

Abstract
Human induced pluripotent stem cells (hiPSCs) derived from patient samples have tremendous potential for innovative approaches to disease pathology investigation and regenerative medicine therapies. However, most hiPSC derivation techniques utilize integrating viruses, which may leave residual transgene sequences as part of the host genome, thereby unpredictably altering cell phenotype in downstream applications. Here we describe a protocol for hiPSC derivation by transfection of a simple, nonviral minicircle DNA construct into human adipose stromal cells (hASCs). Minicircle DNA vectors are free of bacterial DNA and thereby capable of high expression in mammalian cells. Their repeated transfection into hASCs, an abundant somatic cell source that is amenable to efficient reprogramming, results in transgene-free hiPSCs. This protocol requires only readily available molecular biology reagents and expertise, and produces hiPSC colonies from an adipose tissue sample in ~4 weeks.
Keywords: induced pluripotent stem (iPS) cells, reprogramming, minicircle DNA, human adipose stem cells
INTRODUCTION
Human induced pluripotent stem cells (hiPSCs) can be derived from somatic cells through a reprogramming process driven by overexpression of a defined set of transcription factors [1, 2]. These hiPSCs share the properties of self-renewal and pluripotency with human embryonic stem cells (hESCs), and can therefore be used to generate unlimited quantities of differentiated cell types of all three germ layers, including cardiac cells, neural cells, and hepatic cells. hiPSCs can be generated from patients of virtually any genetic background, including those with disease-conferring genetic mutations [3–5]. In contrast, the derivation of hESCs from different genetic backgrounds is challenging because human embryo use is limited and ethically debated. The production of patient-specific and disease-specific hiPSCs enables a variety of downstream applications, including drug screening, disease modeling, pathogenesis studies, and regenerative medicine therapies.
However, traditional approaches to deriving hiPSCs require use of retroviruses or lentiviruses that integrate reprogramming genes into the host genome. Random integration of the reprogramming genes may result in insertional mutagenesis that causes malignant transformation of a clonal cell population [6]. In addition, some of the genes used in the reprogramming process are known proto-oncogenes, and incomplete silencing of these transgenes may result in unknown adverse effects. These challenges have been partially circumvented in mice by the development of non-integrating viral [7], non-viral episomal [8], and excisional [9, 10] techniques for reprogramming. Yet clinical translation of these safer iPSC derivation techniques is challenging because human cells are relatively more resistant to non-viral transfection and are not immediately available in large quantities. Although hiPSCs may be generated by lentiviral transduction with subsequent Cre-loxP excision of reprogramming factors [3], residual vector sequences will be left behind in the genome. Transgene-free hiPSCs have been derived from neonatal foreskin fibroblasts using a combination of three episomal plasmids expressing seven reprogramming factors [11]. Alternatively, transgene-free hiPSCs can be derived from fetal or neonatal cells by repeated transduction of proteins in the presence of chemical treatments (e.g., valproic acid) [12]. However, none of the aforementioned techniques for transgene-free hiPSC derivation have been demonstrated using adult donors, a more clinically relevant population. Here, we describe in detail a protocol for the derivation of transgene-free hiPSCs with a non-viral minicircle DNA reprogramming construct used in conjunction with human adipose stromal cells (hASCs) [13]. This technique is advantageous in translational studies because somatic cells from human adults can be reprogrammed in the absence of genomic modification, viral sequences, or proto-oncogenes (such as c-Myc), effectively mitigating safety concerns [14]. This protocol can be used to derive hiPSCs from human samples in ~4 weeks using standard molecular biology reagents and cell culture expertise (Figure 1).
Figure 1.
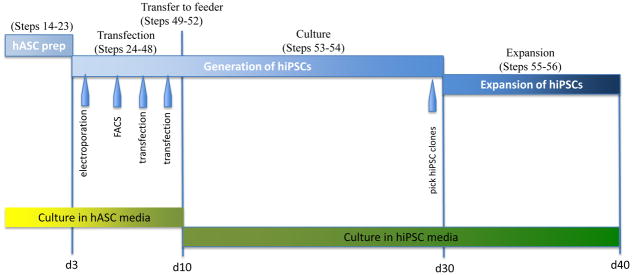
Schematic of hiPSC derivation protocol. Approximate time table of the hiPSC derivation process is shown with numbered steps above and cell culture media below.
Limitations of the protocol
Cells are not transduced with infectious viral particles in this protocol, ensuring a high likelihood of generating transgene-free hiPSCs. However, reprogramming efficiency using this protocol is substantially lower (~0.005%) compared to lentiviral techniques for overexpression of the transcription factors OCT4, SOX2, NANOG, and LIN-28. Further improvement of reprogramming efficiency may be achieved by treatment with small molecules (e.g. valproic acid) [15] or cell signaling peptides (Wnt) [16]. Also, users should be aware that the protocol as described here has not yet been successfully applied to the reprogramming of human dermal fibroblasts derived from adult sources. We have found that minicircle-based reprogramming of hASCs as described here is advantageous for deriving transgene-free hiPSCs from adult human donors, a clinically relevant cell source. Such methods offer the ability to develop patient-specific or disease-specific cell lines for exciting new translational and disease modeling studies.
EXPERIMENTAL DESIGN
Adipose Tissue Harvest
hASCs are an attractive source for hiPSC derivation because they are available in large numbers and are amenable to reprogramming [17, 18]. While skin biopsies are necessarily minimal in size, adipose tissue can be harvested in very large quantities during lipoaspiration procedures, allowing quick expansion to a large starting population of hASCs. For example, reprogramming can start as early as 24 hours after the liposuction procedure with a large starting cell population of up to 10 million cells, whereas 3–4 weeks are required following punch skin biopsy for expansion of the dermal fibroblasts. hASCs are a heterogeneous group of multipotent progenitor cells that can differentiate into adipogenic, osteogenic, chondrogenic, and myogenic cell lineages [19, 20]. As such, hASCs express relatively high levels of genes such as c-Myc and Klf4, which may underlie the relative ease of their induction to pluripotency. Users should note that the protocol described here has been most successfully applied when using hASCs as the starting cell source, although neonatal fibroblasts (IMR90) can also be used but with lower efficiency.
