

Abstract
BACKGROUND
We investigated whether the antihypertensive actions of the angiotensin II (Ang II) receptor (AT1-R) blocker, olmesartan medoxomil, may in part be mediated by increased Ang-(1–7) in the absence of significant changes in plasma Ang II.
METHODS
mRen2.Lewis congenic hypertensive rats were administered either a vehicle (n = 14) or olmesartan (0.5mg/kg/day; n = 14) by osmotic minipumps. Two weeks later, rats from both groups were further randomized to receive either the mas receptor antagonist A-779 (0.5mg/kg/day; n = 7 per group) or its vehicle (n = 7 per group) for the next 4 weeks. Blood pressure was monitored by telemetry, and circulating and tissue components of the renin–angiotensin system (RAS) were measured at the completion of the experiments.
RESULTS
Antihypertensive effects of olmesartan were associated with an increase in plasma renin concentration, plasma Ang I, Ang II, and Ang-(1–7), whereas serum aldosterone levels and kidney Ang II content were reduced. Preserved Ang-(1–7) content in kidneys was associated with increases of ACE2 protein but not activity and no changes on serum and kidney ACE activity. There was no change in cardiac peptide levels after olmesartan treatment. The antihypertensive effects of olmesartan were not altered by concomitant administration of the Ang-(1–7) receptor antagonist except for a mild further increase in plasma renin concentration.
CONCLUSIONS
Our study highlights the independent regulation of RAS among plasma, heart, and kidney tissue in response to AT1-R blockade. Ang-(1–7) through the mas receptor does not mediate long-term effects of olmesartan besides counterbalancing renin release in response to AT1-R blockade.
Keywords: angiotensin II, angiotensin-(1–7), AT1 receptor, blood pressure, heart, hypertension, kidney, mas receptor, olmesartan.
Although the mechanism of action of angiotensin II (Ang II) receptor (AT1-R) blockers (ARBs) is well established, data suggest that their antihypertensive effects may be limited in part by the overriding effect of compensatory increases in plasma renin activity (PRA) and Ang II.1 Contrasting with the mechanism of action of other ARBs, Agata et al. 2 reported that the antihypertensive and cardiac antihypertrophic actions of 4-week olmesartan medoxomil administration were not associated with the expected increases in circulating Ang II in adult stroke-prone spontaneously hypertensive rats (SHR). Agata et al. 2 speculated that the failure of plasma Ang II to rise in response to blockade of AT1-R could be explained by angiotensin-(1–7) [Ang-(1–7)]–induced inhibition of angiotensin converting enzyme (ACE) activity, given previous observations of increased Ang-(1–7) after treatment with ARBs,3,4 as well as an inhibitory role of the heptapeptide at the C- and N-terminus catalytic activity of the enzyme.5,6 The intriguing possibility of a differential effect of olmesartan on plasma Ang II levels compared with other ARBs was also consistent with 2 other studies in human essential hypertensive subjects in whom prolonged therapy with olmesartan was accompanied by either decreases or no changes in plasma Ang II levels.7,8
The effects of olmesartan on Ang II levels may also result from increased conversion of the octapeptide to Ang-(1–7) because ACE2 has been shown to be upregulated by AT1-R blockade.4,9,10 This has been demonstrated in vivo by recent work showing that the infusion of soluble human recombinant ACE2 efficiently lowered plasma Ang II while increasing Ang-(1–7).10 Furthermore, in isolated cardiac myocytes, ACE2 messenger RNA expression and activity were not affected by Ang-(1–7); however, the inhibitory effects of Ang II on ACE2 were blocked by Ang-(1–7).11 The heptapeptide modulatory effect was prevented by the Ang-(1–7) mas receptor antagonist [D-ALA7]-Ang-(1–7) (A-779), indicating that the Ang-(1–7) response was mediated by a specific Ang-(1–7) receptor. A-779 is a selective blocker of the mas receptor that has been identified to mediate vasodilatory, antitrophic, and antiproliferative effects of Ang-(1–7).12–14
The long-term effects of Ang-(1–7) antagonism in the presence of concomitant Ang II receptor blockade have not been determined. With this in mind, we investigated the Ang-(1–7)–mediated effects of olmesartan on blood pressure, plasma, renal, and cardiac Ang II as well as ACE2 in mRen2.Lewis congenic hypertensive rats. This monogenetic hypertensive rat strain was developed in our laboratory through a backcross of the hypertensive (mRen2)27 transgenic rats with normotensive Lewis rats. The aim of this backcross was to offset the heterogeneity of the parent strain that contributed to the genetic variability found within the original transgenic strain.15,16 Because the malignant phase of hypertension is not observed in mRen2.Lewis rats, the longer life span of this experimental model provides a better opportunity to investigate the function and regulation of tissue renin–angiotensin system (RAS) and its contribution to the etiology of hypertension and target organ damage.
