

Abstract
Increased levels of nicotinamide/nicotinic acid mononucleotide adenylyltransferase (NMNAT) act as a powerful suppressor of Wallerian degeneration and ataxin- and tau-induced neurodegeneration in flies and mice. However, the nature of the suppression mechanism/s remains controversial. Here, we show that in yeast models of proteinopathies, overexpression of the NMNAT yeast homologs, NMA1 and NMA2, suppresses polyglutamine (PolyQ) and α-synuclein-induced cytotoxicities. Unexpectedly, overexpression of other genes in the salvage pathway for NAD+ biosynthesis, including QNS1, NPT1 and PNC1 also protected against proteotoxicity. Our data revealed that in all cases, this mechanism involves extensive clearance of the non-native protein. Importantly, we demonstrate that suppression by NMA1 does not require the presence of a functional salvage pathway for NAD+ biosynthesis, SIR2 or an active mitochondrial oxidative phosphorylation (OXPHOS) system. Our results imply the existence of histone deacetylase- and OXPHOS-independent crosstalk between the proteins in the salvage pathway for NAD+ biosynthesis and the proteasome that can be manipulated to achieve cellular protection against proteotoxic stress.
INTRODUCTION
Nicotinamide/nicotinic acid mononucleotide adenylyl transferase (NMNAT or NMA) is a key enzyme in both de novo and salvage pathways for NAD+ biosynthesis (1) (Fig. 1A). NMNAT can additionally function as a stress chaperone (2,3), and over the last 5 years, several groups have reported that NMNAT is a key neuronal maintenance factor (4–6). NMNAT is essential for maintaining physiologic neuronal integrity, and when overexpressed, it has been shown to protect against several neurodegenerative conditions in fly and mouse models, including Wallerian degeneration (WD) (3,7), exotoxicity (8), spinocerebellar ataxia 1-induced neurodegeneration (3,9) and tau-induced neurodegeneration (10,11). The mechanism by which NMNAT exerts neuroprotection remains to be fully characterized. Two recent reports have suggested that NMNAT prevents axonal degeneration in two distinct Drosophila models through modulation of mitochondria-related functions (12,13).
Figure 1.

Overexpression of NMA1 and other genes in the NAD+ salvage pathway suppress mutant PolyQ- and α-synuclein-induced proteotoxicity in yeast. (A) Diagram depicting the salvage and de novo pathways for NAD+ biosynthesis in S. cerevisiae. Note that whereas in higher eukaryotes (not depicted here), NMNAT acts as a nicotinamide/nicotinic acid mononucleotide adenylyltransferase, its S. cerevisiae homologs, the products of NMA1/2, act exclusively as nicotinic acid mononucleotide adenylyltransferases. The products of PNC1, NPT1 and QNS1 are nicotinamidase, nicotinic acid phosphoribosyltransferase and NAD synthetase, respectively. Several histone deacetylases, including Sir2, break down NAD+ into nicotinamide. BNA1 through BNA7 are biosynthesis of nicotinic acid genes in the de novo NAD+ biosynthesis pathway. (B and C) Serial dilutions of indicated strains were spotted on YPD and YPGAL plates. The expression of 103Q, α-synuclein and all the indicated genes are under the control of a galactose inducible promoter. Pictures were taken after 2 days of incubation at 30°C. YEp352 is an empty episomal vector.
Owing to the high degree of conservation of cellular activities from yeast to humans, we and others have successfully used yeast Saccharomyces cerevisiae as a model organism to study the molecular mechanisms underlying human neurodegenerative diseases, including Huntington's disease (HD) (14–16) and Parkinson's disease (PD) (17–19). Yeast models of human neurodegenerative diseases are based on the expression of ‘gain of function’ mutations in proteins responsible for the human disorders [e.g. huntingtin with mutant polyglutamine (PolyQ) domains in the case of HD and α-synuclein in the case of PD]. Yeast models recapitulate the crucial events preceding cell death that are manifested during the course of the human condition, including protein misfolding and aggregation, and we have, therefore, exploited them to gain insight into the mechanism of suppression by NMNAT and functionally related proteins.
RESULTS
Overexpression of genes in the salvage pathway for NAD+ biosynthesis suppresses proteotoxicity in yeast
In yeast, heterologous expression of either mutant PolyQ domains (103Q) or α-synuclein fused to green fluorescent protein (GFP) under the control of a GAL1 promoter induced by growth in media containing galactose (YPGAL) produces significant growth defects (Fig. 1B and C) as we and others have previously described (15,17,20). There are two homologs of NMNAT in S. cerevisiae, NMA1 and NMA2, that are more than 90% identical and enzymatically act exclusively as nicotinic acid mononucleotide adenylyltransferases [Fig. 1A, (21,22)]. When NMA1/2 were overexpressed from an episomal plasmid and under the control of a galactose-inducible promoter, they strongly suppressed the growth defect induced by both 103Q and α-synuclein (Fig. 1B and C), in agreement with previous reports in other neurodegenerative disease models in higher organisms (3). Several enzymes in the NAD+ salvage pathway, including NMA1/2, NPT1 and PNC1 (Fig. 1A), have been shown to delay replicative aging in yeast (22,23). We then tested whether other components of the pathway could suppress the 103Q- and α-synuclein-induced cytotoxicity and observed that NPT1, QNS1 and PNC1 were each able to exert a strong suppressive effect (Fig. 1B and C). In contrast, overexpression of TNA1, encoding the nicotinic acid plasma membrane permease, had no effect on the proteotoxic phenotype (Fig. 1B and C). The suppression mechanisms by NPT1, QNS1 and PNC1 do not involve compensatory induction of the expression of other genes in the pathway, including NMA1/2, as their mRNA steady-state levels were not increased (Supplementary Material, Fig. S1A and B). These results constitute the first report that several components of the NAD+ salvage pathway have the ability to suppress the proteotoxic effect of mutant PolyQ domains and α-synuclein in yeast.
