

Abstract
Mutations in gigaxonin are responsible for Giant Axonal Neuropathy (GAN), a progressive neurodegenerative disorder associated with abnormal accumulations of Intermediate Filaments (IFs). Gigaxonin is the substrate-specific adaptor for a new Cul3-E3-ubiquitin ligase family that promotes the proteasome dependent degradation of its partners MAP1B, MAP8 and TBCB. Here, we report the generation of a mouse model with targeted deletion of Gan exon 1 (GanΔexon1;Δexon1). Analyses of the GanΔexon1;Δexon1 mice revealed increased levels of various IFs proteins in nervous system and the presence of IFs inclusion bodies in the brain. Despite deficiency of full length gigaxonin, the GanΔexon1;Δexon1 mice do not develop overt neurological phenotypes and giant axons reminiscent of the human GAN disease. We propose that the existence of a short gigaxonin isoform expressed in the spinal cord could underlie the mitigation of GAN-phenotypes in GanΔexon1;Δexon1 mice. Nonetheless, the GanΔexon1;Δexon1 mice exhibited modest increase in axon calibers and 27% axonal loss in the L5 ventral roots. This new mouse model should provide a useful tool for testing potential therapeutic approaches for GAN disease.
Keywords: intermediate filaments, inclusion bodies, neuropathy
Introduction
Intermediate filaments (IFs), along with microtubules (MTs) and actin microfilaments, are the basic components of the cytoskeletal network (Chang and Goldman, 2004). The major function of IFs is to maintain structural integrity of the cell in response to mechanical and non-mechanical stress (Fuchs and Cleveland, 1998). The three neurofilaments (NF-L, NF-M and NF-H), α-internexin and peripherin are the components of the neuronal IFs network. In neurons, IFs are thought to be involved in development, response to an injury, determination of axonal caliber and conduction velocities (Xiao et al., 2006). The abnormal accumulation of IFs is a pathological hallmark of many neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS), Charcot Marie Tooth (CMT), Parkinson Disease (PD) and Giant Axonal Neuropathy (GAN) (Lariviere and Julien, 2004). Neurofilament accumulations may be caused by neurofilament gene mutations (Tomkins et al., 1998; Georgiou et al., 2002; Kruger et al., 2003), by kinesin mutations (Xia et al., 2003) or by a disorganization of other cytoskeletal components.
Giant Axonal Neuropathy (GAN, OMIM #256850) is an neurodegenerative autosomal recessive disorder that affects both the central and peripheral nervous system (Asbury et al., 1972; Berg et al., 1972). GAN patients first develop deficits in the sensori- and motor-tracts which progress with areflexia, loss of deep/superficial sensitivity and loss of ambulation. The disorder evolves rapidly with a deterioration of the central nervous system functions and leads to death within 10-30 years (Asbury et al., 1972; Berg et al., 1972). GAN is characterized by the presence of giant axons in nervous tissues and by the systematic accumulation of IFs in a variety of cell types (Donaghy et al., 1988; Bomont and Koenig, 2003).
Since the discovery of the GAN gene (Bomont et al., 2000) more than 40 mutations have been found in GAN patients, including deletion, insertion, missense and nonsense mutations (Bomont et al., 2000; Kuhlenbaumer et al., 2002; Bomont et al., 2003; Bruno et al., 2004; Demir et al., 2005; Houlden et al., 2007; Koop et al., 2007; Leung et al., 2007). These mutations are localised throughout the GAN gene and are thought to lead to loss of function of the encoded protein called gigaxonin. With a N-terminal BTB domain and a C-terminal Kelch domain (Bomont et al., 2000), gigaxonin has been shown to be the substrate-adaptor of a Cul3-E3-ubiquitin ligase complex (Furukawa et al., 2003; Pintard et al., 2003; Xu et al., 2003). By interacting with the E3 ligase complex through its BTB domain, GAN promotes the proteaseome dependent degradation of microtubules associated proteins including MAP1B, MAP8 and the tubulin chaperone TBCB, by interaction with its Kelch domain (Ding et al., 2002; Wang et al., 2005; Ding et al., 2006). It has been reported that the gigaxonin interacts with MAP1B to increase the MTs stability (Ding et al., 2002) whereas it controls protein degradation of TBCB a function critical for the maintenance of MTs (Wang et al., 2006).
