

Abstract
Higher resting heart rate is associated with increased cardiovascular disease and mortality risk. Though heritable factors play a substantial role in population variation, little is known about specific genetic determinants. This knowledge can impact clinical care by identifying novel factors that influence pathologic heart rate states, modulate heart rate through cardiac structure and function or by improving our understanding of the physiology of heart rate regulation. To identify common genetic variants associated with heart rate, we performed a meta-analysis of 15 genome-wide association studies (GWAS), including 38 991 subjects of European ancestry, estimating the association between age-, sex- and body mass-adjusted RR interval (inverse heart rate) and ∼2.5 million markers. Results with P < 5 × 10−8 were considered genome-wide significant. We constructed regression models with multiple markers to assess whether results at less stringent thresholds were likely to be truly associated with RR interval. We identified six novel associations with resting heart rate at six loci: 6q22 near GJA1; 14q12 near MYH7; 12p12 near SOX5, c12orf67, BCAT1, LRMP and CASC1; 6q22 near SLC35F1, PLN and c6orf204; 7q22 near SLC12A9 and UfSp1; and 11q12 near FADS1. Associations at 6q22 400 kb away from GJA1, at 14q12 MYH6 and at 1q32 near CD34 identified in previously published GWAS were confirmed. In aggregate, these variants explain ∼0.7% of RR interval variance. A multivariant regression model including 20 variants with P < 10−5 increased the explained variance to 1.6%, suggesting that some loci falling short of genome-wide significance are likely truly associated. Future research is warranted to elucidate underlying mechanisms that may impact clinical care.
INTRODUCTION
Higher resting heart rate is associated with increased risk of cardiovascular disease (1), cardiovascular mortality (2,3), including sudden death (4), and all-cause mortality independent of traditional risk factors (5–9). However, it is not known whether heart rate directly impacts mortality or merely reflects unrecognized subclinical disease (1,7,8). Recently it was shown that physical exercise reduces the increased cardiovascular mortality risk associated with higher heart rate (3). This suggests that heart rate is a clinically relevant and potentially modifiable risk factor.
Heart rate is a complex trait, determined by multiple environmental, genetic and other endogenous factors. Heredity plays a substantial role in the inter-individual variation of resting heart rate, accounting for 26–32% of heart rate variation in prior studies (10–13). Twin studies report even higher heritability estimates up to 55–63% (14,15). Candidate gene approaches have identified multiple loci associated with heart rate (12,13,16–20), but the results have been inconsistent and difficult to replicate. Genome-wide genotyping arrays of single-nucleotide polymorphisms (SNPs) assay common variation in the human genome and can identify genetic variants with modest influences on a complex trait such as heart rate, as shown by two recent genome-wide association studies (GWAS) that identified common variation at or near MYH6, GJA1 and CD34 associated with heart rate (21,22). These chromosomal loci identified in an unbiased genome-wide study may represent novel risk factors for cardiovascular disease outcomes. This knowledge may also have an impact on clinical care (i) by identifying novel factors that cause pathologic heart rate states (such as sick sinus syndrome or other arrhythmias), (ii) by identifying factors that influence cardiac structure or function (e.g. stroke volume) and thereby modulate heart rate (since cardiac output = heart rate × stroke volume) or (iii) by improving our understanding of the physiologic basis of heart rate regulation. Altogether this will generate insights into the underlying mechanisms of heart rate as a well-established, but poorly understood, risk indicator for cardiovascular disease and mortality.
To identify additional genetic determinants of heart rate, we performed a meta-analysis of GWAS of resting heart rate, measured as the RR interval on the electrocardiogram (ECG) in 38 991 individuals of European ancestry derived from 15 studies in the RRGEN consortium.
RESULTS
There were 38 991 individuals available for genotype–phenotype association analysis after exclusions. Subject characteristics are shown in Table 1. While cohort-specific quantile–quantile plots of P-value distributions approximated expectations under the null, the meta-analysis of all results showed a clear excess of low P-values (Supplementary Material, Fig. S1). Study-specific genomic inflation lambda values ranged from 0.98 to 1.05, suggesting that population stratification or other technical artifacts were minimal (Supplementary Material, Table S1).
Table 1.
Baseline characteristics of samples included by cohort
| Sample size | Male [n (%)] | Age [mean (SD), years] | Body mass index [mean (SD), kg/m2] | Heart rate (SD, bpm) | RR interval (SD, ms) | SD of RR-residual | |
|---|---|---|---|---|---|---|---|
| AGES | 1651 | 622 (37.7) | 75.9 (5.5) | 26.8 (4.4) | 68.3 (10.2) | 897.1 (131.0) | 128.5 |
| ARIC | 6308 | 2855 (45.3) | 53.9 (5.6) | 26.7 (4.7) | 67.3 (9.0) | 907.6 (118.2) | 116.1 |
| CHS | 2544 | 951 (37.4) | 72.2 (5.3) | 26.2 (4.4) | 65.7 (9.2) | 930.1 (123.6) | 121.3 |
| ERF | 1275 | 508 (39.8) | 47.1 (14.0) | 26.5 (4.5) | 64.5 (9.2) | 948.5 (128.8) | 126.9 |
| FHS | 7243 | 3305 (45.6) | 40.2 (10.5) | 26.1 (5.0) | 69.3 (11.1) | 888.2 (139.2) | 121.8 |
| KORA F3 | 995 | 480 (48.2) | 60.0 (10.1) | 27.3 (4.4) | 65.6 (9.7) | 933.8 (130.8) | 128.1 |
| KORA S4 | 1398 | 654 (46.8) | 52.8 (8.7) | 27.3 (4.4) | 66.4 (9.4) | 921.1 (125.6) | 122.5 |
| MICROS | 919 | 399 (44.4) | 44.8 (16.0) | 25.6 (4.8) | 68.8 (11.7) | 897.0 (151.4) | 92.0 |
| NESDA | 1456 | 437 (30.0) | 39.8 (12.2) | 25.1 (4.7) | 68.1 (9.6) | 898.7 (125.4) | 125.1 |
| ORCADES | 546 | 240 (44.0) | 52.6 (14.9) | 27.6 (4.9) | 62.5 (8.1) | 975.2 (119.6) | 118.5 |
| RS-I | 3781 | 1441 (38.1) | 68.5 (8.6) | 26.1 (3.6) | 71.2 (10.2) | 860.6 (126.4) | 124.1 |
| RS-II | 1589 | 695 (43.7) | 64.8 (7.4) | 27.0 (4.0) | 70.3 (10.1) | 871.1 (123.9) | 122.5 |
| SardiNIA | 3977 | 1678 (42.1) | 42.9 (17.3) | 25.3 (4.7) | 64.5 (10.1) | 907.4 (130.0) | 127.4 |
| SHIP | 2582 | 1260 (48.8) | 46.8 (15.7) | 26.8 (4.7) | 72.1 (11.4) | 852.7 (134.4) | 133.5 |
| TwinsUK | 2727 | 117 (4.3) | 51.7 (12.5) | 25.7 (4.4) | 67.1 (9.6) | 911.5 (126.3) | 125.5 |
SD, standard deviation; RR-residual, residuals are from linear regression models adjusting for age, sex and body mass index.