Suppression by NAD+ biosynthetic genes does not require a functional NAD+ salvage pathway and is independent of histone deacetylase activity
In yeast, expression of different components of the NAD+ salvage pathway has been shown to delay replicative aging through a mechanism involving regeneration of NAD+ and increased activity of the histone deacetylase SIR2 (22,23). In addition, this has been proposed as one of the mechanisms underlying lifespan extension by caloric restriction (23,24). To test whether a fully functional salvage pathway for NAD+ regeneration is necessary for NMA1/2-mediated suppression of proteotoxicities, we first tested the effect of disrupting this pathway by deletion of an essential component, NPT1 (Fig. 1A). We found that the absence of NPT1 had no effect on the suppression of 103Q or α-synuclein toxicities by NMA1/2 or other components of the pathway (Fig. 2A), indicating that the suppression mechanism is independent of the integrity of the NAD+ salvage pathway. Although NMA1 overexpression in the 103Q strain increases total intracellular NAD (both NAD+ and NADH) levels, PNC1 overexpression [Supplementary Material, Fig. (S2A) and (25)] or additional copies of NPT1 (22) did not. In addition, the npt1 null strain accumulates ∼2.2-fold less NAD+ than strains with an intact NAD+ salvage pathway [Supplementary Material, Fig. S2B and (26)]. The NAD+ levels in the npt1 mutant 103Q strain were restored by overexpressing NPT1 as expected, but did not increase by overexpression of NMA1, QNS1 or PNC1 (Supplementary Material, Fig. S2B), further suggesting that the presumably common mechanism of proteotoxicity suppression by overexpression of these genes is independent of NAD+ biosynthesis. Similarly, SIR2 is not required for NMA1/2-mediated suppression of proteotoxicity because deletion of SIR2 in combination with overexpression of different components of the NAD+ salvage pathway did not alter their suppression capacity (Fig. 2B). Furthermore, the suppression mechanism does not involve the activity of other histone deacetylases (HDAC) because supplementation of the growth media with the class III HDAC inhibitors nicotinamide (27) or splitomicin (28) had no negative effect on suppression of 103Q or α-synuclein toxicities. If any effect could be claimed, exogenous nicotinamide produced an extremely mild positive effect on suppressing proteotoxicity (Fig. 2C and Supplementary Material, Fig. S3).
Figure 2.

Suppression by NAD+ biosynthetic genes does not require the presence of the intact salvage pathway for NAD+ biosynthesis or HDAC activity. (A–C) Serial dilutions of the indicated strains were spotted on YPD, YPGal and YPGal plates supplemented with or without 20 mm nicotinamide or 20 μm splitomicin to inhibit HDAC activity. Pictures were taken after 2 days of incubation at 30°C.
Mutations in the de novo pathway for NAD+ biosynthesis have been identified as strong suppressors of mutant PolyQ toxicity in yeast (16). Deletion of either BNA4 or BNA1 in the tryptophan degradation pathway (Fig. 1A) has been shown to prevent the accumulation of the toxic metabolites 3-hydroxykynurenine and quinolinic acid, respectively, and suppress PolyQ toxicity (16). Given the direct connection between the NAD+ salvage and de novo pathways, we wanted to investigate whether the NMA1/2 suppression mechanism could involve changes in components of the de novo pathway or vice versa. Overexpression of NMA1/2 had no effect on the expression levels of BNA4 or BNA1 (Supplementary Material, Fig. S1C and D), and deletion of BNA4 had no effect on the expression level of NMA2 (Supplementary Material, Fig. S1B). Similarly, overexpression of PNC1 did not alter BNA4 expression, and overexpression of PNC1, QNS1 or NPT1 did not affect BNA1 expression (Supplementary Material, Fig. S1C and D). Our observations suggest that the suppression by NMA1/2 and by PNC1, QNS1 and NPT1 proceed via a mechanism independent of the de novo NAD+ biosynthesis pathway. However, although unlikely, we cannot rule out the possibility that expression of NMA1/2 or other components of the salvage pathway affects the levels of 3-hydroxykynurenine and quinolinic acid without affecting the expression of BNA4 and BNA1.