Ding et al. reported a mouse model deficient for gigaxonin after disruption of Gan exons 3-5 (GanΔexon3-5). These mice exhibited progressive decline of motor function with onset between 6 to 10 months and with occasional spasticity. However, a subset of these null mice did not develop overt neurological defects.
Here, we report a new mouse model with targeted disruption of the GAN gene based on deletion of exon 1 (GanΔexon1;Δexon1). Despite a lack of the 65 kDa Gan protein, the GanΔexon1;Δexon1 mice do not develop the severe neurological phenotypes anticipated from the human GAN disease. Yet these mice do exhibit accumulations of IF proteins in the nervous system. The alleviation of GAN phenotypes in the Gan mutant mice may be explained by the existence of a spinal cord-specific gigaxonin isoform.
Materials and methods
Knockout mice
A 10.4 kb fragment of the GAN gene, including exon 1 and part of the upstream promoter, were subcloned into a pQZ1 cloning vector. The 0.9 kb AcsI-XmaI fragment containing exon 1 and part of the 3’ promoter was replaced with a Neo cassette. The vector was then digested by NotI and PmaCI. The targeting fragment was isolated and electroporated into embryonic stem (ES) cells. Positive clones were picked up and amplified. The homologous recombination event was detected by Southern blot using an external 5’-EcoRV probe. The use of animals and all surgical procedures described in this article were carried out according to The Guide to the Care and Use of Experimental Animals of the Canadian Council on Animal Care.
Southern blots and PCR
DNA was extracted from mouse tails with phenol-chloroform procedure. A PCR-amplified fragment of the Gan promoter was used as a 500bp probe. Genomic DNA digested with EcoRV and analyzed according to Southern blotting methods (Couillard-Despres et al., 1998). The probe detected a 10-kb wild-type band and a 5-kb band for the knockout mice. For PCR genotyping, wild type primers chosen inside the deleted region amplified a sequence of 190bp. Standard Neo primers were used for genotyping of the knockout allele and yielded to an amplification product of 280bp. Primers sequence is described table 1.
Table 1.
List of the primers used for genotyping and RT-PCR
| Target sequence | Foward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| Genotyping | ||
| neo cassette | CTTGGGTGGAGAGGCTATTC | AGGTGAGATGACAGGAGATC |
| exon 1 WT | GTGTCCGACCCTCAGCAC | GCCAGGATGTTCTTCTGCAC |
| RT-PCR | ||
| exon 1–2 | GTGTCCGACCCTCAGCAC | GTGGATCCGTCATCTTTTGG |
| exon 2–7 | CATCTTCAGTGGGCAGATCA | CAAAGTTGTGTCTTGCCTCATGC |
RT-PCR
Total RNA from brain and spinal cord was extracted with trizol reagent according to the manufacturing instructions (Invitrogen, Burlington, ON). RNA concentration was determined at 260nm and aliquots of 5μg were stored at -80°C. Pairs of primers were chosen to target Gan cDNA and are described in table 1. RT-PCR was performed in one step with Superscript-Taq RT-PCR one step kit (Invitrogen, Burlington, ON) according to the manufacturer’s protocol. RT-PCR products were separated by electrophoresis on agarose gel.
Western blot/ Dot blot
After dissection, tissues were homogenized in a SUB denaturing buffer (0,5% SDS/8 M urea in 7.4 phosphate buffer) with a pool of protease inhibitors [phenylmethylsulfonyl fluoride (PMSF), protease inhibitor cocktail (Sigma, Saint-Louis, MI)]. Homogenates were centrifuged at 13 000 rpm 20 min at room temperature. The protein concentration of the supernatant was determined by the method of Bradford. Equal amount of proteins were loaded on SDS-PAGE and transferred to a nitrocellulose membrane, or directly loaded on membrane through dot blot apparatus. Membrane was blocked in PBSMT (PBS1X; tween 0.1%; dry milk 5%) and then incubated with a dilution of different primary antibodies in PBSMT overnight at 4°C. The different antibodies were gigA/gigB (gigaxonin; Bomont et al., manuscript in preparation), N52 (NF-H), NN18 (NF-M), NR4 (NF-L), Tau1, α-internexin, Vimentin, GFAP, α-tubulin, β-tubulin, γ-tubulin and actin (Chemicon, Temecula, CA), except for tubulins (Sigma, Saint-Louis, MI), and NFL (Novocastra, Newcastle Upon Tyne, UK). Incubation with the secondary antibody diluted 1:5000 (Jackson Immunoresearch, West Grove, PA) was done at room temperature 30 min. Detection was done by chemiluminescence (Perkin and Elmer, Waltham, MA).