Meta-analysis of results from all studies resulted in 156 SNPs reaching the pre-specified genome-wide statistical significance level (P < 5 × 10−8; Supplementary Material, Table S2) before applying post-meta-analysis genomic control. In total, nine independent genome-wide significant signals were observed across seven chromosomal loci (Table 2, Fig. 1), with all but one locus harboring multiple SNPs reaching the genome-wide significance threshold (Supplementary Material, Table S2; Fig. 2A–E).
Table 2.
Association analysis results for independent index SNPs from loci with P < 5 × 10−8 in the meta-analysis
| Chr | Basepair position (kb) | SNP | Correlation with index SNPa | Function/gene | Coded/non-coded allele | Allele frequency | Effective sample | Effect estimate | SE | Two-sided P | Two-sided Pmeta-gc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 6q22 | 122 187 | rs9398652 | – | Intergenic, 400 kb from GJA1 | A/C | 0.10 | 37 050 | −12.6 | 1.56 | 7.7 × 10−16 | 3.8 × 10−15 |
| 6q22 | 121 790 | rs11154022 | 0.006 | Intergenic, 8 kb from GJA1 | A/G | 0.33 | 31 676 | 5.8 | 1.05 | 3.5 × 10−8 | 7.2 × 10−8 |
| 14q12 | 22 935 | rs452036 | – | Intronic MYH6 | A/G | 0.36 | 34 640 | −7.8 | 1.00 | 8.1 × 10−15 | 3.8 × 10−14 |
| 14q12 | 22 931 | rs365990 | 0.96 | Non-synonymous coding MYH6 (Ala-1101-Val) | G/A | 0.37 | 32 627 | −7.7 | 1.02 | 5.4 × 10−14 | 2.1 × 10−13 |
| 14q12 | 23 046 | rs223116 | 0.08 | Intergenic, nearest to MYH7, NDNG | A/G | 0.24 | 26 899 | −7.4 | 1.30 | 1.1 × 10−8 | 2.5 × 10−8 |
| 12p12 | 24 662 | rs17287293 | – | Intergenic | G/A | 0.15 | 37 988 | 8.6 | 1.31 | 5.7 × 10−11 | 1.6 × 10−10 |
| 6q22 | 118 680 | rs281868 | – | Intronic SLC35F1 | G/A | 0.50 | 32 109 | −6.3 | 0.99 | 1.5 × 10−10 | 4.3 × 10−10 |
| 7q22 | 100 291 | rs314370 | – | Intronic SLC12A9 | C/T | 0.19 | 35 170 | −7.6 | 1.21 | 2.3 × 10−10 | 6.1 × 10−10 |
| 7q22 | 100 324 | rs12666989 | 0.88 | Non-synonymous coding UfSp1 (Leu-41-Val) | C/T | 0.18 | 35 750 | −7.0 | 1.21 | 9.4 × 10−9 | 2.1 × 10−8 |
| 11q12 | 61 327 | rs174547 | – | Intronic FADS1 | C/T | 0.33 | 34 907 | −6.2 | 1.01 | 8.2 × 10−10 | 2.1 × 10−9 |
| 1q32 | 206 195 | rs2745967 | – | Intergenic near CD34 | G/A | 0.37 | 34 913 | 5.4 | 0.98 | 3.2 × 10−8 | 6.6 × 10−8 |
Chromosomal positions and coded alleles are given relative to the forward strand of NCBI build 36. Effect sizes (on the millisecond scale) are shown as beta estimates from linear regression models for each additional copy of the coded allele. The effective sample size reflects the imputation quality-adjusted sample size. Final column shows the P-value from inverse variance weighted meta-analyses. Chr, chromosome; SE, standard error. Bold values indicate P < 5 × 10−8
aCEU HapMap population linkage disequilibrium r2 values to the index SNP.
Figure 1.
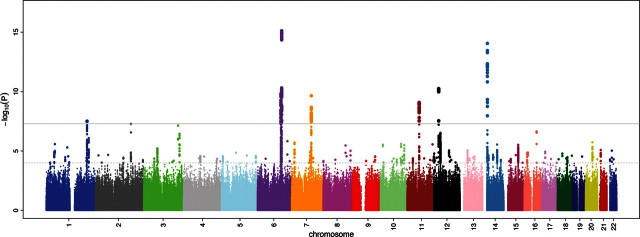
RR interval association results for ∼2.5 million imputed autosomal SNPs in 38 991 individuals from 15 cohorts. Results are shown on the −log10(P) scale (y-axis). The x-axis depicts chromosomal position. The gray horizontal line corresponds to the genome-wide significance threshold of P = 5 × 10−8.
Figure 2.
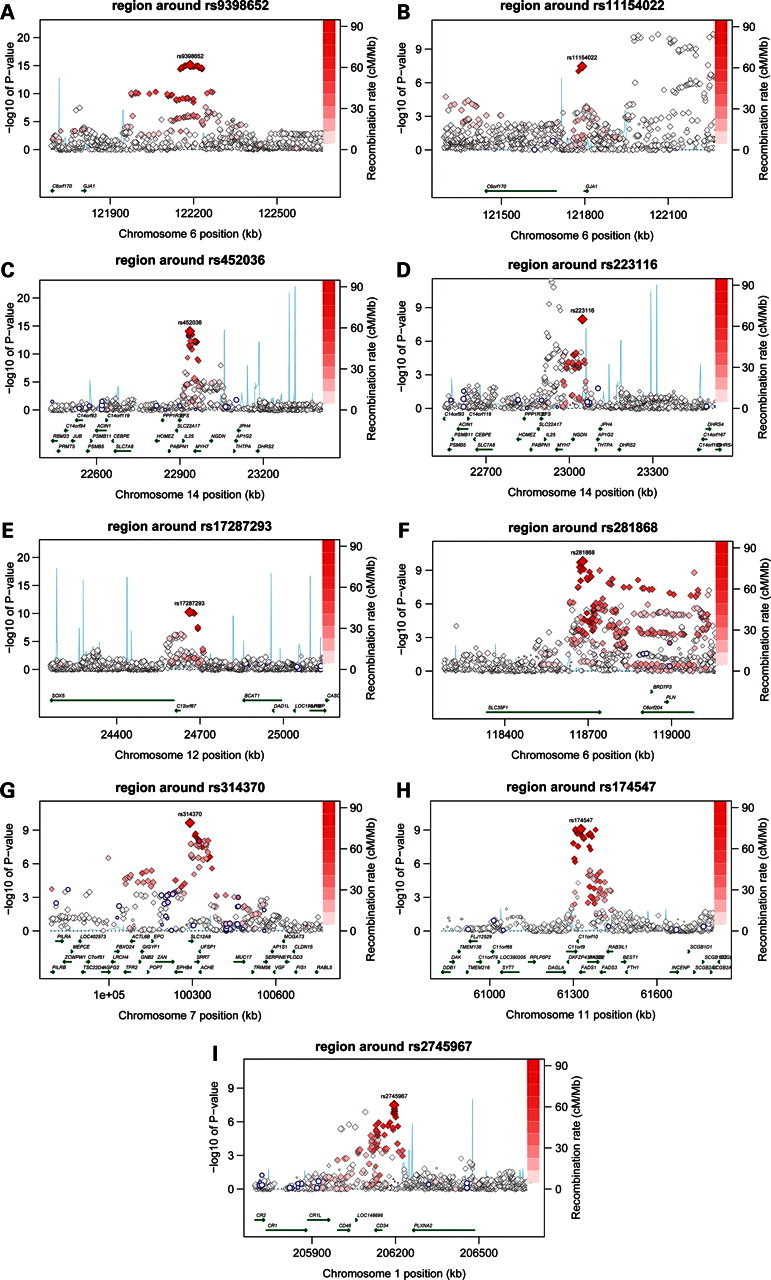
(A–I) Regional association plots covering 1 Mb surrounding the index SNP. Statistical significance of SNPs is shown on the −log10(P) scale as a function of chromosomal position. The primary SNP is annotated by rs number. Correlation of each plotted SNP to the primary SNP is indicated on a color scale from white (minimal correlation) to red (maximal correlation). Non-synonymous coding SNPs resulting in amino acid changes in the encoded protein are plotted as blue circles. Estimated recombination rates from HapMap (blue lines) and RefSeq gene annotations (green arrows) are shown.