Suppression by NAD+ biosynthetic genes does not involve mitochondrial oxidative phosphorylation (OXPHOS) function
The role of mitochondrial function in the suppression of WD and proteotoxicity by NMNAT is highly relevant in the field. This is justified because (i) NAD (including NAD+ and NADH) plays an essential role in mediating energy metabolism and mitochondrial functions and (ii) one of the human isoforms of NMNAT (NMNAT3) has been localized to mitochondria (29). Recently, two reports have proposed that changes in the calcium buffering function, motility and stability of mitochondria mediate the suppression of axonal degeneration by NMNAT (12,13). Therefore, we decided to exploit our yeast models to address whether mitochondrial respiratory function is required for the suppression of mutant PolyQ and α-synuclein toxicities by NMA1/2 and other components of the NAD+ salvage pathway.
It is important to emphasize that the facultative aerobe/anaerobe property of S. cerevisiae offers a unique opportunity for the study of biologic and biomedical problems related to mitochondrial oxidative phosphorylation (OXPHOS) function because respiratory-deficient yeast strains can survive in media containing fermentable carbon sources such as raffinose and galactose. Relevant to the present study, we have previously shown that mutant PolyQ toxicity in yeast involves mitochondrial dysfunction (20) exerted by direct interaction of misfolded PolyQ domains with mitochondrial membranes (30). Additionally, mutant PolyQ-induced cytotoxicity is extensively suppressed by enhancement of mitochondrial biogenesis and OXPHOS function by overexpression of HAP4 (30), the catalytic subunit of the Hap2,3,4,5 complex, essential for the regulation of mitochondrial biogenesis during the transition from fermentation to respiration (31).
Here, we abolished mitochondrial OXPHOS function by genetic and pharmacologic manipulations and determined the efficacy of NMA1/2-mediated suppression of 103Q or α-synuclein-induced cytotoxicities. First, we treated the 103Q and α-synuclein yeast strains overexpressing either NMA1 or HAP4 (as a negative control) with ethidium bromide to generate rho0 strains devoid of mitochondrial DNA (mtDNA) as reported (32). The mtDNA encodes essential components of the OXPHOS system, and, therefore, rho0 yeast are unable to grow in the presence of respiratory substrates such as ethanol and glycerol (YPEG). Because rho0 cells do not grow efficiently in galactose-containing media, we used raffinose as the fermentable carbon source and induced protein expression when required by supplementing the plates with 0.25% galactose. As shown in Figures 3A and B, HAP4 overexpression suppresses both mutant PolyQ and α-synuclein-induced toxicities. As expected, the suppression was only effective in rho+ strains, containing mtDNA, but not in rho0 strains inasmuch as HAP4-mediated suppression requires functional mitochondrial respiration (30). On the contrary, suppression by NMA1 was equally effective in rho+ and rho0 strains, thus demonstrating an OXPHOS-independent mechanism.
Figure 3.

Suppression by NAD+ biosynthetic genes does not require mitochondrial OXPHOS function. (A–D) Serial dilutions of indicated strains were plated on solid media containing YPRaf, YPEG or YPRaf, supplemented with 0.25% galactose (Gal) with or without 50 μm antimycin A (AMA). Pictures were taken after 2 days of incubation at 30°C.
In addition to the genetic approach, we also pharmacologically inhibited mitochondrial respiratory complex III activity with antimycin A (AMA) that prevents growth in respiratory media (YPEG). AMA treatment did not affect the efficacy of suppression by NMA1/2, NPT1 or PNC1 and only slightly lowered the suppression efficacy of QNS1 (Fig. 3C and D). Similar outcomes were obtained when mitochondrial OXPHOS was impaired by supplementation of the growth media with oligomycin, an inhibitor of the F1F0 ATPase (Supplementary Material, Fig. S4). These results further confirm that mitochondrial respiration is not an essential component of the suppression mechanism.
Suppression by NAD+ biosynthetic genes acts through the clearance of proteotoxic protein species
In human neurodegenerative proteinopathies, including HD and PD, the mechanism of neuronal cell toxicity involves the accumulation of misfolded mutant proteins that are targeted for degradation, overload proteasome function and induce downstream toxic effects in several subcellular compartments (33,34). Toxicity can be partially attenuated by enhancing the biogenesis and function of some of the downstream target organelles such as mitochondria (30). More effective suppression of proteotoxicity can be achieved by enhancing protein clearance pathways by increasing autophagy (35), activating the ubiquitin-proteasome system (36) or repairing the endoplasmic reticulum-associated degradation pathway (37). Toxicity can also be attenuated by modulating the chaperone systems involved in protein refolding and disaggregating misfolded protein complexes, thus shifting the balance toward non-toxic protein species (9). NMNAT has been previously shown to be able to act as a chaperone and increase the clearance of toxic proteins in several models of neurodegenerative diseases (3,10). Therefore, we tested whether NMA1/2 and other genes in the NAD+ salvage pathway could suppress protein toxicity by regulating misfolded protein degradation and aggregation.