METHODS
Experimental protocol
Twenty-eight hemizygous male mRen2.Lewis hypertensive rats were obtained from the congenic colony founded at the Wake Forest University Hypertension and Vascular Research Center. Rats were housed in an American Association of Laboratory Animal Care–approved facility in a temperature-controlled room (22±2 °C) with a 12:12-hour light/dark cycle (lights on from 6:00 am to 6:00 pm) and were allowed free access to food and water. The rats were handled in accordance with National Institute of Health guidelines; our Institutional Animal Care and Use Committee approved the study in advance. At age 10 weeks and under aseptic conditions, radiotelemetry probes (PA-C40; DSI, St. Paul, MN) were chronically implanted under anesthesia for continuous monitoring of arterial pressure and heart rate, as described elsewhere.17 After a 2-week recovery period, animals were randomized to receive either vehicle (2.5% sodium bicarbonate; n = 14) or olmesartan (Daiichi Sankyo, Inc., Parsippany, NJ; 0.5mg/kg/day dissolved in 2.5% sodium bicarbonate; n = 14) by osmotic minipumps implanted subcutaneously for the ensuing 2 weeks (Figure 1). Thereafter, rats from both groups were randomized to receive either the Ang-(1–7) antagonist A-779 (Bachem, Torrance, CA; 0.5mg/kg/day in mili-Q water; n = 7) or its vehicle (mili-Q water; n = 7) for the next 4 weeks. Two-week pumps implanted initially at the beginning of the therapeutic period were replaced at the same time with new pumps to cover the remaining 4 weeks of the experiment. As shown in Figure 1, the design of the study allowed us to assess the effects of vehicle or olmesartan alone or in combination with A-779. After 6 weeks on the respective treatment, animals were decapitated, and trunk blood was collected for measurements of renin–angiotensin–aldosterone system components. In addition, their heart and kidneys were removed and weighed, and the tissue samples were quickly frozen on dry ice for later measurement of angiotensin peptides and ACE, ACE2, and neprilysin (NEP) activities.
Figure 1.
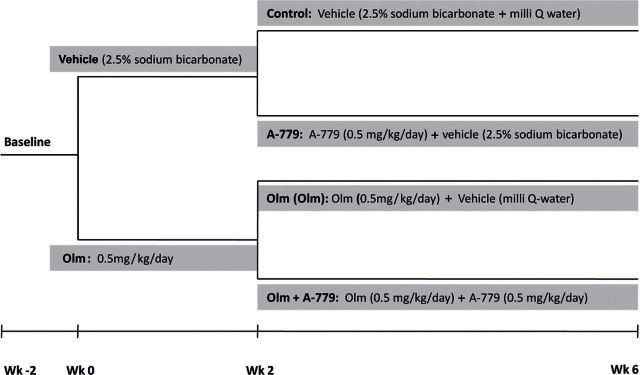
Outline of experimental design. Male mRen2.Lewis congenic hypertensive rats were implanted with radio-telemetry probes at age 10 weeks. After 2-week recovery period, rats were randomized to receive either vehicle (2.5% sodium bicarbonate; n = 14) or olmesartan (Olm; 0.5mg/kg/day dissolved in 2.5% sodium bicarbonate; n = 14) by osmotic minipumps implanted subcutaneously for the ensuing 2 weeks. After the 2-week period on the respective treatment, rats from both groups were randomized to receive either the mas receptor antagonist A-779 (0.5mg/kg/day in mili Q water; n = 7) or its vehicle (mili Q water; n = 7) for the next 4 weeks. Two-week pumps implanted initially at the beginning of the therapeutic period were replaced at the same time with new pumps to cover the remaining 4 weeks of the experiment.
Plasma and tissue hormone assays
Plasma renin concentration (PRC) and plasma and tissue Ang I, Ang II, and Ang-(1–7) were measured by radioimmunoassays, as previously described (details provided in Supplementary Methods).3
Enzyme assays
Serum and tissue ACE activity was determined with the synthetic substrate Hip-His-Leu.18 Tissue ACE2 and NEP activity were measured by high-performance liquid chromatography (HPLC), as previously described (details provided in Supplementary Methods).3
Western blotting
ACE2 protein was analyzed by Western blot, as previously described (details provided in Supplementary Methods).3
Statistics
All values are expressed as the mean ± 1 SEM. Data were analyzed by use of analysis of variance followed by Newman–Keuls posttest. A value of P < 0.05 was considered to be of statistical significance.
RESULTS
Body and organs weight
Body and kidney weights were not different between experimental groups, whereas heart weight and the heart weight–to–body weight ratios were significantly lower in both olmesartan and olmesartan plus A-779 groups when compared with the control and A-779–treated animals (Table 1).
Table 1.
Effect of treatments on body, kidney, and heart weights in congenic mRen2.Lewis rats
| Variables | Control | A-779 | OLM | OLM + A-779 |
|---|---|---|---|---|
| Body weight, g | 436±7 | 447±5 | 436±9 | 442±11 |
| Left kidney weight, g | 1.46±0.01 | 1.52±0.03 | 1.48±0.02 | 1.48±0.05 |
| Right kidney weight, g | 1.53±0.02 | 1.56±0.03 | 1.53±0.02 | 1.53±0.04 |
| Heart weight, g | 1.35±0.02 | 1.41±0.02 | 1.03±0.04* | 1.01±0.02*,‡ |
| Heart weight/body weight ratio, mg/kg | 3.09±0.05 | 3.16±0.04 | 2.37±0.10* | 2.29±0.05*,‡ |
Values are means ± SEM. A-779, mas receptor antagonist [D-ALA7]-Ang-(1–7). Abbreviation: OLM, olmesartan.
