senior Fellowship to A.D.H. (grant number FS/2005/125), Chair for S.E.H.; by the National Heart Lung and Blood Institute (grant number HL36310) for M.Kivimaki's and M.Kumari's contributions to this work; by the Medical Research Council (grant number G0802432) Population Health Scientist Fellowship to M.V.H.; by the Health and Safety Executive; by the Department of Health; by the National Institute on Aging in the USA (grant number AG13196); by the Agency for Health Care Policy Research (grant number HS06516); by the John D. and Catherine T. MacArthur Foundation Research Networks on Successful Midlife Development and Socioeconomic Status and Health.
Conflict of Interest statement. C.M.M. and S.C. are employees of Merck Pharmaceuticals, USA.
AUTHORS' CONTRIBUTIONS
Conceived and designed the experiment, alphabetically F.A., P.deB., S.G., B.K., D.L., X.Z. Performed the experiments: individual cohorts conducted genotyping and cohort-level analyses, except for the NHLBI CARe program samples were genotyped under a contract with the Broad/MIT. Analyzed the data: F.A., P.deB., S.G., W.G., B.K., D.L., V.T., X.Z. Contributed reagents/materials/analysis tools: all authors. Wrote the paper: F.A., P.deB., S.G., W.G., B.K., D.L., V.T., X.Z., with contributions from all authors.
FUNDING
The grants and contracts that have supported CARe are listed at http://www.nhlbi.nih.gov/resources/geneticsgenomics/programs/care.htm. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
ACKNOWLEDGEMENTS
We thank the National Heart, Lung and Blood Institute and the research institutions, study investigators and field staff for their support in creating the CARe program for biomedical research. We also thank the study participants, without whom this endeavor would not have been possible. DNA samples were genotyped in part through the Broad Institute (N01-HC-65226).
REFERENCES
- 1.Lewington S., Clarke R., Qizilbash N., Peto R., Collins R. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–1913. doi: 10.1016/s0140-6736(02)11911-8. doi:10.1016/S0140-6736(02)11911-8. [DOI] [PubMed] [Google Scholar]
- 2.Turnbull F. Effects of different blood-pressure-lowering regimens on major cardiovascular events: results of prospectively-designed overviews of randomised trials. Lancet. 2003;362:1527–1535. doi: 10.1016/s0140-6736(03)14739-3. doi:10.1016/S0140-6736(03)14739-3. [DOI] [PubMed] [Google Scholar]
- 3.Franklin S.S., Larson M.G., Khan S.A., Wong N.D., Leip E.P., Kannel W.B., Levy D. Does the relation of blood pressure to coronary heart disease risk change with aging? The Framingham Heart Study. Circulation. 2001;103:1245–1249. doi: 10.1161/01.cir.103.9.1245. doi:10.1161/01.CIR.103.9.1245. [DOI] [PubMed] [Google Scholar]
- 4.Franklin S.S., Khan S.A., Wong N.D., Larson M.G., Levy D. Is pulse pressure useful in predicting risk for coronary heart Disease? The Framingham heart study. Circulation. 1999;100:354–360. doi: 10.1161/01.cir.100.4.354. doi:10.1161/01.CIR.100.4.354. [DOI] [PubMed] [Google Scholar]
- 5.Franklin S.S., Gustin W.T., Wong N.D., Larson M.G., Weber M.A., Kannel W.B., Levy D. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation. 1997;96:308–315. doi: 10.1161/01.cir.96.1.308. doi:10.1161/01.CIR.96.1.308. [DOI] [PubMed] [Google Scholar]
- 6.Ehret G.B., Munroe P.B., Rice K.M., Bochud M., Johnson A.D., Chasman D.I., Smith A.V., Tobin M.D., Verwoert G.C., Hwang S.J., et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–109. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy D., Ehret G.B., Rice K., Verwoert G.C., Launer L.J., Dehghan A., Glazer N.L., Morrison A.C., Johnson A.D., Aspelund T., et al. Genome-wide association study of blood pressure and hypertension. Nat. Genet. 2009;41:677–687. doi: 10.1038/ng.384. doi:10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newton-Cheh C., Johnson T., Gateva V., Tobin M.D., Bochud M., Coin L., Najjar S.S., Zhao J.H., Heath S.C., Eyheramendy S., et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet. 2009;41:666–676. doi: 10.1038/ng.361. doi:10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newton-Cheh C., Larson M.G., Vasan R.S., Levy D., Bloch K.D., Surti A., Guiducci C., Kathiresan S., Benjamin E.J., Struck J., et al. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat. Genet. 2009;41:348–353. doi: 10.1038/ng.328. doi:10.1038/ng.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salvi E., Kutalik Z., Glorioso N., Benaglio P., Frau F., Kuznetsova T., Arima H., Hoggart C., Tichet J., Nikitin Y.P., et al. Genomewide association study Using a high-density single nucleotide polymorphism array and case–control design identifies a novel essential hypertension susceptibility locus in the promoter region of endothelial NO synthase. Hypertension. 2012;59:248–255. doi: 10.1161/HYPERTENSIONAHA.111.181990. doi:10.1161/HYPERTENSIONAHA.111.181990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wain L.V., Verwoert G.C., O'Reilly P.F., Shi G., Johnson T., Johnson A.D., Bochud M., Rice K.M., Henneman P., Smith A.V., et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat. Genet. 2011;20:2273–2284. doi: 10.1038/ng.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Padmanabhan S., Melander O., Johnson T., Di Blasio A.M., Lee W.K., Gentilini D., Hastie C.E., Menni C., Monti M.C., Delles C., et al. Genome-wide association study of blood pressure extremes identifies variant near UMOD associated with hypertension. PLoS Genet. 2010;6:e1001177. doi: 10.1371/journal.pgen.1001177. doi:10.1371/journal.pgen.1001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson A.D., Newton-Cheh C., Chasman D.I., Ehret G.B., Johnson T., Rose L., Rice K., Verwoert G.C., Launer L.J., Gudnason V., et al. Association of hypertension drug target genes with blood pressure and hypertension in 86,588 individuals. Hypertension. 2011;57:903–910. doi: 10.1161/HYPERTENSIONAHA.110.158667. doi:10.1161/HYPERTENSIONAHA.110.158667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho J.E., Levy D., Rose L., Johnson A.D., Ridker P.M., Chasman D.I. Discovery and replication of novel blood pressure genetic loci in the Women's Genome Health Study. J. Hypertens. 2011;29:62–69. doi: 10.1097/HJH.0b013e3283406927. doi:10.1097/HJH.0b013e3283406927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox E.R., Young J.H., Li Y., Dreisbach A.W., Keating B.J., Musani S.K., Liu K., Morrison A.C., Ganesh S., Kutlar A., et al. Association of genetic variation with systolic and diastolic blood pressure among African Americans: the Candidate Gene Association Resource study. Hum. Mol. Genet. 2011;20:2273–2284. doi: 10.1093/hmg/ddr092. doi:10.1093/hmg/ddr092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu X., Young J.H., Fox E., Keating B.J., Franceschini N., Kang S., Tayo B., Adeyemo A., Sun Y.V., Li Y., et al. Combined admixture mapping and association analysis identifies a novel blood pressure genetic locus on 5p13: contributions from the CARe consortium. Hum. Mol. Genet. 2011;20:2285–2295. doi: 10.1093/hmg/ddr113. doi:10.1093/hmg/ddr113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehne B., Lewis C.M., Schlitt T. Exome localization of complex disease association signals. BMC Genomics. 2011;12:92. doi: 10.1186/1471-2164-12-92. doi:10.1186/1471-2164-12-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang W., de las Fuentes L., Davila-Roman V.G., Charles Gu C. Variable set enrichment analysis in genome-wide association studies. Eur. J. Hum. Genet. 2011;19:893–900. doi: 10.1038/ejhg.2011.46. doi:10.1038/ejhg.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson T., Gaunt T.R., Newhouse S.J., Padmanabhan S., Tomaszewski M., Kumari M., Morris R.W., Tzoulaki I., O'Brien E.T., Poulter N.R., et al. Blood pressure loci identified with a gene-centric array. Am. J. Hum. Genet. 2011;89:688–700. doi: 10.1016/j.ajhg.2011.10.013. doi:10.1016/j.ajhg.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keating B.J., Tischfield S., Murray S.S., Bhangale T., Price T.S., Glessner J.T., Galver L., Barrett J.C., Grant S.F., Farlow D.N., et al. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS One. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. doi:10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhuiyan M.E., Waki H., Gouraud S.S., Takagishi M., Kohsaka A., Maeda M. Histamine receptor H1 in the nucleus tractus solitarii regulates arterial pressure and heart rate in rats. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H523–H529. doi: 10.1152/ajpheart.00263.2011. doi:10.1152/ajpheart.00263.2011. [DOI] [PubMed] [Google Scholar]