Using fluorescence microscopy, we followed GFP-tagged mutant PolyQ or α-synuclein expression and oligomerization in strains overexpressing NMA1, NMA2, PNC1 or just carrying an empty vector (YEp352). After 2 h of protein expression, the 103Q-GFP signal was primarily diffuse in the cytoplasm, forming a few small aggregates. The number and size of the aggregates progressed with time, and after only 4 h of induction, the protein was mainly localized in large aggregates (Fig. 4A). In the case of α-synuclein, it initially localized to the plasma membrane as previously observed (17), and at later times, it aggregated in a pattern similar to that of mutant PolyQ (Supplementary Material, Fig. S5A). We found that overexpression of NMA1, NMA2 or PNC1 was able to attenuate the fluorescence signal of both soluble and aggregated mutant PolyQ and α-synuclein as well as the number of aggregates (Fig. 4A). Flow cytometry analyses showed a reduction in GFP signal of ∼40 and 50% in the NMA1-overexpressing strains after 2 and 4 h of protein expression, respectively (Supplementary Material, Fig. S6). Approximately 25 and 75% of the 103Q cells contained at least one 103Q-GFP punctuate focus after 2 and 4 h of 103Q-GFP expression, respectively. These values were reduced to ∼5–10% and 20–30%, respectively, in cells overexpressing NMA1 or PNC1 (Fig. 4B). Overexpression of NMA1, NMA2 or PNC1 also reduced the number of cells containing at least one α-synuclein focus/aggregate after 4 h of protein expression from ∼90 to ∼15–30% (Supplementary Material, Fig. S5B). Overexpression of NMA1, NMA2 or PNC1 did not affect the level of expression of mutant PolyQ and α-synuclein as measured by real-time PCR (Supplementary Material, Fig. S1E), thus suggesting that the lower steady-state levels of these toxic proteins could be the result of increased protein degradation. To test this hypothesis, we performed western blot analysis of protein extracts from cells expressing mutant PolyQ alone or in combination with the suppressor genes to follow the kinetics of 103Q-GFP oligomerization. In agreement with our fluorescence microscope analyses, we observed a time-dependent pattern of protein aggregation, as we and others previously reported (15,20). After 2 h of expression, 103Q-GFP proteins were found both at their expected size and forming SDS-insoluble oligomers (Fig. 4C). At later time points, 103Q-GFP was detected only as part of SDS-insoluble large oligomers trapped in the higher portions of the stacking gel (Fig. 4C). Noticeably, when NMA1 was overexpressed, we detected lower amounts of SDS-insoluble large oligomers at the several time points, accompanied by increased signal corresponding to either degradation products or intermediate states of protein oligomerization (Fig. 4C).
Figure 4.
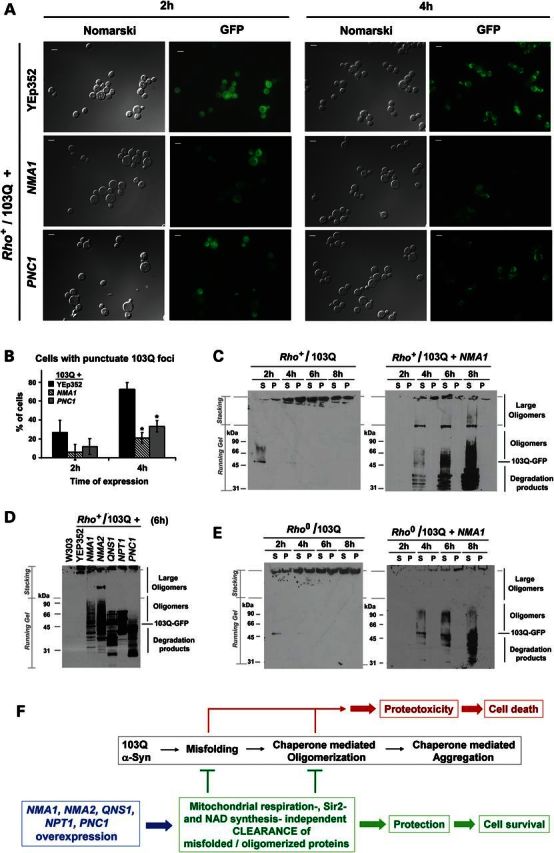
Suppression by NAD+ biosynthetic genes does involve clearance of cytotoxic protein species. (A) Visualization of PolyQ-GFP fusion protein expression and aggregation. The indicated strains were induced for 2 and 4 h in complete medium containing 2% galactose, mounted on slides and visualized by fluorescence microscopy. Bar represents 5 μm. (B) Quantification of cells containing at least one 103Q aggregation focus in the indicated strains. At least 50 cells were counted in each case. Error bars represent the mean ± SD of two independent assessments with P-values from comparisons to 103Q + YEp352 (vector) strain denoted by *P < 0.05. (C–E) Cells were converted into spheroplasts, lyzed with 1% Triton X-100, and the lysates were clarified of cell debris by gravity sedimentation and then fractionated into the pellet (P) and supernatant (S) fractions by centrifugation at 800g for 10 min as reported (20). P and S fractions from extracts obtained from cells expressing 103Q-GFP after 2, 4, 6 and 8 h of induction in YPGal medium, respectively, were analyzed by western blotting. 103Q-GFP fusion proteins were detected using an anti-GFP polyclonal antibody. (F) Diagram depicting a model of the mechanism of suppression of mutant PolyQ- or α-synuclein-induced toxicities by NAD+ biosynthetic proteins.