*P < 0.05 vs. control; ‡P < 0.05 vs. A-779.
Blood pressure and heart rate
Mean blood pressure was lower in rats treated with olmesartan throughout the whole experimental period, whereas addition of A-779 had no effect on either the mean blood pressure or the heart rate of rats medicated with olmesartan or the vehicle (Figure 2). Heart rate was not different between the groups except for the first few days after olmesartan administration when higher heart rate followed the profound reduction in blood pressure in groups treated with the AT1-R antagonist (Figure 2).
Figure 2.

Weekly arterial blood pressure and heart rate profiling by telemetry in congenic mRen2.Lewis rats. Olmesartan (Olm) reduced blood pressure in congenic rats from the first week of the treatment (P < 0.05 vs. control and mas receptor antagonist [D-ALA7]-Ang-(1–7) (A-779)). Coadministration of A-779 did not affect the blood pressure–lowering effects of olmesartan nor did the treatment with A-779 alone induce any change in blood pressure. The profound antihypertensive effect of olmesartan was associated with an increase in heart rate in olmesartan and olmesartan + A-779 (P < 0.05 vs. control and A-779) that returned to the pretreatment values after the first week of treatment. Values are mean ± SEM. n = 7 per group. *P < 0.0.5 vs. control; ‡P < 0.0.5 vs. A-779.
Plasma and tissue RAS
PRC and plasma concentrations of Ang I, Ang II, and Ang-(1–7) were significantly higher in olmesartan and olmesartan plus A-779 groups compared with the control and A-779 groups (Figure 3a). Concomitant treatment with A-779 further raised PRC in olmesartan-treated rats. Serum levels of aldosterone were lower in both olmesartan-treated groups, although they reached the level of statistical significance only when compared with A-779. Serum ACE activity was not different among the experimental groups (Figure 3a). Neither olmesartan nor A-779 treatment changed angiotensin peptide concentrations in heart tissue (Figure 3b). In contrast, renal cortical Ang I increased significantly whereas Ang II decreased in both olmesartan and olmesartan plus A-779 groups when compared with the control and A-779 groups. There was no difference in Ang-(1–7) concentration in kidney cortex among the experimental groups.
Figure 3.

Effects of AT1 and mas receptor antagonists on circulating (a), heart (b), and kidney cortex (b) renin–angiotensin–aldosterone system components. Values are expressed as mean ± SEM. n=7 per group. Abbreviations: Ang I, angiotensin I; Ang II, angiotensin II; Ang-(1–7), angiotensin-(1–7); A-779, mas receptor antagonist [D-ALA7]-Ang-(1–7); Olm, olmesartan; RAS, renin–angiotensin system. *P < 0.05 vs. control; +P < 0.05 vs. Olm; ‡P < 0.05 vs. A-779.
To further elucidate the effects of AT1 and mas receptor antagonism on renal angiotensin peptide levels we measured in kidney cortex membranes protein expression and activity of ACE2, the enzyme that preferentially cleaves Ang II into Ang-(1–7).19 ACE2 protein expression, but not enzymatic activity, was increased significantly in both groups treated with olmesartan, whereas A-779 had no effect on ACE2 in both control and olmesartan-treated groups (Figure 4 and Table 2). In addition, neither treatment significantly affected ACE or NEP activities (Table 2).
Figure 4.

Effects of AT1 and mas receptor antagonists on renal cortical angiotensin converting enzyme 2 (ACE2) protein expression. Values are expressed as mean ± SEM. n=7 per group. Abbreviations: A-779, mas receptor antagonist [D-ALA7]-Ang-(1–7); Olm, olmesartan; * P < 0.05 vs. control; ‡ P < 0.05 vs. A-779.
Table 2.
Angiotensin converting enzyme (ACE), ACE2, and neprilysin (NEP) activities in kidney of mRen2.Lewis rats treated with AT1 and mas receptor antagonists
| Enzyme | Control | A-779 | OLM | OLM + A-779 |
|---|---|---|---|---|
| ACE, nmol/mg/min | 1.9±0.6 | 2.5±0.3 | 2.4±0.9 | 2.0±0.8 |
| ACE2, fmol/mg/min | 32.0±1.7 | 30.6±1.0 | 36.7±1.7 | 34.4±1.9 |
| NEP, fmol/mg/min | 95.5±2.9 | 105.0±4.1 | 100.9±4.7 | 97.5±4.3 |
Values are mean ± SEM. A-779, mas receptor antagonist [D-ALA7]-Ang-(1–7). Abbreviation: OLM, olmesartan.
DISCUSSION
The results from this study add additional weight to the concept of independent regulation of angiotensin peptides content among plasma, heart, and kidney tissue in response to AT1-R blockade. In addition, we showed that the antihypertensive effects of olmesartan treatment are associated with marked suppression of serum aldosterone levels despite the presence of elevated plasma Ang I and Ang II levels. The reactive hyperreninemia associated with the antihypertensive actions of olmesartan treatment and consequent AT1-R blockade induced differential changes in the expression of Ang I and Ang II content in heart and renal cortical tissues that were characterized by prominent and opposing changes in renal Ang I and Ang II, respectively, and no changes in their content in heart tissue. Reduced AT1-R–mediated uptake seems to predominantly contribute to decreased renal Ang II content, although the 2-fold increases in the ACE2 protein suggest an additional contribution through increased Ang II metabolism. However, we could not demonstrate an increase in ACE2 enzymatic activity despite increase in protein levels in this study. Although the AT1-R blockade increased plasma Ang-(1–7) levels and preserved the renal content of the heptapeptide, the absence of the effects of A-799 suggests that the mas receptor does not mediate long-term effects of olmesartan on blood pressure or profile of angiotensin peptides in congenic mRen2.Lewis rats. The augmentation in PRC due to the combined effect of both AT1 and mas receptor blockade is a new finding implicating a counterbalancing role for Ang-(1–7) in the control of renin release.