Immunohistochemistry
Mice were perfused with NaCl 0.9% and fixed with 4% PFA (paraformaldehyde) pH7.4. Brain and spinal cord were dissected and then post-fixed in PFA 4% pH7.4; sucrose 10%. Brains and spinal cords were cut on microtom in 25μ sections. Tissues were stocked at -20°C in a cryopreservative solution and used for immunodetection. Tissue samples were washed in 50 mM potassium PBS and preincubated for 30 min at room temperature in blocking solution (potassium PBS containing 0.4% Triton X-100, 1% BSA, and 4% goat serum). The slices were incubated overnight at 4°C with the appropriate dilution of primary antibody in blocking serum. The slices were then washed and incubated for 90 min at room temperature in the secondary biotinylated Ab solution 1/1500 in potassium PBS containing 0.4% Triton X-100 and 1% BSA. After washing, the sections were incubated in ABC complex 1h at room temperature. Staining was developed by incubating the samples in 0.5mg/ml DAB + 0.003% H2O2 in potassium PBS. The rinsed tissues were mounted on superfrost slides (Fisher, Ottawa ON), counterstained with hematoxylin, dehydrated in graded concentration of EtOH and xylene, and coverslipped with DPX.
Immunofluorescence
As previously described for immunohistochemistry until incubation with primary Ab (NF-H poly; α-internexin, Chemicon, Temecula CA), the tissues sections were washed in KPBS and then incubated for 90 min in secondary Ab (Alexa fluor, Invitrogen, Burlington, ON) diluted 1:500 in potassium PBS containing 0.4% Triton X-100 and 1% BSA. The sections were mounted on superfrosst slides (Fisher, Ottawa, ON) and finally coverslipped with PVA-DABKO.
Axon and neuronal cell count
Dorsal root ganglions from 6 months old mice were dissected after perfusion with PFA and then post fixed in glutaraldehyde 3%. Tissue samples were washed three times in 0.1M NaHPO4 pH 7.4 and then treated with osmium tetroxyde 2% in NaHPO4 0.1M for 2 hours at room temperature. The samples were then deshydrated in increased concentration of EtOH and in Acetone. The final deshydratation was performed 1 hour RT with 50% epoxy resin in acetone. Ventral (VR) and dorsal roots (DR) were properly separated and embedded in epoxy resin at least 2 hour RT before cooking overnight at 60°C. Resulting blocks were cut in 1μ semi-thin section and stained with toluidine blue. Axon calibers were evaluated with stereomiscrocopy (Neurolucida Tablet).
Grip strength test
Hind limb grip strength testing was done by using a Chatillon DFIS-2 digital force gauge (model DFIS 2, Ametek, Paoli, PA). Briefly, mice were allowed to grip wire mesh of the apparatus by their hind limbs. The animal was moved away from the bar slowly and apparatus measured if the animal exerted active force against the movement. Readings were taken in T-peak and measured in grams of force. Each animal was given three trials per examining period (Kerr et al., 2003).
Results
Generation of gigaxonin-deficient mice
The Gan gene (~46 kb) is composed of 11 exons separated by 10 introns in mice (Bomont et al, 2000; Gene ID: 209239, NCBI). Exons 1 and 2 are separated by over 20 kb. Our strategy to disrupt expression of Gan was to remove a 1 kb sequence containing part of the promoter with the translation initiation site in the first exon. A targeting vector was generated to replace exon 1 and part of the 3’-promoter by a neo cassette (Fig. 1A). The vector was digested with restriction enzymes to yield a 1.5 kb targeting fragment that was then electroporated in ES cells. Neomycin-resistant colonies were picked up for Southern blot analysis. ES cell clones positive for homologous recombination were then microinjected into mouse blastocysts to generate chimeric founder mice. Male chimeras were then mated with C57BL/6 females to generate mice heterozygous for Gan exon 1 deletion. Genotyping of DNA extracted from mouse tails was determined either by Southern blot analysis using a 500-bp 5’-EcoRV probe (Fig. 1C) or by PCR using primers flanking exon 1 (Fig. 1B). Mendelian transmission of the disrupted Gan gene was obtained by the breeding of heterozygous F1 mice.
Figure 1.