- 22.Ma R.Z., Gao J., Meeker N.D., Fillmore P.D., Tung K.S., Watanabe T., Zachary J.F., Offner H., Blankenhorn E.P., Teuscher C. Identification of Bphs, an autoimmune disease locus, as histamine receptor H1. Science. 2002;297:620–623. doi: 10.1126/science.1072810. doi:10.1126/science.1072810. [DOI] [PubMed] [Google Scholar]
- 23.Lu C., Diehl S.A., Noubade R., Ledoux J., Nelson M.T., Spach K., Zachary J.F., Blankenhorn E.P., Teuscher C. Endothelial histamine H1 receptor signaling reduces blood–brain barrier permeability and susceptibility to autoimmune encephalomyelitis. Proc. Natl Acad. Sci. USA. 2010;107:18967–18972. doi: 10.1073/pnas.1008816107. doi:10.1073/pnas.1008816107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rozenberg I., Sluka S.H., Rohrer L., Hofmann J., Becher B., Akhmedov A., Soliz J., Mocharla P., Boren J., Johansen P., et al. Histamine H1 receptor promotes atherosclerotic lesion formation by increasing vascular permeability for low-density lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2010;30:923–930. doi: 10.1161/ATVBAHA.109.201079. doi:10.1161/ATVBAHA.109.201079. [DOI] [PubMed] [Google Scholar]
- 25.Okunade G.W., Miller M.L., Azhar M., Andringa A., Sanford L.P., Doetschman T., Prasad V., Shull G.E. Loss of the Atp2c1 secretory pathway Ca(2+)-ATPase (SPCA1) in mice causes golgi stress, apoptosis, and midgestational death in homozygous embryos and squamous cell tumors in adult heterozygotes. J. Biol. Chem. 2007;282:26517–26527. doi: 10.1074/jbc.M703029200. doi:10.1074/jbc.M703029200. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi Y., Hirawa N., Tabara Y., Muraoka H., Fujita M., Miyazaki N., Fujiwara A., Ichikawa Y., Yamamoto Y., Ichihara N., et al. Mice Lacking Hypertension Candidate Gene ATP2B1 in Vascular Smooth Muscle Cells Show Significant Blood Pressure Elevation. Hypertension. 2012;59:854–860. doi: 10.1161/HYPERTENSIONAHA.110.165068. [DOI] [PubMed] [Google Scholar]
- 27.Parant J., Chavez-Reyes A., Little N.A., Yan W., Reinke V., Jochemsen A.G., Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 2001;29:92–95. doi: 10.1038/ng714. doi:10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 28.Tyner S.D., Venkatachalam S., Choi J., Jones S., Ghebranious N., Igelmann H., Lu X., Soron G., Cooper B., Brayton C., et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. doi:10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura Y., Suzuki S., Suzuki T., Ono K., Miura I., Satoh F., Moriya T., Saito H., Yamada S., Ito S., et al. MDM2: a novel mineralocorticoid-responsive gene involved in aldosterone-induced human vascular structural remodeling. Am. J. Pathol. 2006;169:362–371. doi: 10.2353/ajpath.2006.051351. doi:10.2353/ajpath.2006.051351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biderman L., Poyurovsky M.V., Assia Y., Manley J.L., Prives C. MdmX is required for p53 interaction with and full induction of the Mdm2 promoter after cellular stress. Mol. Cell Biol. 2012;32:1214–1225. doi: 10.1128/MCB.06150-11. doi:10.1128/MCB.06150-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toledo F., Krummel K.A., Lee C.J., Liu C.W., Rodewald L.W., Tang M., Wahl G.M. A mouse p53 mutant lacking the proline-rich domain rescues Mdm4 deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory network. Cancer Cell. 2006;9:273–285. doi: 10.1016/j.ccr.2006.03.014. doi:10.1016/j.ccr.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 32.Toledo F., Wahl G.M. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007;39:1476–1482. doi: 10.1016/j.biocel.2007.03.022. doi:10.1016/j.biocel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bielinski S.J., Lynch A.I., Miller M.B., Weder A., Cooper R., Oberman A., Chen Y.D., Turner S.T., Fornage M., Province M., et al. Genome-wide linkage analysis for loci affecting pulse pressure: the Family Blood Pressure Program. Hypertension. 2005;46:1286–1293. doi: 10.1161/01.HYP.0000191706.41980.29. doi:10.1161/01.HYP.0000191706.41980.29. [DOI] [PubMed] [Google Scholar]