Telemetric measurements of arterial pressure, by providing high precision and accuracy, showed that mas receptor blockade had no effect on the 24-hour blood pressure of either vehicle- or olmesartan-treated rats. The absence of an effect of A-779 on blood pressure cannot be due to insufficient blockade because the doses used in these experiments are within the range reported by others10,20,21 and the same as those used by Agata et al. 2 As first pointed out by Ferrario,22 the direct effects of Ang-(1–7) on blood pressure can be demonstrated only in conditions in which compensatory mechanisms are rendered ineffective or in the presence of significant RAS overactivity. Although direct antihypertensive effects of Ang-(1–7) administration have been documented in SHR23 and SHR treated with NG-nitro-L-arginine methyl ester (L-NAME)24, a chronic infusion of Ang-(1–7) did not modify the course of 2K-1C hypertension.25 Although A-779 has been reported to reverse the blood pressure–lowering effects of Ang-(1–7) or an ACE inhibitor in an experimental model of diabetes,24,26 neither stimulation nor blockade of the mas receptor influences blood pressure in WKY–23 or Ang II–infused Sprague–Dawley rats.27 Moreover, in mice receiving an acute infusion of human recombinant ACE2 and Ang II, neither the infusion of Ang-(1–7) nor the infusion of a mas receptor blocker had any effect on blood pressure.10 Our study further shows in a chronic setting that blockade of AT1-R–dependent changes in blood pressure predominates over the potential impact of additional mas-R–mediated vasodilator mechanisms related to the observed increase in Ang-(1–7) levels. This conclusion does not negate a contribution of mas receptor–mediated Ang-(1–7) actions in conditions of reduced circulating Ang II, such as those resulting from ACE inhibition, because we showed that the blood pressure increase in response to acute Ang-(1–7) neutralization was larger in lisinopril- vs. losartan-treated SHR.28 In addition, in the presence of prolonged AT1-R blockade, circulating Ang II may increase to an extent that the peptide interacts with a D-[Ala7]-Ang-(1–7)–sensitive site.29 It is possible that in that way Ang II prevents the effects of Ang-(1–7) on mas receptor without any particular effects of its own. Moreover, previous studies also showed that Ang-(1–7) effects are mediated through receptors other then mas, for example AT2.20 Finally, Ang-(1–7), like olmesartan,1,30,31 may elicit inverse agonist effect on AT1-Rs. Thus, Ang-(1–7) and olmesartan may have overlapping activities at the AT1-Rs and that may be why A-779 has no apparent blood pressure effect when given together with olmesartan. Further experiments are necessary to confirm the attractive hypothesis that AT1-Rs must be unblocked for Ang-(1–7) to show an antihypertensive potential.
In 2006, Ichikawa and Takayama7 reported in hypertensive patients that olmesartan either did not change or decreased circulating Ang II in association with decreases in serum aldosterone after longer-term administration (2 years). Those findings were in sharp contrast with the effects of other AT1-R antagonists that largely increase PRC and consequently plasma Ang II. Comparative findings reported by Tsutamoto et al. 8 showed that a 12-month exposure to olmesartan caused a modest decrease in plasma Ang II. Our findings in mRen2.Lewis hypertensive rats do not corroborate those studies or the Agata et al. 2 previous report in stroke-prone SHR. In the Agata et al. study, a 4-week administration of olmesartan reversed cardiac hypertrophy and lowered blood pressure in association with nonsignificant increases in circulating Ang II levels. Although the duration of the treatment period and the doses of olmesartan employed in our study and the Agata et al. 2 study were similar, differences in findings may be related to the use of stroke-prone SHR vs. congenic mRen2.Lewis hypertensive rats. Although Agata et al. 2 attributed the effects on circulating Ang II to an inhibitory effect of increased Ang-(1–7) on ACE activity, their hypothesis was not substantiated by our current study through direct measure of ACE activity. The majority of studies implicating Ang-(1–7) inhibitory effects on ACE activity were in vitro studies employing concentrations of Ang-(1–7) (µmol/L) greater than physiological levels5,6 or the concentration of the heptapeptide reached in our studies after AT1-R blockade (pmol/L). Furthermore, the relative increase of Ang II to Ang I as well as the relative decrease in Ang-(1–7) to Ang I in olmesartan-treated rats argues against a significant role of Ang-(1–7) in inhibiting the conversion of Ang I to Ang II by ACE in the circulation. In addition, a major difference between the 2 studies relates to their observation that A-799 caused a small but significant increase in the blood pressure (measured by tail-cuff) of olmesartan-treated rats that was associated with a 312% increases in plasma Ang II.2 The more precise use of blood pressure telemetry in our study demonstrated that blockade of mas receptors does not oppose the antihypertensive effects induced by olmesartan and that A-799 has no additional effect on plasma and tissue Ang II levels or serum ACE activities. Therefore, our data demonstrate that the major antihypertensive mode of action of Ang II blockade with olmesartan is mediated through the AT1-R.