Genome-wide significant associations were observed at 6q22 nearest to GJA1; 14q12 near MYH6 and MYH7; 12p12 near SOX5, c12orf67, BCAT1, LRMP and CASC1; 6q22 near SLC35F1, 6orf204 and PLN (>3 Mb away from 6q22 GJA1); 7q22 near SLC12A9; 11q12 near FADS1; and 1q32 near CD34. The genomic inflation lambda value of the meta-analysis was 1.05. After applying post-meta-analysis genomic control, the signal at 1q34 near CD34 and a second independent signal at 6q22 GJA1 lost genome-wide significance.
The strongest association was observed for an intergenic SNP 370 kb upstream of GJA1 at 6q22 rs9398652 [minor allele frequency (MAF) = 0.10] with a 12.6 ms shorter RR interval per minor A allele, which is equivalent to a 0.95 bpm higher heart rate based on the baseline mean heart rate of 66.8 bpm, across all studies (P = 8.0 × 10−16, Pmeta-gc = 3.8 × 10−15; Table 2, Fig. 2A). Cho et al. (21) observed genome-wide significant association between rs12110693 and pulse rate in an Asian population-based GWAS. This SNP is in perfect linkage disequilibrium (r2 = 1) with rs939862 in both Caucasian and Asian HapMap reference populations and thus reflects the same signal 370 kb from GJA1. A second SNP only 8 kb away from GJA1 also reached genome-wide significance pre-genomic control, rs11154022 (MAF = 0.33) with a 5.8 ms longer RR interval (0.43 bpm lower heart rate) per A allele (P = 3.5 × 10−8, Pmeta-gc = 7.2 × 10−8; Fig. 2B, Table 2). This SNP had low correlation with rs9398652 (r2 = 0.006 in HapMap CEU), suggesting a novel-independent association signal. Since HapMap is limited to 90 subjects, we assessed the linkage disequilibrium in our data. All observed r2 values ranged between 0.0001 and 0.004, which is lower than seen in the HapMap CEU reference population. Conditional analysis confirmed that these two signals are independent with Pconditional = 2.4 × 10−11 and Pconditional = 3.3 × 10−8, respectively, for rs9398652 and rs1154022 in the subset (n ≤ 33 846) used for this analysis.
The second locus with two signals in low correlation reaching genome-wide significance is located on chromosome 14q12. The strongest association at this locus was observed for rs452036 located in intron 19 of MYH6 [MAF = 0.36, with a 7.8 ms shorter RR interval (0.58 bpm higher heart rate) per C allele, P = 8.1 × 10−15, Pmeta-gc = 3.8 × 10−14, Fig. 2C]. This replicates the finding by Holm et al. (22) who previously described an association between rs452036 and heart rate. The non-synonymous coding variant rs365990, which results in an amino acid change at position 1101 (Alanine > Valine) of the MYH6 gene product, is a possible functional variant since it showed strong correlation with (r2 = 0.96 in HapMap CEU) and association results are indistinguishable from rs452036 (Table 2). A second SNP located near MYH7 rs223116 (MAF = 0.24), associated with a 7.4 ms shorter RR interval per A allele (0.55 bpm higher heart rate, P = 1.1 × 10−8, Pmeta-gc = 2.5 × 10−8, Fig. 2D) was in low correlation with rs452036 (r2 = 0.08 in HapMap CEU) and reflects a novel association. We observed r2 values similar to HapMap CEU with values ranging from 0.03 to 0.08. Conditional analysis confirmed the presence of two independent signals with Pconditional = 7.9 × 10−14 and Pconditional = 3.7 × 10−4 for rs452036 and rs223116, respectively.
For the other five loci, only a single signal of association met our genome-wide significance threshold, meaning that all other genome-wide significant SNPs have an r2 > 0.1 in HapMap CEU to the most significant SNP at that locus. For these loci, the MAF of the index SNPs ranged from 15 to 50%, effect sizes ranged between 5.4 and 8.6 ms and Pmeta-gc ranged between 2.1 × 10−9 and 1.6 × 10−10 (Table 2, Fig. 2C–H). The CD34 locus lost genome-wide significance upon meta-analytic genomic control but replicates the association reported by Cho et al. (21). Of these five loci, only the index SNP at 7q22 SLC12A9 shows strong correlation with a non-synonymous coding SNP. This coding SNP (rs12666989, r2 = 0.88 to rs314370 in HapMap CEU) results in a Leucine > Valine substitution at amino acid position 41 in the UfSp1 gene product.
Additionally, an association result that just missed our genome-wide significance threshold (P = 5.2 × 10−8, Pmeta-gc = 1.1 × 10−7) was observed for a non-synonymous coding SNP in CCDC141 at 2q31. This SNP, rs17362588 (MAF = 0.12, with a 8.3 ms shorter RR interval per A allele), results in an amino acid substitution at position 360 (Tryptophan > Arginine) of the encoded protein.
Evidence for additional causal loci not reaching genome-wide significance
We used polygenic modeling methods to quantify the genetic variance explained and to indicate if loci falling short of genome-wide significance are likely to harbor additional variants that influence heart rate. The explained variance of resting heart rate in RS-II (first extended Rotterdam Study cohort; n = 1589) was ∼0.7% when the score was calculated based on genome-wide significant signals. Inclusion of 20 independent variants with P < 1 × 10−5 resulted in the maximal proportion of explained variance of 1.6% (Supplementary Material, Fig. S2). These 20 variants included signals from the 7 genome-wide significant loci and signals from an additional 12 loci, including several loci with genes of potential cardiac relevance (see Supplementary Material, Table S3, for full list).
DISCUSSION
The application of genome-wide association methods in a large sample of subjects of European ancestry identified common variants at multiple loci associated with inter-individual variation in resting heart rate. Genetic determinants of heart rate could alter the function of the sinus node (the dominant pacemaker in the normal heart) either directly through altered pacemaking activity (23) or indirectly through sympathetic or parasympathetic inputs to the heart. We were therefore encouraged to find that genes at some of the loci identified here are cardiac ion channels or their regulatory proteins. Besides a direct effect on sinus node function, effects on cardiac structure—either developmental or through remodeling—and function could underlie the observed associations.