We subsequently tested whether all the suppressor genes identified here could act through a mechanism similar to NMA1 overexpression. As shown in Figure 4D, following 6 h of induction, the overexpression of NMA2, NPT1, QNS1 and PNC1 affected mutant 103Q-GFP oligomerization and degradation in a manner comparable to NMA1 (Fig. 4D). We detected a similar effect on extracts from cells expressing α-synuclein-GFP. In this case, the protein forms large aggregates that failed to enter the gel. However, the suppressors promote the accumulation of smaller oligomers and the accumulation of some degradation products (Supplementary Material, Fig. S5C). Interestingly, the pattern of oligomerization/degradation was specific to each of the different suppressors tested (Fig. 4D), although no clear correlation with the efficacy of suppression was apparent. In a hypothetical model in which the suppressor proteins could act as chaperones promoting protein refolding, oligomer disruption and targeting of misfolded proteins for degradation, the patterns could result from the way in which each suppressor interacts with the misfolded and oligomerized proteins (Fig. 4D). We have tested whether NMA1/2 overexpression alters the steady-state levels of several Hsp proteins in wild-type and 103Q cells. Although there is no clear pattern of alteration in the steady-state levels of Hsp104, Hsp82, Hsp42 and Hsp26 chaperones (Supplementary Material, Fig. S7), we cannot discard that NMA1/2 and the other suppressors could physically and/or functionally interact with these chaperones promoting or enhancing their activities.
Finally, to further test that the suppression mechanism by NMA1 is independent of mitochondrial function, we analyzed the pattern of 103Q oligomerization in a rho0 strain. In 103Q rho0cells, mutant PolyQ aggregated in a time-dependent manner, and formation of large PolyQ oligomers was delayed by NMA1 overexpression as in rho+ cells (Fig. 4E). Similarly, we detected bands corresponding to degradation products and intermediate states of protein oligomerization (Fig. 4E). These results indicate that the mechanism of proteotoxicity suppression by NMA1 involves the clearance of misfolded/oligomerized toxic proteins and does not necessarily require mitochondrial OXPHOS function (Fig. 4F).
DISCUSSION
In this study, we have used S. cerevisiae models of human PolyQ- and α-synuclein-induced proteinopathies to demonstrate that the yeast homologs of NMNAT (NMA1/2) and other genes in the NAD+ salvage pathway protect cells through a mechanism involving the targeting of toxic misfolded and oligomerized proteins for degradation and clearance.
We have established that the mechanism described here is independent of NAD+ levels, the integrity of the NAD+ salvage pathway, histone deacetylase activity and mitochondrial OXPHOS function. Therefore, the mechanism of proteotoxicity suppression by NMA1/2, QNS1, NPT1 and PNC1 is different from their previously described replicative life extension properties, where NAD+ and SIR2 activity are essential players (22,23). The mechanism described here also differs from the recently reported mechanisms of suppression of axonal degeneration in Drosophila models by NMNAT, where mitochondria-related functions were proposed to be essential (12,13). Specifically, mitochondrial calcium buffering and mitochondrial motility were reported to be altered upon axotomy of larva peripheral nerves and enhanced by NMNAT overexpression (13), and wing nerve axotomy in the adult fly was reported to induce mitochondrial loss that was suppressed by upregulation of NMNAT (13). However, in a model of motor neuron axonal degeneration, following axotomy in Drosophila larva, Kitay and colleagues have demonstrated that WD proceeds independently of axonal mitochondria, and NMNAT is capable of delaying WD in axons devoid of mitochondria (38). This has suggested that mitochondria preservation could likely be a consequence of NMNAT-mediated axon protection, rather than its cause. Despite the difference in the complexity of our model systems, and in agreement with the report by Kitay et al., our data show that NMA1/2 and other proteins in the NAD+ salvage pathway have the ability to suppress proteotoxicities in cells that are unable to respire.
Our data indicate that the mechanism of proteotoxicity suppression by NMA1/2 and other NAD+ salvage pathway genes involves an increase in the refolding and/or clearance of misfolded and oligomerized toxic proteins. Mitochondrial aerobic energy production would be expected to have a positive impact on mutant polyQ and α-synuclein clearance pathways and refolding chaperone systems that require ATP for functioning. However, overexpressed NMA1/2 and the other suppressors do not require mitochondrial OXPHOS to effectively induce clearance of misfolded/oligomerized toxic proteins. This function could be performed through the direct or indirect action of the suppressor proteins. NMNAT has already been shown to have the capacity to act as a chaperone in vivo and in vitro (3) and interacts with phosphorylated tau and promotes the ubiquitination and clearance of toxic tau species in a Drosophila model of tauopathy (10). Therefore, a possible direct interaction of NMA1/2 with mutant PolyQ and α-synuclein might be responsible for the increase in protein degradation. Whether Qns1, Npt1 and Pnc1 have the ability to act as molecular chaperones themselves or enhance the chaperone activity of Nma1/2 is an open question. As an alternative mechanism, we could envision Nma1/2 enhancing the function of other chaperones from the Hsp70 or Hsp90 families that have previously been shown to modify the aggregation and toxicity of mutant PolyQ in yeast (15,39,40). The results we have obtained so far show that at least the steady state levels of Hsp104, Hsp72, Hsp40 and Hsp26 are not consistently altered by overexpression of NMA1/2. The investigation of these exciting possibilities warrants future research efforts.