In addition, we showed that the antihypertensive effects of olmesartan treatment are associated with marked suppression of serum aldosterone levels despite the presence of elevated plasma Ang I and Ang II levels. Thus, olmesartan appears to confer sustained aldosterone suppression over several weeks of administration, which is consistent with the effect of valsartan in patients with heart failure32 but not diabetes mellitus.33
Concomitant blockade of AT1 and mas receptors resulted in an additional augmentation in PRC when compared with AT1-R antagonism alone. This is a new finding implicating a counterbalancing role for Ang-(1–7) in the control of renin release. Furthermore, the effect of mas receptor blockade on renin release was not observed in rats not treated with olmesartan, suggesting that renin activation may be essential for initiation of mas receptor–mediated effects. Importantly, AT2-Rs inhibit renal renin production in salt-depleted rats treated with valsartan.34 The inhibitory effect of AT2-R on renin production was mediated through nitric oxide (NO), and the combined inhibition of AT2-R and NO synthase causes no additional increase in renal renin. Because the majority of Ang-(1–7) renal actions are mediated through the NO pathway, it is possible that renal mas receptor activation leads to NO production and renin inhibition under the condition of concomitant AT1-R blockade. In support of this hypothesis, we point to our recent findings of reduced renal mas protein expression in SHR fed a high-salt diet that was associated with increased plasma renin concentration.35 Moreover, β-1 receptor antagonism reduced renin release and corrected the reduced mas receptor expression due to a high-salt diet while increasing expression of neuronal NO synthase.
We further demonstrated that kidney Ang II decreased following AT1-R antagonism in both the absence and presence of mas receptor blockade. Numerous reports demonstrated AT1-R–mediated Ang II uptake as well as stimulation of angiotensinogen36,37 in the kidneys, which explains, at least in part, the dissociation between the kidney and plasma Ang II levels after AT1-R blockade. Interestingly, we observed no significant change in renal Ang-(1–7) levels despite a significant fall in renal Ang II in the olmesartan-treated rats. That prompted us to investigate the activity of enzymes involved in the Ang-(1–7) formation and metabolism. We first demonstrated a significant increase in ACE2 protein in kidney cortex membranes of both groups treated with olmesartan, confirming tonic downregulation of ACE2 mediated by AT1-R in rats harboring the mRen2 gene. However, HPLC measurement of ACE2 activities revealed only a tendency for an increase in ACE2-mediated conversion of Ang II to Ang-(1–7) in olmesartan-treated groups when compared with either vehicle- or A-779–treated rats. The uncoupling between the ACE2 gene, protein, and activity has been reported earlier.3,38,39 However, previous studies from our laboratory3,16 and others40 showed that short-term blockade of AT1-R induces an increase in ACE2 protein and activity. It is noteworthy to mention that Lew et al. 41 observed the presence of an endogenous inhibitor in plasma of healthy volunteers, upon which removal of the ACE2 activity was detected. In addition, the presence of endogenous antibodies with inhibitory ACE2 activity has been shown in patients with vasculopathy and increased ACE2 enzyme protein.38 Thus, it is tempting to speculate that increased ACE2 in response to longer exposure to AT1-R antagonist may stimulate the production of ACE2 antibodies or inhibitors, which may eventually reduce its activity. In addition, previous in vitro reports showed that Ang-(1–7) prevented Ang II–induced decrease in ACE2 mRNA expression and activity. The effects were abolished by the Ang-(1–7) mas receptor antagonist A-799.11 In this study on renal tissue, we did not observe any effects of mas receptor blockade with respect to ACE2 protein and activity in either vehicle- or olmesartan-treated rats. Further studies are thus required to determine whether absence of ACE2 change in kidney in response to A-799 administration implies a differential involvement of mas receptors in ACE2 regulation in diverse organs.
We also investigated the activity of ACE and NEP because ACE metabolizes Ang-(1–7) whereas NEP forms it from Ang I and metabolizes it to smaller fragments, mainly Ang-(1–4) in the kidney.18 Thus, inhibition of ACE or NEP could lead to Ang-(1–7) accumulation in the kidney of olmesartan-treated animals. However, there was no change in these enzyme activities in the kidney cortex membranes after olmesartan treatment, and therefore other enzymes relevant for Ang-(1–7) formation, which were not examined in this study, may contribute to the preserved renal Ang-(1–7). More specifically, prolyl endopeptidase, whose activity was confirmed in different region of nephron, uses both Ang I and Ang II to form Ang-(1–7).42–44 In this connection, a detailed study by Velez et al. 19 on angiotensin metabolism in cultured human glomerular podocytes and mesangial cells showed that prolyl endopeptidase rather than NEP contributes to Ang-(1–7) formation.