- 34.Need A.C., Attix D.K., McEvoy J.M., Cirulli E.T., Linney K.L., Hunt P., Ge D., Heinzen E.L., Maia J.M., Shianna K.V., et al. A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Hum. Mol. Genet. 2009;18:4650–4661. doi: 10.1093/hmg/ddp413. doi:10.1093/hmg/ddp413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musunuru K., Lettre G., Young T., Farlow D.N., Pirruccello J.P., Ejebe K.G., Keating B.J., Yang Q., Chen M.H., Lapchyk N., et al. Candidate gene association resource (CARe): design, methods, and proof of concept. Circ. Cardiovasc. Genet. 2010;3:267–275. doi: 10.1161/CIRCGENETICS.109.882696. doi:10.1161/CIRCGENETICS.109.882696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. doi:10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen M.H., Yang Q. GWAF: an R package for genome-wide association analyses with family data. Bioinformatics. 2010;26:580–581. doi: 10.1093/bioinformatics/btp710. doi:10.1093/bioinformatics/btp710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shuldiner A.R., O'Connell J.R., Bliden K.P., Gandhi A., Ryan K., Horenstein R.B., Damcott C.M., Pakyz R., Tantry U.S., Gibson Q., et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA. 2009;302:849–857. doi: 10.1001/jama.2009.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willer C.J., Li Y., Abecasis G.R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. doi:10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Bakker P.I., Ferreira M.A., Jia X., Neale B.M., Raychaudhuri S., Voight B.F. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 2008;17:R122–128. doi: 10.1093/hmg/ddn288. doi:10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. doi:10.1111/j.0006-341X.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 42.Talmud P.J., Drenos F., Shah S., Shah T., Palmen J., Verzilli C., Gaunt T.R., Pallas J., Lovering R., Li K., et al. Gene-centric association signals for lipids and apolipoproteins identified via the HumanCVD BeadChip. Am. J. Hum. Genet. 2009;85:628–642. doi: 10.1016/j.ajhg.2009.10.014. doi:10.1016/j.ajhg.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. 2011;7:e1002260. doi: 10.1371/journal.pgen.1002260. doi:10.1371/journal.pgen.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lo K.S., Wilson J.G., Lange L.A., Folsom A.R., Galarneau G., Ganesh S.K., Grant S.F., Keating B.J., McCarroll S.A., Mohler E.R., III, et al. Genetic association analysis highlights new loci that modulate hematological trait variation in Caucasians and African Americans. Hum. Genet. 2011;129:307–317. doi: 10.1007/s00439-010-0925-1. doi:10.1007/s00439-010-0925-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson A.D., Handsaker R.E., Pulit S.L., Nizzari M.M., O'Donnell C.J., de Bakker P.I. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. doi:10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goring H.H., Curran J.E., Johnson M.P., Dyer T.D., Charlesworth J., Cole S.A., Jowett J.B., Abraham L.J., Rainwater D.L., Comuzzie A.G., et al. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat. Genet. 2007;39:1208–1216. doi: 10.1038/ng2119. doi:10.1038/ng2119. [DOI] [PubMed] [Google Scholar]
- 47.Idaghdour Y., Czika W., Shianna K.V., Lee S.H., Visscher P.M., Martin H.C., Miclaus K., Jadallah S.J., Goldstein D.B., Wolfinger R.D., et al. Geographical genomics of human leukocyte gene expression variation in southern Morocco. Nat. Genet. 2010;42:62–67. doi: 10.1038/ng.495. doi:10.1038/ng.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heap G.A., Trynka G., Jansen R.C., Bruinenberg M., Swertz M.A., Dinesen L.C., Hunt K.A., Wijmenga C., Vanheel D.A., Franke L. Complex nature of SNP genotype effects on gene expression in primary human leucocytes. BMC Med. Genomics. 2009;2:1. doi: 10.1186/1755-8794-2-1. doi:10.1186/1755-8794-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dixon A.L., Liang L., Moffatt M.F., Chen W., Heath S., Wong K.C., Taylor J., Burnett E., Gut I., Farrall M., et al. A genome-wide association study of global gene expression. Nat. Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. doi:10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 50.Stranger B.E., Nica A.C., Forrest M.S., Dimas A., Bird C.P., Beazley C., Ingle C.E., Dunning M., Flicek P., Koller D., et al. Population genomics of human gene expression. Nat. Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. doi:10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kwan T., Benovoy D., Dias C., Gurd S., Provencher C., Beaulieu P., Hudson T.J., Sladek R., Majewski J. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008;40:225–231. doi: 10.1038/ng.2007.57. doi:10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- 52.Heinzen E.L., Ge D., Cronin K.D., Maia J.M., Shianna K.V., Gabriel W.N., Welsh-Bohmer K.A., Hulette C.M., Denny T.N., Goldstein D.B. Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol. 2008;6:e1. doi: 10.1371/journal.pbio.1000001. doi:10.1371/journal.pbio.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeller T., Wild P., Szymczak S., Rotival M., Schillert A., Castagne R., Maouche S., Germain M., Lackner K., Rossmann H., et al. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. doi:10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emilsson V., Thorleifsson G., Zhang B., Leonardson A.S., Zink F., Zhu J., Carlson S., Helgason A., Walters G.B., Gunnarsdottir S., et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. doi:10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 55.Greenawalt D.M., Dobrin R., Chudin E., Hatoum I.J., Suver C., Beaulaurier J., Zhang B., Castro V., Zhu J., Sieberts S.K., et al. A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome Res. 2011;21:1008–1016. doi: 10.1101/gr.112821.110. doi:10.1101/gr.112821.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fehrmann R.S., Jansen R.C., Veldink J.H., Westra H.J., Arends D., Bonder M.J., Fu J., Deelen P., Groen H.J., Smolonska A., et al. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 2011;7:e1002197. doi: 10.1371/journal.pgen.1002197. doi:10.1371/journal.pgen.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kompass K.S., Witte J.S. Co-regulatory expression quantitative trait loci mapping: method and application to endometrial cancer. BMC Med. Genomics. 2011;4:6. doi: 10.1186/1755-8794-4-6. doi:10.1186/1755-8794-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Webster J.A., Gibbs J.R., Clarke J., Ray M., Zhang W., Holmans P., Rohrer K., Zhao A., Marlowe L., Kaleem M., et al. Genetic control of human brain transcript expression in Alzheimer disease. Am. J. Hum. Genet. 2009;84:445–458. doi: 10.1016/j.ajhg.2009.03.011. doi:10.1016/j.ajhg.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schadt E.E., Molony C., Chudin E., Hao K., Yang X., Lum P.Y., Kasarskis A., Zhang B., Wang S., Suver C., et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. doi:10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Innocenti F., Cooper G.M., Stanaway I.B., Gamazon E.R., Smith J.D., Mirkov S., Ramirez J., Liu W., Lin Y.S., Moloney C., et al. Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet. 2011;7:e1002078. doi: 10.1371/journal.pgen.1002078. doi:10.1371/journal.pgen.1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grundberg E., Kwan T., Ge B., Lam K.C., Koka V., Kindmark A., Mallmin H., Dias J., Verlaan D.J., Ouimet M., et al. Population genomics in a disease targeted primary cell model. Genome Res. 2009;19:1942–1952. doi: 10.1101/gr.095224.109. doi:10.1101/gr.095224.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ding J., Gudjonsson J.E., Liang L., Stuart P.E., Li Y., Chen W., Weichenthal M., Ellinghaus E., Franke A., Cookson W., et al. Gene expression in skin and lymphoblastoid cells: Refined statistical method reveals extensive overlap in cis-eQTL signals. Am. J. Hum. Genet. 2010;87:779–789. doi: 10.1016/j.ajhg.2010.10.024. doi:10.1016/j.ajhg.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dimas A.S., Deutsch S., Stranger B.E., Montgomery S.B., Borel C., Attar-Cohen H., Ingle C., Beazley C., Gutierrez Arcelus M., Sekowska M., et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. doi:10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]