The most strongly related locus on chromosome region 6q22 included two independent association signals 8 and 370 kb away from GJA1. The latter finding is in line with a recent GWAS that described an association between rs12110693 and pulse rate in an Asian sample (21). rs9398652 described in the present study is in perfect linkage disequilibrium (r2 = 1) with rs12110693 in both the Caucasian and Asian HapMap reference populations and thus reflects the same signal 370 kb from GJA1. GJA1 encodes Cx43, a connexin family protein and a major component of the cardiac gap junction, crucial in electrical coupling of myocytes (24). Mutations in GJA1 cause a Mendelian inherited hypoplastic left heart syndrome (25). To our knowledge, a role for Cx43 in pacemaker function in the adult sinus node has not been reported.
The 14q12 MYH6 locus was also associated with resting heart rate in the RRGEN study. Its gene product, the myosin heavy chain-6 protein, is a component of the hexameric myosin protein. Mutations in MYH6 have been related to Mendelian forms of hypertrophic cardiomyopathy (26), atrial septal defect (27) and dilated cardiomyopathy (28). The index SNP was strongly correlated with an amino acid-altering common variant in MYH6. The association of the coding SNP with heart rate raises the possibility that it is the causal variant and MYH6 the causal gene. This finding replicates the result of Holm et al. (22) who performed a GWAS on heart rate in an Icelandic population-based sample. We do report a novel-independent signal located near MYH7. Future research is warranted to define the allelic architecture of this locus in relation to heart rate. Of note, cardiac-specific microRNAs encoded within intronic regions of MYH6 (miR-208a) and MYH7 (miR-208b) have regulatory effects on cardiac conduction (29,30).
The locus on chromosome 12p12 includes several genes, but without a clear candidate for association with heart rate in the associated interval. The closest genes are BCAT1, which encodes a cytosolic form of the branched-chain amino acid transaminase enzyme that catalyzes transamination of branched-chain amino acids to their respective alpha-keto acids essential for cell growth and protein synthesis and SOX5, a member of the Sox family (31) and of incompletely understood function. Sox genes play a major role in cell fate modulation through transcriptional activity but without a clear cardiac role for SOX5 (32). The variant at 12p12 we describe here, rs17287293, is a perfect proxy for rs11047543 which was associated with the PR interval (reflecting atrial depolarization duration and atrioventricular nodal conduction time) in a GWAS (33), independent of heart rate, suggesting pleiotropic electrophysiological effects.
The second locus on 6q22 is located near SLC35F1 and PLN, which encodes phospholamban. This locus is located >3 Mb away from the loci near GJA1 on 6q22. We have previously described the common variation at this locus to be associated with heart rate corrected QT interval (34–36). The index SNP reported here was in moderate linkage disequilibrium with two SNPs associated with heart rate-adjusted QT interval duration [rs11756438 (35), r2 = 0.43 in HapMap CEU, RR interval P = 6.5 × 10−6 and rs11970286 (34), r2 = 0.58 in HapMap CEU, RR interval P = 2.7 × 10−8], resulting in overlapping signals in independent phenotypes. In addition to higher heart rate and longer QT interval, this locus has also been associated with decreased end-diastolic left ventricular diameter in the EchoGen Study (37). In humans, the sinoatrial node shows comparable expression of phospholamban compared with atrial myocytes (23). However, basal cAMP-mediated, protein kinase A (PKA)-dependent phosphorylation of phospholamban is elevated in sinoatrial nodal pacemaker cells compared with other cardiac cell types (38). Basal PKA-dependent phosphorylation is obligatory for spontaneous basal pacemaking activity, and graded changes in the phosphorylation of phospholamban cause graded changes of the pacemaker cell basal heart rate (38,39). The location of the top SNP at this locus in an intron of SLC35F1, which is mainly expressed in the brain (40), raises the possibility that causal variation influencing this gene in fact underlies the association at the locus. However, the observation that heart rate, QT interval and left ventricular structure are all associated with genetic variation at this locus makes phospholamban the best candidate in light of its role in excitation–contraction coupling and intracellular calcium signaling, critical to action potential development in both the sinoatrial node and ventricles.
A locus without a clear candidate to explain its strong association with resting heart rate is located on chromosome 7q22 with the index SNP in SLC12A9 encoding a cation-Cl− co-transporter-interacting protein. However, this SNP was strongly correlated with genome-significant SNPs in TRIP6 encoding a thyroid receptor-interacting protein, AChE, which encodes acetylcholinesterase and a non-synonymous-coding SNP in UfSp1. This gene encodes an ubiquitin-fold modifier protease (41,42).
The exact same signal that we report from the FADS1 locus on 11q12 was previously associated with cholesterol levels (43–45) and fatty acid metabolism (46,47). The direct product of the reaction catalyzed by FADS1 is arachidonyl-CoA, which has been shown to release Ca2+ from the sarcoplasmic reticulum (48).
The 1q32 CD34/C1orf132 locus has no clear potential mechanism through which it is related to heart rate and it loses genome-wide significance after applying post-meta-analysis genomic control. However, it is likely to be truly associated with pulse rate since it was previously identified in a non-Caucasian sample (21), and our study thus represents a replication of this finding in a different ethnic group.
The 2q31 CCDC141 locus just missed our genome-wide significance threshold, but the most significant SNP is a non-synonymous coding variant within CCDC141, a coiled-coil domain containing protein of unknown function. Interestingly, the nearby TTN gene encodes Titin, which is expressed in cardiac and skeletal myocytes (49) and plays a key role in muscle assembly, force transmission and maintenance of resting tension (50).
In additional analysis, we have shown that based on the results of this GWAS, the explained variance could be increased to 1.6% with the inclusion of additional signals with P < 10−5. The goal of this analysis was to describe whether additional variants associated with heart rate are likely to be present. Although true positives are among these loci, we cannot discriminate them from the false-positives. We did observe that the loci contributing to the maximal explained variance include loci of specific interest. Three loci that stand out are 3q26 near GNB4, 12p13 near CACNA1C and 14q11 near PRKD1. GNB4 encodes Gβ4 known to influence G-protein-activated inwardly rectifying K+ channels (GIRK) that play an important role in heartbeat regulation (51–54) and are activated on binding of acetylcholine to the muscarinic M2-receptor present in the sinoatrial node (23,54). CACNA1C encodes the alpha-1 subunit of the voltage-dependent calcium channel and is related to Timothy's syndrome including, among other traits, prolonged QT interval, high arrhythmia risk and with a single observation of in utero bradycardia (55). Lastly, the PRKD1 gene product is relevant for calcium-/calmodulin-dependent kinase affecting cardiac remodeling and contraction (56). Validation studies in independent samples have to indicate whether these loci are truly associated with heart rate.
Strengths and limitations
The large sample derived from several population-based cohort studies allowed us to identify common variants with modest effects. In addition, these cohort studies have extensive data on covariates and disease status that allowed us to harmonize exclusion criteria and phenotype modeling prior to analyzing the data.