The absence of any changes in plasma or tissue Ang-(1–7) content in our experiments using a specific mas receptor antagonist in both vehicle and olmesartan treatment suggests that the mas receptor does not mediate tissue Ang-(1–7) uptake. Thus, it is less likely that renal mas receptor–mediated Ang-(1–7) uptake is responsible for the preserved renal Ang-(1–7) levels in kidneys in which less Ang II is available for its conversion through ACE2. In keeping with this interpretation, a recent study suggests that megalin, the receptor expressed abundantly in the brush border of the proximal tubules, mediates the nonspecific uptake of different peptides, including Ang-(1–7).45 Moreover, AT1-R regulates megalin expression in cultured proximal tubular cells,46 whereas 3 weeks of AT1-R blockade upregulated reduced megalin expression in hypertensive kidney of transgenic m[Ren2]27 hypertensive rats.47 Although further studies will validate the pathophysiological relevance of megalin in renal uptake of Ang-(1–7) in response to AT1-R blockade, the higher renal Ang-(1–7)/Ang II ratio found in our study in response to RAS blockade supports the concept that renal Ang-(1–7) contributes to the well-documented beneficial renal effects of ARBs.
In summary, this study shows that the potent antihypertensive and antihypertrophic effects of olmesartan are comparable with those obtained with other AT1-R antagonists that are not capable of effectively suppressing serum aldosterone in the face of increased circulating Ang II. Suppression of renal Ang II content without changes in renal Ang-(1–7) after AT1-R blockade may be a mechanism associated with the known renoprotective effect of AT1-R blockade. The primary effects of olmesartan are not altered by concomitant administration of an Ang-(1–7) receptor antagonist, a finding that underscores a primary role of the AT1-R rather than the mas receptor in the modulation of blood pressure. In addition, we show a mild but significant effect of Ang-(1–7) in the modulation of renal renin secretion that may be mediated by a non-AT1-R.
SUPPLEMENTARY MATERIAL
Supplementary materials are available at American Journal of Hypertension (http://ajh.oxfordjournals.org).
DISCLOSURE
C.M. Ferrario received investigator-initiated support from Daiichi-Sankyo. All other authors reported no conflict of interest.
ACKNOWLEDGMENTS
This study was supported by grants from Daichi Daiichi-Sankyo (to C.M.F.), the American Heart Association (2300114 to J.V.), and the National Institutes of Health (2PO1 HL-051952 to C.M.F.). We also acknowledge partial support provided by the Farley-Hudson Foundation, Jacksonville, NC.
REFERENCES
- 1. Schindler C, Ferrario CM. Olmesartan for the treatment of arterial hypertension. Future Cardiol 2008; 4: 357–372 [DOI] [PubMed] [Google Scholar]
- 2. Agata J, Ura N, Yoshida H, Shinshi Y, Sasaki H, Hyakkoku M, Taniguchi S, Shimamoto K. Olmesartan is an angiotensin II receptor blocker with an inhibitory effect on angiotensin-converting enzyme. Hypertens Res 2006; 29: 865–874 [DOI] [PubMed] [Google Scholar]
- 3. Ferrario CM, Jessup J, Gallagher PE, Averill DB, Brosnihan KB, Tallant EA, Smith RD, Chappell MC. Effects of renin-angiotensin system blockade on renal angiotensin-(1–7) forming enzymes and receptors. Kidney Int 2005; 68: 2189–2196 [DOI] [PubMed] [Google Scholar]
- 4. Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, Diz DI, Gallagher PE. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005; 111: 2605–2610 [DOI] [PubMed] [Google Scholar]
- 5. Deddish PA, Marcic B, Jackman HL, Wang HZ, Skidgel RA, Erdos EG. N-domain-specific substrate and C-domain inhibitors of angiotensin-converting enzyme: angiotensin-(1–7) and keto-ACE. Hypertension 1998; 31: 912–917 [DOI] [PubMed] [Google Scholar]
- 6. Roks AJ, van Geel PP, Pinto YM, Buikema H, Henning RH, de ZD, van Gilst WH. Angiotensin-(1–7) is a modulator of the human renin-angiotensin system. Hypertension 1999; 34: 296–301 [DOI] [PubMed] [Google Scholar]
- 7. Ichikawa S, Takayama Y. Long-term effects of olmesartan, an Ang II receptor antagonist, on blood pressure and the renin-angiotensin-aldosterone system in hypertensive patients. Hypertens Res 2001; 24: 641–646 [DOI] [PubMed] [Google Scholar]
- 8. Tsutamoto T, Nishiyama K, Yamaji M, Kawahara C, Fujii M, Yamamoto T, Horie M. Comparison of the long-term effects of candesartan and olmesartan on plasma angiotensin II and left ventricular mass index in patients with hypertension. Hypertens Res 2010; 33: 118–122 [DOI] [PubMed] [Google Scholar]
- 9. Igase M, Strawn WB, Gallagher PE, Geary RL, Ferrario CM. Angiotensin II AT1 receptors regulate ACE2 and angiotensin-(1–7) expression in the aorta of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 2005; 289: H1013–H1019 [DOI] [PubMed] [Google Scholar]