Finally, our data suggest that in higher eukaryotes, NAD+ salvage pathway proteins could act as neuronal maintenance factors and, together with NMNAT, constitute possible relevant targets for therapeutic interventions aiming to combat neuronal stress and degeneration.
MATERIALS AND METHODS
Yeast strains and culture conditions
The S. cerevisiae strains used are listed in Supplementary Material, Table S1. The S. cerevisiae strains used were all in the W303 background and included the wild-type W303-1A (MATa ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1) and the previously reported W303-1A strains containing chromosomally integrated N-terminus of human huntingtin with 103Q or wild-type α-synuclein fused to GFP expressed under the control of the Gal1 promoter (17,20). The plasmids overexpressing NMA1, NMA2, NPT1, QNS1, TNA1 and ATG32 were obtained from Open Biosystems (Thermo Scientific, Lafayette, CO, USA) and transformed into the indicated strains. SIR2 and NPT1 were deleted by replacing the open reading frame with a KANMAX4 cassette. The following complete YP (1% yeast extract, 2% peptone) and minimum WO (0.67% nitrogen base w/o amino acids) media were used routinely to grow yeast: YPD (+2% glucose) YPGal (+2% galactose), YPRaf (+2% raffinose), YPEG (+2% ethanol, 2% glycerol), WOD (+2% glucose) and WOEG (+2% ethanol, 2% glycerol). The following chemicals were used as supplement to solid YPGal plates: 20 mm nicotinamide, 20 μm splitomicin and 50 μm antimycin A (all from Sigma-Aldrich Corp., St Louis, MO, USA).
Fluorescence microscopy
Wide-field fluorescence microscopy was performed to detect polyQ-GFP and α-synuclein-GFP expression. We used an Olympus fluorescence BX61 microscope equipped with Nomarski differential interference contrast optics, a Uplan Apo 100× objective (NA 1.35), a Roper CoolSnap HQ camera and Sutter Lambda10-2 excitation and emission filter wheels and a 175 watt Xenon remote source with liquid light guide. Images were acquired using SlideBook 4.01 (Intelligent Imaging Innovations, Denver, CO, USA). All experiments were done at least in duplicate with a minimum of 100 cells per sample.
Preparation of cell extracts
Fifty milliliter cultures (OD600 = 0.125) of strains carrying either polyQ-GFP or α-synuclein–GFP were induced for 2, 4, 6 and 8 h in complete media containing 2% galactose. Cells were harvested by centrifugation at 1100× g for 5 min and washed once with 1.2 M sorbitol. The cell wall was digested with 1 mg/ml zymolyase for 30 min, and the spheroplasts were re-suspended in lysis buffer (40 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer pH 7.4, 50 mm KCl, 1% Triton X-100, 0.3 mg/ml dithiothreitol, 5 mm EDTA and 0.5 mm phenylmethylsulfonyl fluoride). Samples were collected following gravity sedimentation of cell debris for 1 h on ice. The samples were centrifuged 10 min at 800g at 4°C. The supernatant (S) was collected, and the pellet (P) was washed once in lysis buffer and then resuspended in the same volume of lysis buffer. Protein concentration was quantified by Folin method. Equal amount of protein was re-suspended in Laemmli buffer [1% SDS, 0.1% (v/v) glycerol, 0.01% β-mercaptoethanol and 50 mm Tris–HCl pH 6.8) for western blot analysis.
Cellular NAD measurements
Protein expression was induced in galactose media for 2 h, when the culture reached an OD600 = 2. One milliliter of OD600 = 1 equivalent of cells were harvested, washed once in 1.2 m sorbitol and re-suspended in 300 μl zymolyase buffer (1.2 m sorbitol, 20 mm Tris, pH = 7.4, 10 mg/ml zymolyase 20T) at room temperature for 20 min to digest the cell wall. The spheroplasts were washed once in 1.2 m sorbitol. Approximately 3 × 106 cells were then used for NAD+/NADH extraction and quantification using the BioVision NAD+/NADH quantification kit # K337 (Biovision, Milpitas ,CA, USA), following the manufacturer's recommendations. We constructed an NADH standard curve, to which we applied the sample readings and expressed the concentrations of NAD+ or NADH in the samples in pmol/106 cells.