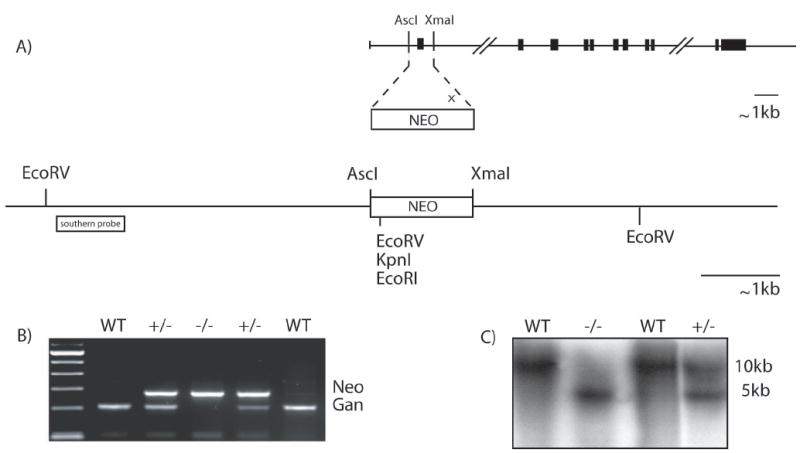
Targeted disruption of the GAN gene.
(A) Schematic representation of the mouse GAN gene. The targeting vector was generated by replacing a 1kb AscI/XmaI fragment including the first exon by a nlsLacZ/Neo cassette. (B,C) PCR genotyping and Southern blot of EcoRV-digested tail DNA probed with a fragment flanking the 5’ region of the targeting vector.
The mice homozygous for exon 1 deletion (GanΔexon1;Δexon1) did not exhibit overt neurological phenotypes. The GanΔexon1;Δexon1 mice were viable and reproduced normally. Their lifespan did not differ significantly from that of normal mice (data not shown).
mRNA and protein analyses
Immunoblotting with monoclonal antibody raised against both the N-terminal and C-terminal gigaxonin domain confirmed the absence of full length 65 kDa protein in brain and spinal cord of GanΔex1;Δex1 mice (Fig. 2A). Intriguingly, these antibodies also detected a prominent 47.5 kDa protein in spinal cord samples of GanΔex1;Δex1 (Fig. 2A). This smaller protein was present in lower amount in the spinal cord of heterozygous GanΔexon1;wt mice and wild type Ganwt;wt mice. The presence of this 47.5 kDa band prompted us to investigate the expression of GAN transcripts in spinal cord of GanΔex1;Δex1 mice.
Figure 2.

(A) Western blots of total protein extracts from brain and spinal cord confirm the absence of the 68kDa Gan protein in knockout mice. However a 47.5 kDa band was detected in spinal cord extract and enriched in the GanΔex1;Δex1 mice. (B) RT-PCR analyses of total RNA from liver, brain and spinal cord show that a Gan mRNA species is still present in the spinal cord but not in the brain of GanΔex1;Δex1 (arrow). (C) Schematic representations of GAN gene and Gan mRNA species. (D) Comparison of Gan and sGig mRNAs. (E) Predicted Gan and sGig proteins.
RT-PCR analyses were performed with primers to amplify the region between exons 1 and 2 as well as the region between exons 2 and 7. Using RT-PCR primers for exons 1 to 2, no band was detected in CNS RNA samples from GanΔex1;Δex1, as expected from exon 1 deletion (Fig. 2B). However, RT-PCR with primers for exons 2 and 7 yielded a band in spinal cord samples from GanΔex1;Δex1 mice but not in other tissues such as brain and liver (Fig. 2B). This transcript spans Gan exon 2 to 11 and it is missing exon 1. Thus, the RT-PCR results confirm the lack of full lenght Gan mRNA in GanΔex1;Δex1 mice but they support the existence of shorter gigaxonin species in the spinal cord. This short gigaxonin variant will be called sGig (Fig 2C, D,E).