- 10. Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K, Schuster M, Loibner H, Brosnihan KB, Ferrario CM, Penninger JM, Batlle D. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension 2010; 55: 90–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gallagher PE, Ferrario CM, Tallant EA. Regulation of ACE2 in cardiac myocytes and fibroblasts. Am J Physiol Heart Circ Physiol 2008; 295: H2373–H2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Benter IF, Yousif MH, Al-Saleh FM, Raghupathy R, Chappell MC, Diz DI. Angiotensin-(1–7) blockade attenuates captopril- or hydralazine-induced cardiovascular protection in spontaneously hypertensive rats treated with NG-nitro-L-arginine methyl ester. J Cardiovasc Pharmacol 2011; 57: 559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raffai G, Durand MJ, Lombard JH. Acute and chronic angiotensin-(1–7) restores vasodilation and reduces oxidative stress in mesenteric arteries of salt-fed rats. Am J Physiol Heart Circ Physiol 2011; 301:H1341–H1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tallant EA, Ferrario CM, Gallagher PE. Angiotensin-(1–7) inhibits growth of cardiac myocytes through activation of the mas receptor. Am J Physiol Heart Circ Physiol 2005; 289: H1560–H1566 [DOI] [PubMed] [Google Scholar]
- 15. Chappell MC, Gallagher PE, Averill DB, Ferrario CM, Brosnihan KB. Estrogen or the AT1 antagonist olmesartan reverses the development of profound hypertension in the congenic mRen2. Lewis rat. Hypertension 2003; 42: 781–786 [DOI] [PubMed] [Google Scholar]
- 16. Jessup JA, Gallagher PE, Averill DB, Brosnihan KB, Tallant EA, Chappell MC, Ferrario CM. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren-2 rats. Am J Physiol Heart Circ Physiol 2006; 291: H2166–H2172 [DOI] [PubMed] [Google Scholar]
- 17. Trask AJ, Groban L, Westwood BM, Varagic J, Ganten D, Gallagher PE, Chappell MC, Ferrario CM. Inhibition of angiotensin-converting enzyme 2 exacerbates cardiac hypertrophy and fibrosis in Ren-2 hypertensive rats. Am J Hypertens 2010; 23: 687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Allred AJ, Diz DI, Ferrario CM, Chappell MC. Pathways for angiotensin-(1–7) metabolism in pulmonary and renal tissues. Am J Physiol Renal Physiol 2000; 279: F841–F850 [DOI] [PubMed] [Google Scholar]
- 19. Velez JC, Ierardi JL, Bland AM, Morinelli TA, Arthur JM, Raymond JR, Janech MG. Enzymatic processing of angiotensin peptides by human glomerular endothelial cells. Am J Physiol Renal Physiol 2012; 302:F1583–F1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walters PE, Gaspari TA, Widdop RE. Angiotensin-(1–7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension 2005; 45: 960–966 [DOI] [PubMed] [Google Scholar]
- 21. Widdop RE, Sampey DB, Jarrott B. Cardiovascular effects of angiotensin-(1–7) in conscious spontaneously hypertensive rats. Hypertension 1999; 34: 964–968 [DOI] [PubMed] [Google Scholar]
- 22. Benter IF, Diz DI, Ferrario CM. Cardiovascular actions of angiotensin-(1–7). Peptides 1993; 14: 679–684 [DOI] [PubMed] [Google Scholar]
- 23. Benter IF, Ferrario CM, Morris M, Diz DI. Antihypertensive actions of angiotensin-(1–7) in spontaneously hypertensive rats. Am J Physiol 1995; 269: H313–H319 [DOI] [PubMed] [Google Scholar]
- 24. Benter IF, Yousif MH, Anim JT, Cojocel C, Diz DI. Angiotensin-(1–7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Am J Physiol Heart Circ Physiol 2006; 290: H684–H691 [DOI] [PubMed] [Google Scholar]
- 25. Burgelova M, Vanourkova Z, Thumova M, Dvorak P, Opocensky M, Kramer HJ, Zelizko M, Maly J, Bader M, Cervenka L. Impairment of the angiotensin-converting enzyme 2-angiotensin-(1–7)-Mas axis contributes to the acceleration of two-kidney, one-clip Goldblatt hypertension. J Hypertens 2009; 27: 1988–2000 [DOI] [PubMed] [Google Scholar]
- 26. Al-Maghrebi M, Benter IF, Diz DI. Endogenous angiotensin-(1–7) reduces cardiac ischemia-induced dysfunction in diabetic hypertensive rats. Pharmacol Res 2009; 59: 263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Collister JP, Nahey DB. Simultaneous administration of Ang(1–7) or A-779 does not affect the chronic hypertensive effects of angiotensin II in normal rats. J Renin Angiotensin Aldosterone Syst 2010; 11: 99–102 [DOI] [PubMed] [Google Scholar]
- 28. Iyer SN, Yamada K, Diz DI, Ferrario CM, Chappell MC. Evidence that prostaglandins mediate the antihypertensive actions of angiotensin-(1–7) during chronic blockade of the renin-angiotensin system. J Cardiovasc Pharmacol 2000; 36: 109–117 [DOI] [PubMed] [Google Scholar]
- 29. Tallant EA, Lu X, Weiss RB, Chappell MC, Ferrario CM. Bovine aortic endothelial cells contain an angiotensin-(1–7) receptor. Hypertension 1997; 29: 388–393 [DOI] [PubMed] [Google Scholar]