Additional signals may have been missed due to random sampling variation, restriction to autosomal SNPs, poor coverage of certain genomic regions or rare alleles by the genotyping platforms used or a lack of power to detect even smaller effects. Lastly, heart rate is a very dynamic trait. Strong environmental influences such as chronic physical activity or training as well as variability in the time at rest or posture at the moment of measurements and other factors, such as anemia or anxiety, which we have not accounted for, would add noise to the phenotype, which would be expected to bias our study toward the null but not toward the false inference of association.
Conclusion
RRGEN identified nine signals at seven loci at which common genetic variation is associated with resting heart rate. Six of these signals at six loci are novel, while the other three signals replicate previous findings from GWAS. Several of these loci include genes encoding ion channel regulator proteins with known involvement in heart rate regulation, or proteins with cardiovascular relevant functions and known associated Mendelian disorders. These variants may impact clinical care by identifying novel factors that influence pathologic heart rate states, are relevant for cardiac structure and thereby modulate heart rate, or by improving our understanding of the physiologic basis of heart rate regulation.
MATERIALS AND METHODS
Study participants
The RRGEN sample consisted of subjects from the five participating studies in the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (57)—comprised of the Age, Gene, Environment Susceptibility (AGES) Study, the Atherosclerosis Risk in Communities (ARIC) Study, the Cardiovascular Health Study (CHS), the Framingham Heart Study (FHS) and the Rotterdam Study (RS)—as well as the Cooperative Health Research in the Region Augsburg (KORA) study, the SardiNIA study, the Study of Health in Pomerania (SHIP), TwinsUK, Netherlands Study of Depression and Anxiety (NESDA) and three population isolate studies in the European Special Populations Network (EUROSPAN), the Erasmus Rucphen Family (ERF), the South Tyrolean Micro-Isolate (MICROS) and the Orkney Complex Disease Study (ORCADES). For RRGEN, both the baseline Rotterdam Study (RS-I) and first extended cohort (RS-II) were used (58,59). KORA subjects in RRGEN were drawn from the F3 and S4 cohorts.
Individuals were excluded if they were non-Caucasian, had atrial fibrillation, a second- or third-degree atrioventricular block, a pacemaker, a diagnosis of prevalent myocardial infarction or prevalent heart failure, used beta-adrenergic blocking agents, non-dihydropyridine calcium antagonists or digoxin, or had a heart rate <50 (RR interval > 1200 ms) or >100 bpm (RR interval < 600 ms).
To adhere to STrenghtening the REporting of Genetic Associations studies (STREGA) statement guidelines (60), we include an online supplement with additional information (Supplementary Material). All studies have approval from their institutional review committee, and the subjects of all cohorts provided written informed consent. A more detailed description of cohorts is given in Supplementary Material, Appendix.
RR interval measurement methods
All cohorts recorded 12-lead ECGs from which the RR interval (which equals the inverse heart rate) was measured. For cohort-specific details on RR interval measurement methods, please refer to the Supplementary Material, Appendix.
Genotyping and imputation
Affymetrix and Illumina arrays were used for genotyping. Using genotype information generated on these platforms, all cohorts imputed genotypes for a common set of ∼2.5 million autosomal SNPs based on linkage disequilibrium patterns observed in HapMap CEU reference samples (Utah residents of Northern and Western European descent). The genetic trait analyzed was the imputed allele dosage, a fractional value between 0 and 2, reflecting the estimated number of allele copies of an SNP for each subject. A more detailed description is given in the Supplementary Material, Appendix and Table S1.
Statistical methods
The resting RR interval was adjusted for age, sex and body mass index. For each SNP, we tested the genotype for association with the covariate-adjusted RR interval under an additive genetic model using linear regression models (Supplementary Material, Appendix). We then conducted a fixed-effects, inverse variance weighted meta-analysis using beta estimates and standard errors from each of the cohorts with applying genomic control on a per study basis and additionally post-meta-analysis (Pmeta-gc). Genomic control refers to the correction made to the test statistics to account for any inflation of the test statistic distribution, which can result from unaccounted population substructure or other technical biases (61,62). We mapped all SNPs to dbSNP build 129, resulting in a unique set of 2 650 552 autosomal SNPs, after confirming the consistency of the coded allele across all studies. Scripts used for this meta-analysis are available online (http://www.broadinstitute.org/~debakker/meta.html).
Genome-wide significance was defined as P < 5 × 10−8, based on the estimated multiple testing burden for all common variants in populations of European ancestry (63). To identify independent signals reaching the genome-wide significance threshold within a locus, genome-wide association meta-analysis results were aggregated into bins by index SNP at a linkage disequilibrium r2 threshold of 0.1, such that all results within a given bin were correlated to the index SNP at r2 ≥ 0.1 but to any index SNP in other bins at r2 < 0.1. Four index SNPs at two loci were subsequently analyzed in conditional regression models (n ≤ 33 846) to assess statistical independence.
Finally, we adopted the polygenic regression modeling approach as recently described by Purcell et al. (64) and implemented in PLINK (http://pngu.mgh.harvard.edu/~purcell/plink) to estimate the genetic variance explained by associated loci at progressively less stringent P-value thresholds within the RS-II sample. The outcome of this analysis is a P-value threshold at which the explained variance is maximized. The list of loci included in the score yielding the maximum explained variance will include non-genome-wide loci that in aggregate contribute to the model's performance and indicate that additional true-positive signals are likely to be present within that list. For this analysis, we removed the RS-I and RS-II data from the discovery meta-analysis to remove the risk of correlation between the discovery and validation sample and overestimation of the explained variance. Subsequently, we obtained a list of independent signals to be included in the model based on their statistical significance from the meta-analysis without RS-I or RS-II (PLINK-clump option, r2 ≥0.05, 1 Mb window) (65). We summarized variation across associated loci (using significance thresholds from P < 5 × 10−8 to P < 0.05) into quantitative scores per individual. The score was then used as a predictor in a linear regression analysis in the RS-II sample (n = 1589) and the resulting r2 is reported as the measure for explained variance for each P-value threshold.
SUPPLEMENTARY MATERIAL
FUNDING
This work has been made possible by the generous financial support of a variety of funding sources to participating studies and individual scientists. A detailed list of acknowledgments and funding sources, as well as an author contribution list, is included in Supplementary Material.