Minicircle vectors
Minicircle vectors are supercoiled DNA molecules free of bacterial plasmid backbone elements, such as an origin of replication and antibiotic resistance gene. They primarily consist of a eukaryotic expression cassette, and therefore do not activate exogenous silencing mechanisms to the same extent as plasmids. Therefore, minicircle vectors benefit from higher transfection efficiences and more stable ectopic transgene expression than plasmid DNA (Figure 2) [21]. The pMC.LGNSO plasmid is the parental DNA construct that is used to produce minicircle reprogramming vector in the initial steps of this protocol (Steps 1–13). Parental plasmid DNA (e.g. pMC.LGNSO) that produces minicircle vectors may be conceptually divided into two parts separated by attB and attP recognition sequences. Intermolecular recombination between the attB and attP sequences catalyzed by the ΦC31 integrase (Step 9) yields a minicircle vector separate from the remainder of the plasmid (Figure 3). The minicircle vector contains the reprogramming genes OCT4, SOX2, NANOG and LIN-28. This expression cassette is isolated and purified from the bacterial suspension (Steps 1–12) before being transfected into somatic cells to induce reprogramming (Step 13–47). The other part of the pMC.LGNSO parental plasmid contains bacterial plasmid backbone elements (e.g., the origin of replication and antibiotic resistance cassette), as well as the ΦC31 integrase and I-SceI restriction enzyme expression cassettes under the control of an L-arabinose inducible promoter. This part of the plasmid is linearized by I-SceI endonucleolytic cleavage and subsequently degraded (Step 9), allowing isolation of pure minicircle DNA by common plasmid purification procedures (e.g., Qiagen Plasmid Purification Kits). Yield and purity of the minicircle DNA preparation may be optimized by use of plasmids encoding multiple copies of ΦC31 integrase [22], or using E. coli strains that encode L-arabinose-inducible I-SceI as part of the bacterial genome (e.g., E. coli strain ZYCY10P3S2T). The purified minicircle expression cassette contains the four reprogramming genes plus GFP separated by 2A self-cleaving peptide sequences, thereby allowing for the equimolar expression of all five proteins from a single RNA transcript (Fig. 3). This protocol requires transfection of hASCs with the minicircle preparation three times; first via electroporation to optimize efficiency, followed by flow cytometry sorting to enrich for successfully transfected cells, followed by two rounds of transfection mediated by cationic lipids (e.g., using Lipofectamine) to optimize cell survival.
Figure 2.

Minicircle DNA vectors give stronger and more persistent transgene expression than regular plasmids. (a) hASCs transfected with either minicircle or regular plasmid carrying identical expression cassettes (CMV promoter driving eGFP – firefly luciferase fusion gene). Representative bioluminescent images are shown of in vitro cell cultures at the indicated time points after transfection. (b) Quantitative photon counts demonstrate increased luciferase expression in minicircle-transfected cells compared to regular plasmid-transfected cells over 72 hours. (c) Quantitative PCR for GFP shows persistent high-level expression of the minicircle transgene compared to plasmid over 12 days. For both experiments, the error bars represent the standard deviation from three independent experiments. Reproduced with permission from reference 13.
Figure 3.

Minicircle expression vector for hiPSC generation. After induction with L-arabinose, expression of ΦC31 integrase catalyzes intermolecular recombination between the attB and attP recognition sites, resulting in a minicircle DNA vector separated from plasmid backbone elements. Expression of I-SceI catalyzes linearization of the plasmid backbone, and it is subsequently degraded. The minicircle vector contains a CMV promoter driving expression of GFP, OCT4, SOX2, NANOG, and LIN-28 cDNAs separated by 2A self cleavage peptide sequences. The SV40 polyA (pA) site ensures transcription termination, polyadenylation and efficient expression of the transcript. Arrows above the minicircle vector show primers #2and #5 (Table 1 ) to be used for screening of the vector’s integration into genomic DNA.
Controls: FACS
Detailed description of the flow cytometric enrichment of GFP+ hASCs after the first electroporation (Step 37) will vary by institution and is beyond the scope of this protocol. Fortunately, well-established protocols for sorting of GFP+ cells are readily available [23, 24]. For the initial sorting procedure, 5 × 105 untransfected hASCs should be trypsinized and resuspended in FACS buffer alongside the transfected hASCs in order to set up the gating parameters. Once set, the gating parameters can be saved and untransfected hASCs are no longer needed. We used a FACSAria equipped with FACSDiva software for cell analysis and sorting (http://facs.stanford.edu). Forward scatter (~82V) and side scatter (~176 V) were adjusted to exclude cell aggregates and debris. The band pass filter for FITC detection was used (~537 V), and cells were analyzed at a flow rate between 200 and 1,000 events per second in a stream formed from a nozzle with a 70 micron orifice.
Picking colonies
hiPSC-like colonies with a tightly-packed, dome-like structure first appear at approximately day 18 (Steps 53–54; Figure 4). Colonies become large enough to be manually picked up and transferred to a separate irradiated mouse embryonic fibroblast (iMEF) feeder layer at approximately day 28 (Steps 55–56). At early stages, colonies remain GFP+, although with continued culture and passaging, bona fide hiPSC clones will become GFP-.
Figure 4.