RNA isolation and analysis
Total RNA was prepared from whole cells by a modified extraction with hot-acidic phenol (41) and used for quantitative real-time polymerase chain reaction (RT-PCR) analyses. For quantitative RT-PCR analysis, total cellular RNA was treated with 6 units of DNase1 (M0303; New England Biolabs, Ipswich, MA, USA) for 1 h at 37°C to eliminate genomic contamination. cDNA was synthesized using Superscript III reverse transcriptase (18080-085; Invitrogen, Life Technologies, Grand Island, NY, USA), following manufacturer's recommendations. RT-PCR analysis of relative expression level of the gene was carried out using Fermentas–Thermo Scientific (Glen Burnie, MD, USA) [Maxima® SYBR Green/ROX qPCR Master Mix (2×)], and data were collected using BioRad CFX96 Touch™ RT-PCR Detection System and analyzed using the manufacturer's program, following the Pfaffl method. Primers used for the RT-PCR analysis are listed in Supplementary Material, Table S2.
Miscellaneous procedures
Standard procedures were used for the preparation and ligation of DNA fragments and for transformation and recovery of plasmid DNA from Escherichia coli (42). Yeast cells were transformed by the method of Schiestl and Gietz (43). Proteins were separated by SDS PAGE in the buffer system of Laemmli (44), and western blots were treated with antibodies against the appropriate proteins, followed by a second reaction with anti-mouse or anti-rabbit IgG conjugated to horseradish peroxidase (Sigma, St Louis, MO, USA). The SuperSignal West Pico was used for the final detection. The one-step gene insertion method (45) was used to integrate linear plasmids at the LEU2 locus of yeast chromosomal DNA.
SUPPLEMENTARY MATERIAL
FUNDING
This research was supported by a Research Challenge grant from the Florida Department of Health/James & Esther King Biomedical Research Program (to A.B.) and NIH-RO1 GM071775-06 (to A.B.).
ACKNOWLEDGEMENTS
We thank Brandon Kitay and Dr Grace Zhai for discussion of the project and sharing the data. We thank Dr Brant Watson and Dr Myriam Bourens for critical reading of the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox. Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- 2.Ali Y.O., McCormack R., Darr A., Zhai R.G. Nicotinamide mononucleotide adenylyltransferase is a stress response protein regulated by the heat shock factor/hypoxia-inducible factor 1 alpha pathway. J. Biol. Chem. 2011;286:19089–19099. doi: 10.1074/jbc.M111.219295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhai R.G., Zhang F., Hiesinger P.R., Cao Y., Haueter C.M., Bellen H.J. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilley J., Coleman M.P. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhai R.G., Cao Y., Hiesinger P.R., Zhou Y., Mehta S.Q., Schulze K.L., Verstreken P., Bellen H.J. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wen Y., Parrish J.Z., Zhai R.G., Kim M.D. NMNAT is required for dendrite maintenance in Drosophila. Mol. Cell. Neurosc. 2011;48:1–8. doi: 10.1016/j.mcn.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sasaki Y., Vohra B.P., Baloh R.H., Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. 2009;29:6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verghese P.B., Sasaki Y., Yang D., Stewart F., Sabar F., Finn M.B., Wroge C.M., Mennerick S., Neil J.J., Milbrandt J., Holtzman D.M. Nicotinamide mononucleotide adenylyl transferase 1 protects against acute neurodegeneration in developing CNS by inhibiting excitotoxic-necrotic cell death. Proc. Acad. Sci. USA. 2011;108:19054–19059. doi: 10.1073/pnas.1107325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coleman M.P., Freeman M.R. Wallerian degeneration, WLD(S), and NMNAT. Annu. Rev. Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ali Y.O., Ruan K., Zhai R.G. NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum. Mol. Genet. 2012;21:237–250. doi: 10.1093/hmg/ddr449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ljungberg M.C., Ali Y.O., Zhu J., Wu C.S., Oka K., Zhai R.G., Lu H.C. CREB-activity and NMNAT2 transcription are down-regulated prior to neurodegeneration, while NMNAT2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum. Mol. Genet. 2012;21:251–267. doi: 10.1093/hmg/ddr492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avery M.A., Rooney T.M., Pandya J.D., Wishart T.M., Gillingwater T.H., Geddes J.W., Sullivan P.G., Freeman M.R. Wld(S) Prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca(2+) buffering. Curr. Biol. 2012;22:596–600. doi: 10.1016/j.cub.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang Y., Soares L., Teng X., Geary M., Bonini N.M. A novel Drosophila model of nerve injury reveals an essential role of NMNAT in maintaining axonal integrity. Curr. Biol. 2012;22:590–595. doi: 10.1016/j.cub.2012.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krobitsch S., Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. USA. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meriin A.B., Zhang X., He X., Newnam G.P., Chernoff Y.O., Sherman M.Y. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 2002;157:997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giorgini F., Guidetti P., Nguyen Q., Bennett S.C., Muchowski P.J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet. 2005;37:526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Outeiro T.F., Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Q., Thorpe J., Keller J.N. Alpha-synuclein alters proteasome function, protein synthesis, and stationary phase viability. J. Biol. Chem. 2005;280:30009–30017. doi: 10.1074/jbc.M501308200. [DOI] [PubMed] [Google Scholar]