ACKNOWLEDGEMENTS
We thank all participants and the study staff of the Age, Gene/Environment Susceptibility Reykjavik Study (AGES); the Atherosclerosis Risk in Communities (ARIC) study; the Cardiovascular Health Study (CHS); the Erasmus Rucphen Family (ERF) study; the Framingham Heart Study (FHS); the Kooperative Gesundheitsforschung in der Region Augsburg (KORA); the Micros Study; the Netherlands Study of Depression and Anxiety (NESDA); the Orkney Complex Disease Study (Orcades); the Rotterdam Study (RS); the SardiNIA study; the Study of Health in Pomerania (SHIP); the TwinsUK study as well as the Cohorts for Heart and Aging Research in Genome Epidemiology (CHARGE) Consortium, for their invaluable contributions.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Fox K., Borer J.S., Camm A.J., Danchin N., Ferrari R., Lopez Sendon J.L., Steg P.G., Tardif J.C., Tavazzi L., Tendera M. Resting heart rate in cardiovascular disease. J. Am. Coll. Cardiol. 2007;50:823–830. doi: 10.1016/j.jacc.2007.04.079. doi:10.1016/j.jacc.2007.04.079. [DOI] [PubMed] [Google Scholar]
- 2.Greenland P., Daviglus M.L., Dyer A.R., Liu K., Huang C.F., Goldberger J.J., Stamler J. Resting heart rate is a risk factor for cardiovascular and noncardiovascular mortality: the Chicago Heart Association Detection Project in Industry. Am. J. Epidemiol. 1999;149:853–862. doi: 10.1093/oxfordjournals.aje.a009901. [DOI] [PubMed] [Google Scholar]
- 3.Nauman J., Nilsen T.I., Wisloff U., Vatten L.J. Combined effect of resting heart rate and physical activity on ischaemic heart disease: mortality follow-up in a population study (the HUNT study, Norway) J. Epidemiol. Community Health. 2010;64:175–181. doi: 10.1136/jech.2009.093088. doi:10.1136/jech.2009.093088. [DOI] [PubMed] [Google Scholar]
- 4.Jouven X., Zureik M., Desnos M., Guerot C., Ducimetiere P. Resting heart rate as a predictive risk factor for sudden death in middle-aged men. Cardiovasc. Res. 2001;50:373–378. doi: 10.1016/s0008-6363(01)00230-9. doi:10.1016/S0008-6363(01)00230-9. [DOI] [PubMed] [Google Scholar]
- 5.Kannel W.B., Kannel C., Paffenbarger R.S., Jr, Cupples L.A. Heart rate and cardiovascular mortality: the Framingham Study. Am. Heart J. 1987;113:1489–1494. doi: 10.1016/0002-8703(87)90666-1. doi:10.1016/0002-8703(87)90666-1. [DOI] [PubMed] [Google Scholar]
- 6.Kristal-Boneh E., Silber H., Harari G., Froom P. The association of resting heart rate with cardiovascular, cancer and all-cause mortality. Eight year follow-up of 3527 male Israeli employees (the CORDIS Study) Eur. Heart J. 2000;21:116–124. doi: 10.1053/euhj.1999.1741. doi:10.1053/euhj.1999.1741. [DOI] [PubMed] [Google Scholar]
- 7.Chang M., Havlik R.J., Corti M.C., Chaves P.H., Fried L.P., Guralnik J.M. Relation of heart rate at rest and mortality in the Women's Health and Aging Study. Am. J. Cardiol. 2003;92:1294–1299. doi: 10.1016/j.amjcard.2003.08.010. doi:10.1016/j.amjcard.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Jouven X., Empana J.P., Escolano S., Buyck J.F., Tafflet M., Desnos M., Ducimetiere P. Relation of heart rate at rest and long-term (>20 years) death rate in initially healthy middle-aged men. Am. J. Cardiol. 2009;103:279–283. doi: 10.1016/j.amjcard.2008.08.071. doi:10.1016/j.amjcard.2008.08.071. [DOI] [PubMed] [Google Scholar]
- 9.Hsia J., Larson J.C., Ockene J.K., Sarto G.E., Allison M.A., Hendrix S.L., Robinson J.G., LaCroix A.Z., Manson J.E. Women's Health Initiative Research Group. Resting heart rate as a low tech predictor of coronary events in women: prospective cohort study. Br. Med. J. 2009;338:b219. doi: 10.1136/bmj.b219. doi:10.1136/bmj.b219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russell M.W., Law I., Sholinsky P., Fabsitz R.R. Heritability of ECG measurements in adult male twins. J. Electrocardiol. 1998;30(suppl.):64–68. doi: 10.1016/s0022-0736(98)80034-4. [DOI] [PubMed] [Google Scholar]
- 11.Singh J.P., Larson M.G., O'Donnell C.J., Tsuji H., Evans J.C., Levy D. Heritability of heart rate variability: the Framingham Heart Study. Circulation. 1999;99:2251–2254. doi: 10.1161/01.cir.99.17.2251. [DOI] [PubMed] [Google Scholar]
- 12.Martin L.J., Comuzzie A.G., Sonnenberg G.E., Myklebust J., James R., Marks J., Blangero J., Kissebah A.H. Major quantitative trait locus for resting heart rate maps to a region on chromosome 4. Hypertension. 2004;43:1146–1151. doi: 10.1161/01.HYP.0000122873.42047.17. doi:10.1161/01.HYP.0000122873.42047.17. [DOI] [PubMed] [Google Scholar]
- 13.Laramie J.M., Wilk J.B., Hunt S.C., Ellison R.C., Chakravarti A., Boerwinkle E., Myers R.H. Evidence for a gene influencing heart rate on chromosome 5p13-14 in a meta-analysis of genome-wide scans from the NHLBI Family Blood Pressure Program. BMC Med. Genet. 2006;7:17. doi: 10.1186/1471-2350-7-17. doi:10.1186/1471-2350-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalageorgou C., Ge D., Jamshidi Y., Nolte I.M., Riese H., Savelieva I., Carter N.D., Spector T.D., Snieder H. Heritability of QT interval: how much is explained by genes for resting heart rate? J. Cardiovasc. Electrophysiol. 2008;19:386–391. doi: 10.1111/j.1540-8167.2007.01030.x. doi:10.1111/j.1540-8167.2007.01030.x. [DOI] [PubMed] [Google Scholar]
- 15.De Geus E.J., Kupper N., Boomsma D.I., Snieder H. Bivariate genetic modeling of cardiovascular stress reactivity: does stress uncover genetic variance? Psychosom. Med. 2007;69:356–364. doi: 10.1097/PSY.0b013e318049cc2d. doi:10.1097/PSY.0b013e318049cc2d. [DOI] [PubMed] [Google Scholar]
- 16.Wilk J.B., Myers R.H., Zhang Y., Lewis C.E., Atwood L., Hopkins P.N., Ellison R.C. Evidence for a gene influencing heart rate on chromosome 4 among hypertensives. Hum. Genet. 2002;111:207–213. doi: 10.1007/s00439-002-0780-9. doi:10.1007/s00439-002-0780-9. [DOI] [PubMed] [Google Scholar]
- 17.Ranade K., Jorgenson E., Sheu W.H., Pei D., Hsiung C.A., Chiang F.T., Chen Y.D., Pratt R., Olshen R.A., Curb D., et al. A polymorphism in the beta1 adrenergic receptor is associated with resting heart rate. Am. J. Hum. Genet. 2002;70:935–942. doi: 10.1086/339621. doi:10.1086/339621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.An P., Rice T., Rankinen T., Leon A.S., Skinner J.S., Wilmore J.H., Bouchard C., Rao D.C. Genome-wide scan to identify quantitative trait loci for baseline resting heart rate and its response to endurance exercise training: the HERITAGE Family Study. Int. J. Sports Med. 2006;27:31–36. doi: 10.1055/s-2005-837628. doi:10.1055/s-2005-837628. [DOI] [PubMed] [Google Scholar]