Characterization of developing and bona fide hiPSCs. (a) Brightfield image of untransfected hASCs. (b) Brightfield (upper) and fluorescence (lower) images of reprogramming hASCs at days 7, 12, and 18 post-transfection. (c) Flow cytometric analysis of sorted hASCs after transfection with the minicircle vector on day 0. Percentage of GFP+ cells is plotted against days post-transfection. (d) Representative images of minicircle-derived hiPSCs immunostained for pluripotency markers as described in steps 52-61. Nuclear antigens requiring permeabilization, such as Oct4, Nanog, and Sox2, were stained using AlexaFluor546-conjugated secondary antibody (red emission), while cell surface antigens Tra-1-60, Tra-1-81, and SSEA4 were stained using AlexaFluor488-conjugated secondary antibody (green emission). Merged images (far right) show localization of Tra-1-60, Tra-1-81, and SSEA4 antigens (green) to the cell surface in contrast to the nuclear DAPI stain (blue). (e) qPCR analysis of three undifferentiated minicircle-derived hiPSC lines compared to H9 hESCs, hASCs, and 293-MC negative control (HEK 293 cells 24 h after transfection with the minicircle vector). Endogenous (endo) OCT4, SOX2, NANOG, and LIN-28 were analyzed with 18S RNA as a control. Genomic DNA was also isolated to screen for integration of the minicircle vector using primers specific for the Oct4-Sox2 transgene as described (bottom). Reproduced with permission from reference 13.
Controls: Screening of the resulting hiPSC lines
Expression of the minicircle-derived reprogramming cassette can be easily monitored throughout the procedure by GFP fluorescence. At the conclusion of the procedure, however, bona fide hiPSC colonies will no longer be dependent on exogenous reprogramming factor expression, and will thereby be GFP negative. The pluripotency of the derived hiPSC lines can be verified using standard protocols, including RT-PCR and immunocytochemistry for pluripotent markers, embryoid body differentiation, and teratoma formation, that are described in further detail elsewhere [25]. In addition to the standard protocols above, the resulting hiPSCs can be screened for rare integration events by a simple PCR-based assay for the presence of the transgene in genomic DNA. The minicircle-derived reprogramming cassette contains the LIN-28, NANOG, SOX2, and OCT4 cDNA sequences in succession (Figure 3), while endogenous genomic DNA loci expressing these genes contain introns and are present on disparate chromosomes. Therefore, PCR of extracted genomic DNA using primers annealing to two contiguous minicircle reprogramming genes (e.g. SOX2 and OCT4) can readily identify rare integration events of the transgene into genomic DNA (Steps 57–60).
MATERIALS
Reagents
pMC.LSNGO plasmid (available upon request or from StemCell Technologies, Vancouver, Canada, cat. no. 05820)
ZYCY10P3S2T E. coli strain (available upon request) or TOP10 Competent cells (Invitrogen, cat. no. 4040) Critical Use these designated E. coli strains to achieve optimal yield of minicircle DNA.
Terrific Broth (TB) Powder (Invitrogen, cat. no. 22711-022)
Luria-Bertani Broth Powder (Invitrogen, cat. no. 12780-052)
LB-Agar Plates for Kanamycin-resistant E. coli strains (Invitrogen, cat. no. 45-0043)
1N NaOH solution (Agilent Technologies, cat. no. 5062-8576) Caution Causes burns; harmful when swallowed; toxic when in contact with skin and eye; use protective gloves and safety glasses when handling.
L-arabinose (Sigma, cat. no. A3256)
Kanamycin Sulfate Solution 10 mg/ml (Invitrogen, cat. no. 15160)
Opti-MEM I reduced-serum medium (Invitrogen, Gibco, cat. no. 31985)
Lipofectamine 2000 (Invitrogen, cat. no. 11668-019)
DMEM, high glucose with L-glutamine (Invitrogen, Gibco, cat. no. 11965)
FBS (Invitrogen, Gibco, cat. no. 10437)
FBS, ES-Cell (Invitrogen, Gibco, cat. no. 16141)
Penicillin/streptomycin (Invitrogen, Gibco, cat. no. 15070)
Nonessential amino acids (Invitrogen, cat. no. 11140-050)
2-mercaptoethanol (Invitrogen, cat. no. 21985-023) Caution Flammable; harmful if swallowed; toxic when in contact with skin and eye; use protective gloves and safety glasses when handling. Gelatin (Sigma, cat. no. G1890)
Knockout DMEM (Invitrogen, Gibco, cat. no. 10829)
PBS, without Ca2+ and Mg2+ (Invitrogen, Gibco, cat. no. 10010)
Hank’s Balanced Salt Solution (HBSS, Invitrogen, cat. no. 14025)
FACS Buffer, PBS-based (Gibco, cat. no. 13150)
Bovine serum albumin (Sigma, cat. no. A9418)
Triton X-100 (Sigma, cat. no. T8787)
Antibiotic/antimycotic solution, 100 × (Mediatech, cat. no. 30-004-CI)
Type II Collagenase (Invitrogen, cat. no. 17101-015)
Irradiated mouse embryonic fibroblast (MEF) feeder cells from CF1 mice (GlobalStem, cat. no. GSC-6001G)
New England Biolabs Restriction Endonuclease HindIII, SphI, NdeI, or XhoI (see Step 1)
BlueJuice 10 × DNA loading dye (Invitrogen, cat. no. 10816-015)
TrackIt 1kb+ DNA Ladder (Invitrogen, cat. no. 10488-085)
Reagent Setup
Minicircle induction broth
First prepare a stock of 20% L-arabinose by dissolving 2 g L- arabinose in 100 ml of ddH2O. Freeze stock at -20°C for up to 6 months. To prepare induction broth, mix 384 ml LB, 16 ml 1N NaOH, and 0.4 ml 20% L-arabinose (final concentration of L- arabinose is 0.01%). Make fresh just prior to use. hASC medium: DMEM High Glucose with L-Glutamine containing 10% (vol/vol) FBS, 0.5% (vol/vol) penicillin/streptomycin (final concentration 50 units/mL penicillin and 50 ug/mL streptomycin). Store at 4 °C for ~1 month.
hiPSC medium
Knockout DMEM, 20% (vol/vol) ES Cell FBS, 0.1 mM nonessential amino acids, 0.1 mM 2-mercaptoethanol, and bFGF to final concentration of 8 ng/mL. Store at 4 °C for ~2 weeks.
0.075% Type II Collagenase
Add 75 mg of Type II Collagenase to 100 ml of ddH2O (0.075% wt/vol) and filter sterilize using a 0.44μm vacuum-driven filter system. Make 10 ml aliquots of solution and store at −20 °C for up to 2 months.