- 30. Ferrario C. Effect of angiotensin receptor blockade on endothelial function: focus on olmesartan medoxomil. Vasc Health Risk Manag 2009; 5: 301–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ferrario CM, Smith RD. Role of olmesartan in combination therapy in blood pressure control and vascular function. Vasc Health Risk Manag 2010; 6: 701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cohn JN, Anand IS, Latini R, Masson S, Chiang YT, Glazer R. Sustained reduction of aldosterone in response to the angiotensin receptor blocker valsartan in patients with chronic heart failure: results from the Valsartan Heart Failure Trial. Circulation 2003; 108: 1306–1309 [DOI] [PubMed] [Google Scholar]
- 33. Yoneda T, Takeda Y, Usukura M, Oda N, Takata H, Yamamoto Y, Karashima S, Yamagishi M. Aldosterone breakthrough during angiotensin II receptor blockade in hypertensive patients with diabetes mellitus. Am J Hypertens 2007; 20: 1329–1333 [DOI] [PubMed] [Google Scholar]
- 34. Siragy HM, Inagami T, Carey RM. NO and cGMP mediate angiotensin AT2 receptor-induced renal renin inhibition in young rats. Am J Physiol Regul Integr Comp Physiol 2007; 293: R1461–R1467 [DOI] [PubMed] [Google Scholar]
- 35. Varagic J, Ahmad S, Brosnihan KB, Habibi J, Tilmon RD, Sowers JR, Ferrario CM. Salt-induced renal injury in spontaneously hypertensive rats: effects of nebivolol. Am J Nephrol 2010; 32: 557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kobori H, Prieto-Carrasquero MC, Ozawa Y, Navar LG. AT1 receptor mediated augmentation of intrarenal angiotensinogen in angiotensin II-dependent hypertension. Hypertension 2004; 43: 1126–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishiyama A, Seth DM, Navar LG. Angiotensin II type 1 receptor-mediated augmentation of renal interstitial fluid angiotensin II in angiotensin II-induced hypertension. J Hypertens 2003; 21: 1897–1903 [DOI] [PubMed] [Google Scholar]
- 38. Takahashi Y, Haga S, Ishizaka Y, Mimori A. Autoantibodies to angiotensin-converting enzyme 2 in patients with connective tissue diseases. Arthritis Res Ther 2010; 12: R85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burchill L, Velkoska E, Dean RG, Lew RA, Smith AI, Levidiotis V, Burrell LM. Acute kidney injury in the rat causes cardiac remodelling and increases angiotensin-converting enzyme 2 expression. Exp Physiol 2008; 93: 622–630 [DOI] [PubMed] [Google Scholar]
- 40. Arumugam S, Thandavarayan RA, Palaniyandi SS, Giridharan VV, Arozal W, Sari FR, Soetikno V, Harima M, Suzuki K, Kodama M, Watanabe K. Candesartan cilexetil protects from cardiac myosin induced cardiotoxicity via reduction of endoplasmic reticulum stress and apoptosis in rats: involvement of ACE2-Ang (1–7)-mas axis. Toxicology 2012; 291: 139–145 [DOI] [PubMed] [Google Scholar]
- 41. Lew RA, Warner FJ, Hanchapola I, Yarski MA, Manohar J, Burrell LM, Smith AI. Angiotensin-converting enzyme 2 catalytic activity in human plasma is masked by an endogenous inhibitor. Exp Physiol 2008; 93: 685–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Casarini DE, Boim MA, Stella RC, Schor N. Endopeptidases (kininases) are able to hydrolyze kinins in tubular fluid along the rat nephron. Am J Physiol 1999; 277: F66–F74 [DOI] [PubMed] [Google Scholar]
- 43. Santos RA, Brosnihan KB, Jacobsen DW, DiCorleto PE, Ferrario CM. Production of angiotensin-(1–7) by human vascular endothelium. Hypertension 1992; 19: II56–II61 [DOI] [PubMed] [Google Scholar]
- 44. Welches WR, Brosnihan KB, Ferrario CM. A comparison of the properties and enzymatic activities of three angiotensin processing enzymes: angiotensin converting enzyme, prolyl endopeptidase and neutral endopeptidase 24.11. Life Sci 1993; 52: 1461–1480 [DOI] [PubMed] [Google Scholar]
- 45. Gonzalez-Villalobos R, Klassen RB, Allen PL, Johanson K, Baker CB, Kobori H, Navar LG, Hammond TG. Megalin binds and internalizes angiotensin-(1–7). Am J Physiol Renal Physiol 2006; 290: F1270–F1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hosojima M, Sato H, Yamamoto K, Kaseda R, Soma T, Kobayashi A, Suzuki A, Kabasawa H, Takeyama A, Ikuyama K, Iino N, Nishiyama A, Thekkumkara TJ, Takeda T, Suzuki Y, Gejyo F, Saito A. Regulation of megalin expression in cultured proximal tubule cells by angiotensin II type 1A receptor- and insulin-mediated signaling cross talk. Endocrinology 2009; 150: 871–878 [DOI] [PubMed] [Google Scholar]
- 47. Whaley-Connell A, Habibi J, Nistala R, Hayden MR, Pulakat L, Sinak C, Locher B, Ferrario CM, Sowers JR. Combination of direct renin inhibition with angiotensin type 1 receptor blockade improves aldosterone but does not improve kidney injury in the transgenic Ren2 rat. Regul Pept 2012; 176: 36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]