- 19.Wilk J.B., Myers R.H., Pankow J.S., Hunt S.C., Leppert M.F., Freedman B.I., Province M.A., Ellison R.C. Adrenergic receptor polymorphisms associated with resting heart rate: the HyperGEN Study. Ann. Hum. Genet. 2006;70:566–573. doi: 10.1111/j.1469-1809.2005.00258.x. doi:10.1111/j.1469-1809.2005.00258.x. [DOI] [PubMed] [Google Scholar]
- 20.Wilton S.B., Anderson T.J., Parboosingh J., Bridge P.J., Exner D.V., Forrest D., Duff H.J. Polymorphisms in multiple genes are associated with resting heart rate in a stepwise allele-dependent manner. Heart Rhythm. 2008;5:694–700. doi: 10.1016/j.hrthm.2008.01.039. doi:10.1016/j.hrthm.2008.01.039. [DOI] [PubMed] [Google Scholar]
- 21.Cho Y.S., Go M.J., Kim Y.J., Heo J.Y., Oh J.H., Ban H.J., Yoon D., Lee M.H., Kim D.J., Park M., et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat. Genet. 2009;41:527–534. doi: 10.1038/ng.357. doi:10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- 22.Holm H., Gudbjartsson D.F., Arnar D.O., Thorleifsson G., Thorgeirsson G., Stefansdottir H., Gudjonsson S.A., Jonasdottir A., Mathiesen E.B., Njolstad I., et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat. Genet. 2010;42:117–122. doi: 10.1038/ng.511. doi:10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 23.Chandler N.J., Greener I.D., Tellez J.O., Inada S., Musa H., Molenaar P., Difrancesco D., Baruscotti M., Longhi R., Anderson R.H., et al. Molecular architecture of the human sinus node: insights into the function of the cardiac pacemaker. Circulation. 2009;119:1562–1575. doi: 10.1161/CIRCULATIONAHA.108.804369. doi:10.1161/CIRCULATIONAHA.108.804369. [DOI] [PubMed] [Google Scholar]
- 24.Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc. Res. 2004;62:309–322. doi: 10.1016/j.cardiores.2003.11.035. doi:10.1016/j.cardiores.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 25.Dasgupta C., Martinez A.M., Zuppan C.W., Shah M.M., Bailey L.L., Fletcher W.H. Identification of connexin43 (alpha1) gap junction gene mutations in patients with hypoplastic left heart syndrome by denaturing gradient gel electrophoresis (DGGE) Mutat. Res. 2001;479:173–186. doi: 10.1016/s0027-5107(01)00160-9. [DOI] [PubMed] [Google Scholar]
- 26.Niimura H., Patton K.K., McKenna W.J., Soults J., Maron B.J., Seidman J.G., Seidman C.E. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002;105:446–451. doi: 10.1161/hc0402.102990. doi:10.1161/hc0402.102990. [DOI] [PubMed] [Google Scholar]
- 27.Ching Y.H., Ghosh T.K., Cross S.J., Packham E.A., Honeyman L., Loughna S., Robinson T.E., Dearlove A.M., Ribas G., Bonser A.J., et al. Mutation in myosin heavy chain 6 causes atrial septal defect. Nat. Genet. 2005;37:423–428. doi: 10.1038/ng1526. doi:10.1038/ng1526. [DOI] [PubMed] [Google Scholar]
- 28.Carniel E., Taylor M.R., Sinagra G., Di Lenarda A., Ku L., Fain P.R., Boucek M.M., Cavanaugh J., Miocic S., Slavov D., et al. Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation. 2005;112:54–59. doi: 10.1161/CIRCULATIONAHA.104.507699. doi:10.1161/CIRCULATIONAHA.104.507699. [DOI] [PubMed] [Google Scholar]
- 29.Callis T.E., Pandya K., Seok H.Y., Tang R.H., Tatsuguchi M., Huang Z.P., Chen J.F., Deng Z., Gunn B., Shumate J., et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Invest. 2009;119:2772–2786. doi: 10.1172/JCI36154. doi:10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Rooij E., Sutherland L.B., Qi X., Richardson J.A., Hill J., Olson E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. doi:10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 31.Wunderle V.M., Critcher R., Ashworth A., Goodfellow P.N. Cloning and characterization of SOX5, a new member of the human SOX gene family. Genomics. 1996;36:354–358. doi: 10.1006/geno.1996.0474. doi:10.1006/geno.1996.0474. [DOI] [PubMed] [Google Scholar]
- 32.Lefebvre V. The SoxD transcription factors—Sox5, Sox6 and Sox13—are key cell fate modulators. Int. J. Biochem. Cell Biol. 2009;42:429–432. doi: 10.1016/j.biocel.2009.07.016. doi:10.1016/j.biocel.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfeufer A., van Noord C., Marciante K.D., Arking D.E., Larson M.G., Smith A.V., Tarasov K.V., Muller M., Sotoodehnia N., Sinner M.F., et al. Genome-wide association study of PR interval. Nat. Genet. 42:153–159. doi: 10.1038/ng.517. doi:10.1038/ng.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfeufer A., Sanna S., Arking D.E., Muller M., Gateva V., Fuchsberger C., Ehret G.B., Orru M., Pattaro C., Kottgen A., et al. Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat. Genet. 2009;41:407–414. doi: 10.1038/ng.362. doi:10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newton-Cheh C., Eijgelsheim M., Rice K.M., de Bakker P.I., Yin X., Estrada K., Bis J.C., Marciante K., Rivadeneira F., Noseworthy P.A., et al. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat. Genet. 2009;41:399–406. doi: 10.1038/ng.364. doi:10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nolte I.M., Wallace C., Newhouse S.J., Waggott D., Fu J., Soranzo N., Gwilliam R., Deloukas P., Savelieva I., Zheng D., et al. Common genetic variation near the phospholamban gene is associated with cardiac repolarisation: meta-analysis of three genome-wide association studies. PLoS One. 2009;4:e6138. doi: 10.1371/journal.pone.0006138. doi:10.1371/journal.pone.0006138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vasan R.S., Glazer N.L., Felix J.F., Lieb W., Wild P.S., Felix S.B., Watzinger N., Larson M.G., Smith N.L., Dehghan A., et al. Genetic variants associated with cardiac structure and function: a meta-analysis and replication of genome-wide association data. JAMA. 2009;302:168–178. doi: 10.1001/jama.2009.978-a. doi:10.1001/jama.2009.978-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vinogradova T.M., Sirenko S., Lyashkov A.E., Younes A., Li Y., Zhu W., Yang D., Ruknudin A.M., Spurgeon H., Lakatta E.G. Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+ releases. Circ. Res. 2008;102:761–769. doi: 10.1161/CIRCRESAHA.107.161679. doi:10.1161/CIRCRESAHA.107.161679. [DOI] [PubMed] [Google Scholar]
- 39.Vinogradova T.M., Lyashkov A.E., Zhu W., Ruknudin A.M., Sirenko S., Yang D., Deo S., Barlow M., Johnson S., Caffrey J.L., et al. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ. Res. 2006;98:505–514. doi: 10.1161/01.RES.0000204575.94040.d1. doi:10.1161/01.RES.0000204575.94040.d1. [DOI] [PubMed] [Google Scholar]
- 40.Nishimura M., Suzuki S., Satoh T., Naito S. Tissue-specific mRNA expression profiles of human solute carrier 35 transporters. Drug Metab. Pharmacokinet. 2009;24:91–99. doi: 10.2133/dmpk.24.91. doi:10.2133/dmpk.24.91. [DOI] [PubMed] [Google Scholar]
- 41.Ha B.H., Ahn H.C., Kang S.H., Tanaka K., Chung C.H., Kim E.E. Structural basis for Ufm1 processing by UfSP1. J. Biol. Chem. 2008;283:14893–14900. doi: 10.1074/jbc.M708756200. doi:10.1074/jbc.M708756200. [DOI] [PubMed] [Google Scholar]