Gelatin solution
Dissolve 0.5 g gelatin powder in 500 ml of distilled water, autoclave and store at 4 °C for up to 2 months.
Gelatin-coated culture dishes
Add a sufficient volume of gelatin solution to cover the surface of the plate or well. Incubate the dish for at least 30 minutes at 37 °C. Prior to use, aspirate excess gelatin solution.
Equipment
Qiagen HiSpeed Plasmid Maxi Kit (Qiagen, cat. no. 12662)
ArchivePure DNA Cell/Tissue Kit (5 Prime, cat. no. 2300810)
Cell culture incubator, 95% air and 5% CO2, humidified
Cell culture hood
Automated cell counter (e.g., Countess, Invitrogen)
Inverted epifluorescence microscope
Fluorescence activated cell sorter (FACSAria, BD Biosciences)
5 ml round-bottom tube (BD Falcon, cat. no. 352063)
1.5 ml microcentrifuge tubes
pH meter
Cuvette spectrophotometer (e.g., Eppendorf BioPhotometer)
2-liter Erlenmeyer glass flask
150-mm tissue culture dish (Falcon, cat. no. 353025)
100-mm tissue culture dish (Falcon, cat. no. 353003)
6-well tissue culture dish (Falcon, cat. no. 353046)
15-ml conical tube (Falcon, cat. no. 352196)
50-ml conical tube (Falcon, cat. no. 352070)
100 μm cell strainer (Falcon, cat. no. 352360)
Lonza Nucleofector® Kit R (cat. no. VCA-1001)
Lonza Nucleofector® Device Critical: The parameters of the nucleofection programs are proprietary. We have been unable to obtain high transfection efficiencies using other electroporation systems (e.g. BioRad GenePulser).
Dissecting forceps. Caution Sterilize by autoclave.
Accuprime High Fidelity Taq Polymerase (Invitrogen, cat. no. 12346-086)
ES Cell Marker Characterization Kit (Millipore, cat. no. SCR002)
Formaldehyde solution (Sigma, cat. no. F8775)
Qiagen RNeasy Mini Kit (Qiagen, cat. no. 74104)
iScript cDNA Synthesis Kit (BioRad, cat. no. 170-8890)
GoTaq qPCR Master Mix (Promega, cat. no. A6001)
StepOnePlus Real-Time PCR System (Applied Biosystems, cat. no. 4376600)
PROCEDURE
Minicircle DNA production (3 days)
-
1)
Verify the identity and integrity of the pMC.LGNSO plasmid by restriction digestion. Incubate a mixture of the following reagents in a 1.5 ml microcentrifuge tube at 37 °C for >1 hour:
pMC.LGNSO plasmid 1 μg NEB Buffer 2 μl 10 × BSA 2 μl restriction endonuclease (HindIII/SphI/NdeI/XhoI) 0.5 μl ddH2O up to 20 μl -
2)
Add 2 μl of 10 × loading dye to the above restriction digestion mixture. Load the entire mixture into the well of a 0.8% agarose gel. Load ~5 μl of a DNA ladder of choice (e.g. TrackItR 1kb+) into a neighboring well. Electrophorese at constant voltage of 100 V for 70 minutes. Verify that the following double cutters produce restriction fragment lengths as shown below. If further confirmation of the identity of the plasmid is desired, it can be sequenced using primer #17 (Table 1).
Restriction endonuclease Fragment size (bp) HindIII 2091, 2659 SphI 1800, 3950 NdeI 1030, 4720 XhoI 369, 5381 -
3)
Begin transforming E. coli strain (ZYCY10P3S2 or TOP10) with pMC.LGNSO plasmid by adding 100 pg −50 ng of plasmid to a 50 μl aliquot containing 1 × 109 cfu of chemically competent E. coli. Place on ice for 10 minutes.
-
4)
Heat shock in 42 °C water bath for 30 seconds. Then, place on ice for 2 minutes.
-
5)
Spread transformed E. coli on a pre-warmed LB-agar kanamycin-selective plate. Incubate overnight 12-16 hours at 37 °C.
-
6)
Next day, inoculate 5 ml of LB Broth containing kanamycin (50 μg/ml) with a colony from the freshly streaked plate. Incubate this starter culture at 37 °C with agitation at 250 rpm for 8 hours.
-
7)
Autoclave 400 ml of TB in a 2 liter Erlenmeyer flask. When flask is cool enough to touch (~55 °C), add 0.2 ml of 10 mg/ml kanamycin sulfate solution to a final concentration of 50 μg/ml.
-
8)
Inoculate 100 μl of the starter culture of step 5 into 400 ml of TB prepared in step 6 and incubate overnight 16-18 hrs at 37 °C with shaking at 250 rpm.
-
9)
Next day, take a 0.5 ml sample of the overnight bacterial culture and confirm that the OD600 reading is between 4-5, and the pH is 6.5.
-
10)
Combine 400 ml of the Minicircle Induction Broth (see Reagent Setup) with 400 ml of the overnight culture into a 2 L Erhlenmeyer flask. Incubate at 32 °C with shaking at 250 rpm for greater than 5 hours.
Critical Step: Longer incubation periods of up to 9 hours will yield minicircle DNA of greater purity.
-
11)
Pellet bacterial cells by centrifugation at 6,000 g for 15 minutes at 4 °C.
-
12)
Discard supernatant and resuspend the bacterial pellet in 100 ml of Qiagen Buffer P1. Follow the Maxi Prep protocol per the manufacturer’s instructions, but use double the volume of buffers P1, P2, and P3 (100 ml each). Elute DNA using 200 μl of Buffer EB or Tris-EDTA buffer pH 8.
-
13)
Measure DNA concentration and purity using a spectrophotometer. Minicircle DNA is now ready for subsequent transfection. For further confirmation of the identity and integrity of minicircle DNA preparation, dilute a sample of 0.5 μg in an appropriate volume of 10 × DNA loading dye and electrophorese on a 0.8% agarose gel at 100 V for 70 minutes alongside a DNA ladder. Verify that a clean, solitary 5.6 kb band is present with no smaller bands indicating nicked or degraded DNA.