- 19.Dixon C., Mathias N., Zweig R.M., Davis D.A., Gross D.S. Alpha-synuclein targets the plasma membrane via the secretory pathway and induces toxicity in yeast. Genetics. 2005;170:47–59. doi: 10.1534/genetics.104.035493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solans A., Zambrano A., Rodriguez M., Barrientos A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum. Mol. Genet. 2006;15:3063–3081. doi: 10.1093/hmg/ddl248. [DOI] [PubMed] [Google Scholar]
- 21.Rongvaux A., Andris F., Van Gool F., Leo O. Reconstructing eukaryotic NAD metabolism. Bioessays. 2003;25:683–690. doi: 10.1002/bies.10297. [DOI] [PubMed] [Google Scholar]
- 22.Anderson R.M., Bitterman K.J., Wood J.G., Medvedik O., Cohen H., Lin S.S., Manchester J.K., Gordon J.I., Sinclair D.A. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J. Biol. Chem. 2002;277:18881–18890. doi: 10.1074/jbc.M111773200. [DOI] [PubMed] [Google Scholar]
- 23.Anderson R.M., Bitterman K.J., Wood J.G., Medvedik O., Sinclair D.A. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin S.J., Defossez P.A., Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 25.McClure J.M., Gallo C.M., Smith D.L., Jr., Matecic M., Hontz R.D., Buck S.W., Racette F.G., Smith J.S. Pnc1p-mediated nicotinamide clearance modifies the epigenetic properties of rDNA silencing in Saccharomyces cerevisiae. Genetics. 2008;180:797–810. doi: 10.1534/genetics.108.091090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith J.S., Brachmann C.B., Celic I., Kenna M.A., Muhammad S., Starai V.J., Avalos J.L., Escalante-Semerena J.C., Grubmeyer C., Wolberger C., Boeke J.D. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. USA. 2000;97:6658–6663. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bitterman K.J., Anderson R.M., Cohen H.Y., Latorre-Esteves M., Sinclair D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 28.Bedalov A., Gatbonton T., Irvine W.P., Gottschling D.E., Simon J.A. Identification of a small molecule inhibitor of Sir2p. Proc. Natl. Acad. Sci. USA. 2001;98:15113–15118. doi: 10.1073/pnas.261574398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yahata N., Yuasa S., Araki T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J. Neurosci. 2009;29:6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ocampo A., Zambrano A., Barrientos A. Suppression of polyglutamine-induced cytotoxicity in Saccharomyces cerevisiae by enhancement of mitochondrial biogenesis. FASEB J. 2010;24:1431–1441. doi: 10.1096/fj.09-148601. [DOI] [PubMed] [Google Scholar]
- 31.Forsburg S.L., Guarente L. Identification and characterization of HAP4: a third component of the CCAAT-bound HAP2/HAP3 heteromer. Genes Dev. 1989;3:1166–1178. doi: 10.1101/gad.3.8.1166. [DOI] [PubMed] [Google Scholar]
- 32.Ocampo A., Liu J., Schroeder E.A., Shadel G.S., Barrientos A. Mitochondrial respiratory thresholds regulate yeast chronological life span and its extension by caloric restriction. Cell Metab. 2012;16:55–67. doi: 10.1016/j.cmet.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bence N.F, Sampat R.M., Kopito R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 34.Bennett E.J., Shaler T.A., Woodman B., Ryu K.Y., Zaitseva T.S., Becker C.H., Bates G.P., Schulman H., Kopito R.R. Global changes to the ubiquitin system in Huntington's disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- 35.Sarkar S., Ravikumar B., Floto R.A., Rubinsztein D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- 36.Bauer P.O., Nukina N. Enhanced degradation of mutant huntingtin by rho kinase inhibition is mediated through activation of proteasome and macroautophagy. Autophagy. 2009;5:747–748. doi: 10.4161/auto.5.5.8704. [DOI] [PubMed] [Google Scholar]
- 37.Duennwald M.L., Lindquist S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008;22:3308–3319. doi: 10.1101/gad.1673408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitay B.M., McCormack R., Wang Y., Tsoulfas P., Zhai R.G. Mislocalization of neuronal mitochondria reveals regulation of Wallerian degeneration and NMNAT/WLDS-mediated axon protection independent of axonal mitochondria. Hum. Mol. Genet. 2013;22:1601–1614. doi: 10.1093/hmg/ddt009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cashikar A.G., Duennwald M., Lindquist S.L. A chaperone pathway in protein disaggregation. Hsp26 alters the nature of protein aggregates to facilitate reactivation by Hsp104. J. Biol. Chem. 2005;280:23869–23875. doi: 10.1074/jbc.M502854200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Behrends C., Langer C.A., Boteva R., Bottcher U.M., Stemp M.J., Schaffar G., Rao B.V., Giese A., Kretzschmar H., Siegers K., Hartl F.U. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol. Cell. 2006;23:887–897. doi: 10.1016/j.molcel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 41.Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Seidman J.G., Smith J.A., Struhl K. Current Protocols in Molecular Biology. Vol. 2. New York: Wiley; 1994. Saccharomyces cerevisiae; p. 13. [Google Scholar]
- 42.Maniatis T., Fritsch E.F., Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press; 1982. pp. 507–520. [Google Scholar]
- 43.Schiestl R.H., Gietz R.D. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr. Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- 44.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 45.Rothstein R.J. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]