- 42.Kang S.H., Kim G.R., Seong M., Baek S.H., Seol J.H., Bang O.S., Ovaa H., Tatsumi K., Komatsu M., Tanaka K., et al. Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. J. Biol. Chem. 2007;282:5256–5262. doi: 10.1074/jbc.M610590200. doi:10.1074/jbc.M610590200. [DOI] [PubMed] [Google Scholar]
- 43.Aulchenko Y.S., Ripatti S., Lindqvist I., Boomsma D., Heid I.M., Pramstaller P.P., Penninx B.W., Janssens A.C., Wilson J.F., Spector T., et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 2009;41:47–55. doi: 10.1038/ng.269. doi:10.1038/ng.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kathiresan S., Willer C.J., Peloso G.M., Demissie S., Musunuru K., Schadt E.E., Kaplan L., Bennett D., Li Y., Tanaka T., et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009;41:56–65. doi: 10.1038/ng.291. doi:10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sabatti C., Service S.K., Hartikainen A.L., Pouta A., Ripatti S., Brodsky J., Jones C.G., Zaitlen N.A., Varilo T., Kaakinen M., et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 2009;41:35–46. doi: 10.1038/ng.271. doi:10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gieger C., Geistlinger L., Altmaier E., Hrabe de Angelis M., Kronenberg F., Meitinger T., Mewes H.W., Wichmann H.E., Weinberger K.M., Adamski J., et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008;4:e1000282. doi: 10.1371/journal.pgen.1000282. doi:10.1371/journal.pgen.1000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schaeffer L., Gohlke H., Muller M., Heid I.M., Palmer L.J., Kompauer I., Demmelmair H., Illig T., Koletzko B., Heinrich J. Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum. Mol. Genet. 2006;15:1745–1756. doi: 10.1093/hmg/ddl117. doi:10.1093/hmg/ddl117. [DOI] [PubMed] [Google Scholar]
- 48.Dettbarn C., Palade P. Arachidonic acid-induced Ca2+ release from isolated sarcoplasmic reticulum. Biochem. Pharmacol. 1993;45:1301–1309. doi: 10.1016/0006-2952(93)90283-3. doi:10.1016/0006-2952(93)90283-3. [DOI] [PubMed] [Google Scholar]
- 49.Bang M.L., Centner T., Fornoff F., Geach A.J., Gotthardt M., McNabb M., Witt C.C., Labeit D., Gregorio C.C., Granzier H., et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 2001;89:1065–1072. doi: 10.1161/hh2301.100981. doi:10.1161/hh2301.100981. [DOI] [PubMed] [Google Scholar]
- 50.Itoh-Satoh M., Hayashi T., Nishi H., Koga Y., Arimura T., Koyanagi T., Takahashi M., Hohda S., Ueda K., Nouchi T., et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2002;291:385–393. doi: 10.1006/bbrc.2002.6448. doi:10.1006/bbrc.2002.6448. [DOI] [PubMed] [Google Scholar]
- 51.Kubo Y., Baldwin T.J., Jan Y.N., Jan L.Y. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. doi:10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- 52.Fleischmann B.K., Duan Y., Fan Y., Schoneberg T., Ehlich A., Lenka N., Viatchenko-Karpinski S., Pott L., Hescheler J., Fakler B. Differential subunit composition of the G protein-activated inward-rectifier potassium channel during cardiac development. J. Clin. Invest. 2004;114:994–1001. doi: 10.1172/JCI15925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosskopf D., Nikula C., Manthey I., Joisten M., Frey U., Kohnen S., Siffert W. The human G protein beta4 subunit: gene structure, expression, Ggamma and effector interaction. FEBS Lett. 2003;544:27–32. doi: 10.1016/s0014-5793(03)00441-1. doi:10.1016/S0014-5793(03)00441-1. [DOI] [PubMed] [Google Scholar]
- 54.Ruiz-Velasco V., Ikeda S.R., Puhl H.L. Cloning, tissue distribution, and functional expression of the human G protein beta 4-subunit. Physiol. Genomics. 2002;8:41–50. doi: 10.1152/physiolgenomics.00085.2001. [DOI] [PubMed] [Google Scholar]
- 55.Splawski I., Timothy K.W., Decher N., Kumar P., Sachse F.B., Beggs A.H., Sanguinetti M.C., Keating M.T. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl Acad. Sci. USA. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. discussion 8086–8088. doi:10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Avkiran M., Rowland A.J., Cuello F., Haworth R.S. Protein kinase d in the cardiovascular system: emerging roles in health and disease. Circ. Res. 2008;102:157–163. doi: 10.1161/CIRCRESAHA.107.168211. doi:10.1161/CIRCRESAHA.107.168211. [DOI] [PubMed] [Google Scholar]
- 57.Psaty B.M., O'Donnell C.J., Gudnason V., Lunetta K.L., Folsom A.R., Rotter J.I., Uitterlinden A.G., Harris T.B., Witteman J.C.M., Boerwinkle E., et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ. Cardiovasc. Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. doi:10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hofman A., Breteler M.M., van Duijn C.M., Krestin G.P., Pols H.A., Stricker B.H., Tiemeier H., Uitterlinden A.G., Vingerling J.R., Witteman J.C. The Rotterdam Study: objectives and design update. Eur. J. Epidemiol. 2007;22:819–829. doi: 10.1007/s10654-007-9199-x. doi:10.1007/s10654-007-9199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hofman A., Breteler M.M., van Duijn C.M., Janssen H.L., Krestin G.P., Kuipers E.J., Stricker B.H., Tiemeier H., Uitterlinden A.G., Vingerling J.R., et al. The Rotterdam Study: 2010 objectives and design update. Eur. J. Epidemiol. 2009;24:553–572. doi: 10.1007/s10654-009-9386-z. doi:10.1007/s10654-009-9386-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Little J., Higgins J.P., Ioannidis J.P., Moher D., Gagnon F., von Elm E., Khoury M.J., Cohen B., Davey-Smith G., Grimshaw J., et al. Strengthening the reporting of genetic association studies (STREGA): an extension of the STROBE statement. Eur. J. Epidemiol. 2009;24:37–55. doi: 10.1007/s10654-008-9302-y. doi:10.1007/s10654-008-9302-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Bakker P.I., Ferreira M.A., Jia X., Neale B.M., Raychaudhuri S., Voight B.F. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 2008;17:R122–R128. doi: 10.1093/hmg/ddn288. doi:10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Devlin B., Roeder K., Wasserman L. Genomic control, a new approach to genetic-based association studies. Theor. Popul. Biol. 2001;60:155–166. doi: 10.1006/tpbi.2001.1542. doi:10.1006/tpbi.2001.1542. [DOI] [PubMed] [Google Scholar]
- 63.Pe'er I., Yelensky R., Altshuler D., Daly M.J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol. 2008;32:381–385. doi: 10.1002/gepi.20303. doi:10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 64.Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O'Donovan M.C., Sullivan P.F., Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. doi:10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]