Pause Point: Minicircle DNA preparations can be frozen in 5 μg aliquots at -20 °C indefinitely. Avoid repeated freeze-thaw cycles.
Table 1.
Primer sequences. Primers #1-16 may be used for qPCR analysis of resulting hiPSC clones. Primer pairs specific for the endogenous transcript (#3–4, 7–8, 11–12, 15–16) anneal to the untranslated region (UTR) of the transcript, which is absent on the minicircle-derived transcript. Used in conjunction, primers #2 and #5 amplify a specific 1.1kb region that is unique to the minicircle construct as shown in Figure 3. These two primers can be used to screen genomic DNA for integration of the minicircle-derived transgene as described (Steps 62-65). Primer #17 anneals to the CMV promoter transcriptional start site and may be used for sequence verification of the pMC.LGNSO plasmid.
| # | Name | Direction | Sequence | Size |
|---|---|---|---|---|
| 1 | Oct4 | 352F | ACCCCTGGTGCCGTGAA | 189 bp |
| 2 | 541R | GGCTGAATACCTTCCCAAATA | ||
| 3 | Oct4 endo | 912F | GCGATCAAGCAGCGACTA | 399 bp |
| 4 | 1311R | TTCACCTTCCCTCCAACC | ||
| 5 | Sox2 | 890F | CAGCGCATGGACAGTTAC | 320 bp |
| 6 | 1210R | GGAGTGGGAGGAAGAGGT | ||
| 7 | Sox2 endo | 1183F | CCCTGTGGTTACCTCTTCC | 258 bp |
| 8 | 1441R | CTCCCATTTCCCTCGTTT | ||
| 9 | Nanog | 433F | AAAGGCAAACAACCCACT | 269 bp |
| 10 | 702R | GCTATTCTTCGGCCAGTT | ||
| 11 | Nanog endo | 1197F | CTCCTCCCATCCCTCATA | 105 bp |
| 12 | 1302R | AGGCTCCAACCATACTCC | ||
| 13 | Lin28 | 270F | GTTCGGCTTCCTGTCCAT | 121 bp |
| 14 | 391R | CTGCCTCACCCTCCTTCA | ||
| 15 | Lin28 endo | 1135F | AGCCAAGCCACTACATTC | 299 bp |
| 16 | 1434R | AGATACGTCATTCGCACA | ||
| 17 | CMV seq | F | AAGCAGAGCTGGTTTAGTGAA |
Derivation of human adipose stromal cells (days 1-3)
-
14)
Obtain an adipose tissue sample by liposuction aspiration.
Caution Adipose tissue should be obtained by a surgeon or other appropriately trained physician. Informed consent should be obtained from the individual in accordance with institutional and international patient protection guidelines.
-
15)
Immediately place the adipose tissue specimen on ice.
-
16)
Prepare four 50 ml conical tubes containing 25 ml of serial dilutions of Povidine-iodine (Betadine) and PBS in ratios of 1:0 (undiluted Betadine), 1:1, 1:2 and 1:5. Wash the specimen by immersing it sequentially in each dilution for 5 seconds each.
-
17)
Wash the specimen twice by immersion in 25 ml PBS in two separate 50 ml conical tubes.
-
18)
Place the specimen in a fresh 50 ml conical tube and measure the volume of the tube occupied by adipose tissue (~10–15 ml). Digest tissue by adding an equal volume of 0.075% Type II collagenase (see REAGENT SETUP). Incubate digestion mixture in a 37 °C water bath with agitation at 125 rpm for 30 minutes.
-
19)
Inactivate collagenase by adding an equal volume of serum-containing hASC growth medium (see REAGENT SETUP).
-
20)
Pellet the stromal vascular fraction containing hASCs via centrifugation at 1,200 g for 5 minutes.
-
21)
Aspirate supernatant. Resuspend the pellet in 5 mL of hASC medium. Count the cells using an automated cell counter to verify that cell yield is ~1–5 × 106 cells.
-
22)
Filter the resuspended cells through a 100 μm cell strainer into a 15 ml conical tube.
-
23)
Add hASC medium to a total volume of 15 mL. Seed entire 15 mL hASC cell suspension (~1–5 × 106 cells) on a 150-mm tissue culture dishes for further expansion in a humidified incubator at 37 °C, 5% CO2.
Critical Step: hASCs can be passaged and expanded using standard cell culture techniques [26]. Our lab uses TrypLE Express reagent for digestion and split ratios of 1:4 every 3–5 days. hASCs may be frozen in a cryopreservation solution (10% DMSO, 90% FBS) in aliquots of 2 × 105 per cryovial and immersed in liquid nitrogen indefinitely.
Critical Step: For optimal hiPSC generation efficiency, electroporate hASCs within three passages to minimize replicative senescence.
Induction of pluripotency: hASC transfection #1 (day 4-6)
-
24)
Aspirate hASC medium from 150-mm dish containing hASCs at ~70% confluency. Add 10 ml of PBS and rock plate to wash. Aspirate PBS.
-
25)
Add 2 ml TrypLE Express. Incubate at 37 °C for 2–5 minutes until cells round up and detach.
-
26)
Add 4 ml of hASC growth medium to stop enzymatic digestion. Pipet cell suspension up and down to wash all hASCs off the plate. Pipet the cell suspension into a 15 ml conical tube. Wash the 150-mm dish with another 4 ml of hASC medium and add to the 15 ml conical tube. Count cells using an automated cell counter.
-
27)
Pellet 1–2 × 106 hASCs by centrifugation at 200 g x 5 minutes.
-
28)
Aspirate supernatant completely. Gently resuspend the hASC pellet in 100 μl of Nucleofector Solution R.
Critical Step: Ensure that Nucleofector Solution R is at room temperature (25 °C). Ensure that supplement has been added to solution per manufacturer’s instructions.
-
29)
Add 5 μg of minicircle DNA from Step 13 to the cell suspension.
-
30)
Gently transfer the cell + DNA suspension to a cuvette using a micropipette. Close the cuvette with the cap. Place the cuvette in the Lonza Nucleofector device and execute program U-023.
-
31)
Gently remove the cuvette from the device after completion of the program.
-
32)
Add ~500 μl of hASC medium (preferably pre-warmed to 37 °C) to the cuvette using the plastic pipette. Then use the same plastic pipette to gently transfer the cell suspension to a 150-mm dish. Avoid repeated aspiration of the sample. Carefully distribute the cells on the 150-mm dish by gentle rocking. Place in 37 °C incubator for expansion.
-
33)
Next day, aspirate hASC medium containing non-viable cells. Add fresh hASC growth medium. Place 150-mm dish in 37 °C incubator.
-
34)
Next day, aspirate hASC medium. Wash with PBS.
-
35)
Add 2 ml of TrypLE Express. Incubate at 37 °C for 2–5 minutes until cells round up and detach from the plate.
-
36)
Add 4 ml of hASC medium. Pipet cell suspension up and down to wash all hASCs off the plate. Collect cell suspension into a 15 ml conical tube. Pellet cells by centrifugation at 200 g for 5 minutes. Aspirate supernatant. Resuspend cell pellet in 1 ml of FACS buffer supplemented with 2% FBS.
-
37)
Isolate 1 × 105 GFP+ cells by flow cytometry [23, 24]. Set the sorting gate specifically for green fluorescence with a 488-nm laser for GFP, and maximize yield of GFP+ cells (approximately 2-10% of the population). Pellet sorted cells by centrifugation at 1,200 g for 5 minutes. Aspirate FACS buffer supernatant. Resuspend GFP+ hASC pellet in 4 ml hASC medium. Split cells into 2 wells of a 6 well tissue culture plate (2 ml per well). Place in 37 °C incubator overnight.
Critical Step: Return hASCs to a humidified 37 °C incubator as quickly as possible to maximize cell viability. Be sure to have added penicillin/streptomycin antibiotic to media to reduce risk of contamination after cell sorting procedure.
?Troubleshooting
Induction of pluripotency: hASC transfection #2 (day 7-8)
-
38)
Dilute 10 μg of minicircle DNA from Step 13 in 500 μl of Opti-MEM in a 1.5 ml microcentrifuge tube.
-
39)
Add 20 μl of Lipofectamine 2000 to 500 μl of Opti-MEM in a separate 1.5 ml microcentrifuge tube. Incubate at room temperature for 5 minutes.
-
40)
Add the Lipofectamine-containing solution to the DNA-containing solution gently and incubate at room temperature for 15–20 minutes.
-
41)
Aspirate hASC medium from the 6-well plate containing hASCs from Step 37. Add 2 ml PBS per well and rock plate back and forth to wash. Aspirate PBS. Then add 1 ml of fresh hASC medium per well.
-
42)
Add 500 μl of the Lipofectamine-DNA solution to each well. Media does not need to be refreshed, but if desired, wait until at least 4 hours have elapsed since addition of the Lipofectamine-DNA solution before refreshing the media. Incubate overnight in a humidified incubator at 37 °C.
-
43)
Next day, aspirate medium from the wells containing hASCs. Replace with 2 ml of fresh hASC medium. Return to incubator overnight.
Induction of pluripotency: hASC transfection #3 (day 9)
-
44)
Repeat steps 38-43. Figure 4B shows representative images of reprogramming hASCs during the transfection procedure. Approximately 20% of the hASCs will remain GFP+ by day 10 of the procedure (Figure 4C).
-
45)
Next day, thaw one vial containing 5 × 106 cryopreserved iMEFs (irradiated MEFs, isolated from CF1 mice) by gently swirling in 37 °C water bath until most, but not all, of the contents have thawed.
-
46)
Transfer 10 ml of hASC medium to an empty 15 ml conical tube. Slowly add iMEF cell suspension to the conical tube using a micropipettor.
-
47)
Centrifuge iMEF cell suspension at 200 g for 5 minutes at room temperature. Aspirate supernatant.
-
48)
Resuspend cells in 1 ml of hASC medium, then count cells using an automated cell counter (or hemacytometer). Add hASC medium to adjust cell concentration to 2 × 106 cells per ml. Plate 1 × 106 cells per 100-mm gelatin-coated dish. 2 × 106 cells is sufficient to prepare 2 × 6-well plates or 2 × 10 cm dishes or 2 × 12-well plate of 16 × 4-well plate (~120 cm2). Place in a humidified incubator at 37 °C overnight.
Induction of pluripotency: transfer to iMEF feeder layer (day 10)
-
49)
Next day, aspirate hASC medium from the 6-well plate containing hASCs from Step 43. Add 2 ml of PBS per well, wash by rocking back and forth, and aspirate PBS. Add 500 μl of TrypLE Express per well and incubate at 37 °C for 2-5 minutes until cells round up and detach.
-
50)
Aspirate hASC medium from the 100-mm gelatin-coated dish containing iMEFs. Add 6 ml of PBS, wash by rocking back and forth, and aspirate PBS. Add 8 ml of hiPSC medium to the dish.
-
51)
Add 500 μl of hiPSC medium to each well of the 6-well plate to stop enzymatic digestion. Gently pipet the solution over the surface of the well 5-6 times to detach all hASCs.
-
52)
Transfer the hASC cell suspension from the 6-well plate to the 100-mm dish containing the iMEF feeder layer. Rock the cell suspension in several quick back and forth motions, then carefully place the 100-mm dish in the humidified incubator at 37 °C overnight.
? Troubleshooting
Culturing reprogramming hASCs on iMEF feeder layer (day 11–30)
-
53)
Aspirate and replace the 100-mm dish with 10 ml of fresh hiPSC media every 24 hours.
-
54)
Between days 23-30, colonies with morphologies similar to hESCs will begin to appear.
Picking and expanding hiPSC colonies (day 30–40)
-
55)
Track hiPSC-like colonies displaying a mosaic green fluorescence pattern under an epifluorescence microscope daily (Figure 4). Surrounding unreprogrammed or partially reprogrammed hASCs may appear GFP+. Five to 15 hiPSC colonies are expected from 2 × 105 GFP+ hASCs isolated during FACS (Step 37).
-
56)
When mosaic GFP colonies reach a size of 0.5-1.0 mm diameter, they can be cut into small clumps using a pulled Pasteur pipet. Subsequently, they can be picked up using a micropipettor set at 2 μl and transferred to the well of a 6-well plate containing fresh iMEFs for subcloning culture. This sublcloning procedure may be repeated until GFP+ cells are completely absent. hiPSC clones can then be cultured and expanded according to standard hESC/hiPSC culture techniques [27, 28].
?Troubleshooting
Screening of resulting hiPSC clones for genomic integration (1 day)
-
57)
Isolate genomic DNA from one well of a 50-70% confluent 6-well plate of hiPSCs using the ArchivePure DNA extraction kit according to the manufacturer’s instructions. Also isolate total DNA from a cell line (e.g., HEK 293 cells) that has been transfected with the minicircle reprogramming cassette as a positive control.
Pause Point: DNA preparations can be frozen at 4 °C for 3 months or −20 °C indefinitely. Avoid repeated freeze-thaw cycles.
-
58)
Add the following components to a 0.2 ml PCR tube:
-
59)
Run the PCR reactions in a thermal cycler programmed with the following settings:
94 °C for 3 minutes
35 cycles of : 94 °C for 15 seconds, 60 °C for 30 seconds, 68 °C for 1 minute
68 °C for 7 minutes
hold at 4 °C
-
60)
Perform electrophoresis with 25 μl of each of the above reactions on a 1% agarose gel. Fragment size for integrated minicircle transgene is 1,102 bp. Select clones that do not contain a band for integrated minicircle plasmid for further culture. Any clones containing the 1,102 bp PCR product have integrated the reprogramming factor transgenes and are therefore not suitable for downstream applications because their genomic integrity has been compromised.
Characterization of hiPSC clones: RT-PCR (1 day)
-
61)
Extract total RNA from one well of 50–70% confluent 6-well plate containing established hiPSC clones using the Qiagen RNeasy Mini Kit following the manufacturer’s instructions.
-
62)
Synthesize cDNA using the iScript cDNA Synthesis Kit per manufacturer’s instructions.
-
63)
Perform qPCR reactions using cDNA template as follows:
-
64)
Run qPCR reactions in thermal cycler or Real-Time PCR system using the following parameters (using SYBR Green settings):
95 °C for 2 minutes
30 cycles of: 95 °C for 15 seconds, 60 °C for 1 minute
hold at 4 °C
-
65)
Analyze CT values for gene expression fold change using the ΔΔCT method [29] or perform gel electrophoresis of PCR products on a 1% agarose gel (Figure 4).
Characterization of hiPSC clones: immunocytochemistry (1 day)
-
66)
Aspirate the medium from one well of 50-70% confluent 6-well plate of established hiPSCs.
-
67)
Wash the cells with 2 ml of PBS.
-
68)
Add 2 ml of PBS containing 10% formalin and fix the cells by incubation at room temperature for 10 minutes.
-
69)
Wash the cells once with 2 ml of PBS.
-
70)
Aspirate the PBS and add 1 ml of PBS containing 1% (w/v) bovine serum albumin (BSA) and 0.1% Triton X-100. Incubate 45 minutes at room temperature.
Critical Step: Exclude Triton X-100 if staining cell surface markers such as Tra-1-60, Tra-1-81, or SSEA-4. Triton X-100 is only necessary for staining of intracellular antigens such as Oct4, Nanog, and Sox2.
-
71)
Aspirate blocking solution and add 1 ml of the primary antibody diluted to the appropriate concentration. Incubate overnight at 4 °C.
-
72)
Wash the cells three times for 5 minutes each with PBS.
-
73)
Add 1 ml of secondary antibody conjugated to AlexaFluor488 or 546 supplemented with DAPI at 1 μg/ml and incubate for 45 minutes in the dark.
-
74)
Wash secondary antibody twice with 2 ml of PBS.
-
75)
Observe the cells with a fluorescent microscope equipped with the appropriate filters.
TIMING
Step 1–13: Preparation of minicircle DNA: 3 days
Step 14–23: Derivation of hASCs: 3 days
Step 24–48: Induction of pluripotency: 7 days
Step 49–52: Transfer to iMEF layer: 1 hour
Step 53–54: Culturing reprogramming hASCs on iMEF feeder layer: 20 days
Step 55–56: Picking and expanding hiPSC colonies: 10 days
Steps 57–60: Screening of resulting hiPSC clones: 1 day
Steps 61–65: Characterization of hiPSC clones: RT-PCR: 1 day
Steps 66–75: Characterization of hiPSC clones: immunocytochemistry: 1 day
TROUBLESHOOTING
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 37 | Low yield of GFP+ cells | Low transfection efficiency | Use high quality DNA preparations. Consider using an endotoxin removal kit to further purify the minicircle DNA preparation of contaminants that may limit efficient transfection. |
| Begin with higher numbers of starting cell source, up to 1 × 107 cells can be used during the initial transfection. | |||
| 52 | Cells do not attach | Trypsin cells damaged | Do not incubate hASCs in TrypLE Express for longer than 5 minutes. Cells can be gently scraped from the surface of the plate to aid detachment. |
| Cells experience excessive shear force | Pipette gently | ||
| 56 | Unstable hiPSC clones | Plasmid integration into genome | Use high quality DNA preparations. Store DNA at −20 °C and avoid repeated freeze- thaw cycles as this may cause strand nicking and degradation. |
| Partial reprogramming | Choose high quality clones. Colonies with good hESC morphology are tightly compacted with sharp borders and contain cells containing prominent, large, round nucleoli and scant cytoplasm. Use freshly prepared medium and ensure that the appropriate amount of bFGF has been added. |
ANTICIPATED RESULTS
Transfection efficiency can be monitored by GFP fluorescence under an inverted epifluorescence microscope or by flow cytometry. During the plasmid transfections, floating cells will appear because of the cytotoxicity of electroporation and Lipofectamine®. The initial nucleofection procedure has a transfection efficiency of 2-10%, and GFP fluorescence of the sorted population declines over time as shown in Figure 4C. Picked hiPSC clones will stain positively for pluripotency markers (Tra-1-60, SSEA-4), express high transcript levels of pluripotency genes (Oct4, Sox2, Nanog, Crypto), and display hESC-like morphology, such as large, round nucleoli and scant cytoplasm (Figure 4). We have successfully derived 22 hiPSC lines using this protocol from three adult donors. We calculate our reprogramming efficiency using this technique as ~0.005%, although any such calculation may be obviated by the splitting of cells during the procedure.
Acknowledgments
We are grateful to Ning Sun for expert assistance with cell culture techniques. We thank Zhi Ying Cheng for help with minicircle production techniques. We thank funding support from Howard Hughes Medical Institute (K.H.N.); Mallinckrodt Foundation, NIH DP2OD004437, RC1HL100490, Burroughs Wellcome Foundation, and American Heart Association 0970394N (J.C.W.); NIH R90 DK 07010301, NIH R21 DE018727, NIH R21 DE019274, NIH RC2DE020771, the Oak Foundation and the Hagey Laboratory for Pediatric Regenerative Medicine (M.T.L.); NIH RC1HL100490-02 (J.C.W. & M.T.L.); U01HL099776 (R.C.R.).
Footnotes
AUTHOR CONTRIBUTIONS
K.N., F.J., and J.C.W. prepared most of the paper. R.C.R., M.A.K., and M.T.L. provided advice and proofread the paper.
COMPETING INTERESTS
The authors declare that they have no competing financial interests.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 3.Soldner F, et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–77. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dimos JT, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–21. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 5.Lee G, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–6. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hacein-Bey-Abina S, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 7.Stadtfeld M, et al. Induced pluripotent stem cells generated without viral integration. Science. 2008;322(5903):945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okita K, et al. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322(5903):949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- 9.Kaji K, et al. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458(7239):771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woltjen K, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458(7239):766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu J, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324(5928):797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou H, et al. Generation of induced pluripotent stem cells using recombinant proteins. 2009;4(5):381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia F, et al. A nonviral minicircle vector for deriving human iPS cells. Nat Meth. 2010;7(3):197–199. doi: 10.1038/nmeth.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miura K, et al. Variation in the safety of induced pluripotent stem cell lines. Nat Biotech. 2009;27(8):743–745. doi: 10.1038/nbt.1554. [DOI] [PubMed] [Google Scholar]
- 15.Huangfu D, et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotech. 2008;26(7):795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marson A, et al. Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell. 2008;3(2):132–135. doi: 10.1016/j.stem.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun N, et al. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc Natl Acad Sci U S A. 2009;106(37):15720–5. doi: 10.1073/pnas.0908450106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aoki T, et al. Generation of induced pluripotent stem cells from human adipose-derived stem cells without c-MYC. Tissue Eng Part A. 2010;6:6. doi: 10.1089/ten.TEA.2009.0747. [DOI] [PubMed] [Google Scholar]
- 19.Guilak F, et al. Clonal analysis of the differentiation potential of human adipose-derived adult stem cells. J Cell Physiol. 2006;206(1):229–37. doi: 10.1002/jcp.20463. [DOI] [PubMed] [Google Scholar]
- 20.Bunnell BA, et al. Adipose-derived stem cells: Isolation, expansion and differentiation. Methods. 2008;45(2):115–120. doi: 10.1016/j.ymeth.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen ZY, et al. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther. 2003;8(3):495–500. doi: 10.1016/s1525-0016(03)00168-0. [DOI] [PubMed] [Google Scholar]
- 22.Chen ZY, He CY, Kay MA. Improved production and purification of minicircle DNA vector free of plasmid bacterial sequences and capable of persistent transgene expression in vivo. Hum Gene Ther. 2005;16(1):126–31. doi: 10.1089/hum.2005.16.126. [DOI] [PubMed] [Google Scholar]
- 23.Galbraith DW, Anderson MT, Herzenberg LA. Flow cytometric analysis and FACS sorting of cells based on GFP accumulation. Methods Cell Biol. 1999;58:315–41. doi: 10.1016/s0091-679x(08)61963-9. [DOI] [PubMed] [Google Scholar]
- 24.Pruitt SC, Mielnicki LM, Stewart CC. Analysis of fluorescent protein expressing cells by flow cytometry. Flow Cytometry Protocols. 2004:239–258. doi: 10.1385/1-59259-773-4:239. [DOI] [PubMed] [Google Scholar]
- 25.Ohnuki M, Takahashi K, Yamanaka S. Current Protocols in Stem Cell Biology. John Wiley & Sons, Inc; 2007. Generation and characterization of human induced pluripotent stem cells. [DOI] [PubMed] [Google Scholar]
- 26.Strutt B, Khalil W, Killinger D. Growth and Differentiation of Human Adipose Stromal Cells in Culture. Human Cell Culture Protocols. 1996:41–51. doi: 10.1385/0-89603-335-X:41. [DOI] [PubMed] [Google Scholar]
- 27.Xu C, et al. Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotech. 2001;19(10):971–974. doi: 10.1038/nbt1001-971. [DOI] [PubMed] [Google Scholar]
- 28.Wagner K, Welch D. Feeder-free adaptation, culture and passaging of human IPS cells using complete Knockout Serum Replacement feeder-free medium. J Vis Exp. 2010;15(41) doi: 10.3791/2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2- CT Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Feng B, et al. Molecules that Promote or Enhance Reprogramming of Somatic Cells to Induced Pluripotent Stem Cells. 2009;4(4):301–312. doi: 10.1016/j.stem.2009.03.005. [DOI] [PubMed] [Google Scholar]