Changes in IF protein levels in the nervous system
It is well established that gigaxonin-deprived tissues from GAN patients present characteristic accumulations of IFs (Asbury et al. 1972, Berg et al., 1972, Prineas et al., 1976). We therefore examined whether the absence of exon 1 in GanΔex1;Δex1 mice resulted in abnormal levels of IFs and of other cytoskeletal components. Western blot analyses of total protein extracts at 3 months of age from the brain, cerebellum and spinal cord revealed modest increases in protein levels of IF proteins including NF-L, NF-M, NF-H and -internexin (Fig. 3A). A two-fold increase in peripherin and vimentin levels were also observed in the spinal cord of GanΔex1;Δex1 mice when compared to normal littermates. The analysis of sciatic nerve sections revealed enhanced NF protein levels along the nerve. The three subunits seem to be more abundant in the nerve proximal region (Fig. 3B). The increased levels of neuronal IF proteins in GanΔex1;Δex1 was not due to increased mRNA expression. RT-PCR analysis for NF-L, NF-M and NF-H showed no differences of transcript levels in the brain, cerebellum or spinal cord of GanΔex1;Δex1 mice and littermate controls (Fig. 3C).
Figure 3.
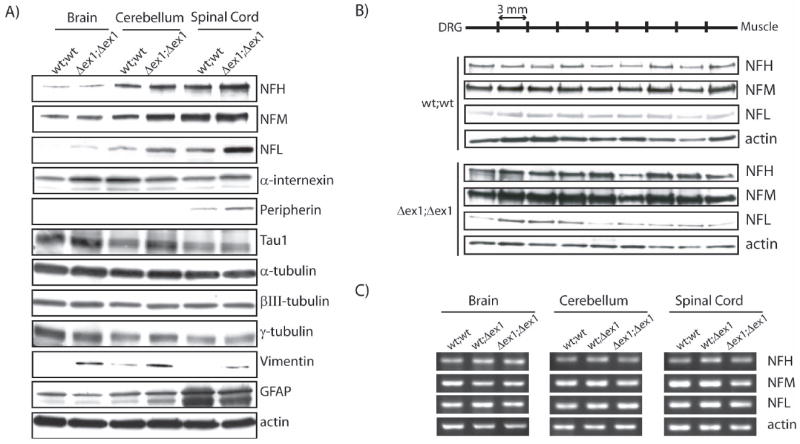
Increased levels of IFs in specific regions of the nervous system.
(A) Western blots for IF and tubulin proteins reveal variations for NF-H, NF-M, NF-L, α-internexin, peripherin, vimentin and GFAP. (B) Western blot of sciatic nerve sections showed that NF proteins are more abundant in proximal than in distal sections in GanΔex1;Δex1 mice unlike normal mice(C) No variation of neurofilament mRNA levels was detected by RT-PCR.
To determine if the variations in IF protein levels were constant during aging, dot blot immunodetection analyses were performed for NF subunits, α-internexin and vimentin at different ages (Fig. 4). The results confirmed a deregulation of all IF protein levels early as 3 months of age that was still present at 24 months of age. In particular, NF-H and NF-L levels were increased up to two folds at all ages in the brain, cerebellum and spinal cord samples of GanΔex1;Δex1mice. NF-M levels remained unchanged at 6 and 12 months of age in cerebellum and spinal cord samples of GanΔex1;Δex1 mice. The α-internexin levels were increased by up to 3.8 fold in the cerebellum of GanΔex1;Δex1 mice at 3 months of age as compared to controls.
Figure 4.

Dot blot analysis of IF content in the CNS. The levels of NF-H, NF-M, NF-L, α-internexin and vimentin were quantified by dot blot in extracts from the brain, cerebellum and spinal cord of GanΔex1;Δex1 mice and of wild-type littermate at 3, 6, 12 and 24 months of age (**p<0.001 and *p<0.01).
In the brain as well as in the ventral part of the lumbar spinal cord, the immunostaining for NF-L was stronger but no inclusions were detected (Fig. 5A, 5E). The most notable changes came from the immunodetection of NF-H and α-internexin. In the cerebral cortex these two proteins formed accumulations in the neuronal cell bodies of GanΔex1;Δex1 mice(Fig. 5B-C). These IF accumulations appear as early as one month of age and are still present at 12 months of age (data not shown). Double immunofluorescence revealed that NF-H and α-internexin accumulations did not always colocalize (Fig. 5D). In the lumbar spinal cord, the NF-H signal was more intense in cell bodies from GanΔex1;Δex1 tissue as compared to wild-type samples (Fig. 5G-H). α-internexin staining showed IF accumulations resembling those detected in the cerebral cortex (Fig. 5I-J). Finally, even though western blots exhibited higher level of peripherin in the spinal cord, immunohistochemistry did not reveal significant peripherin accumulations in GanΔex1;Δex1 spinal motor neurons (Fig. 5K-L).
Figure 5.
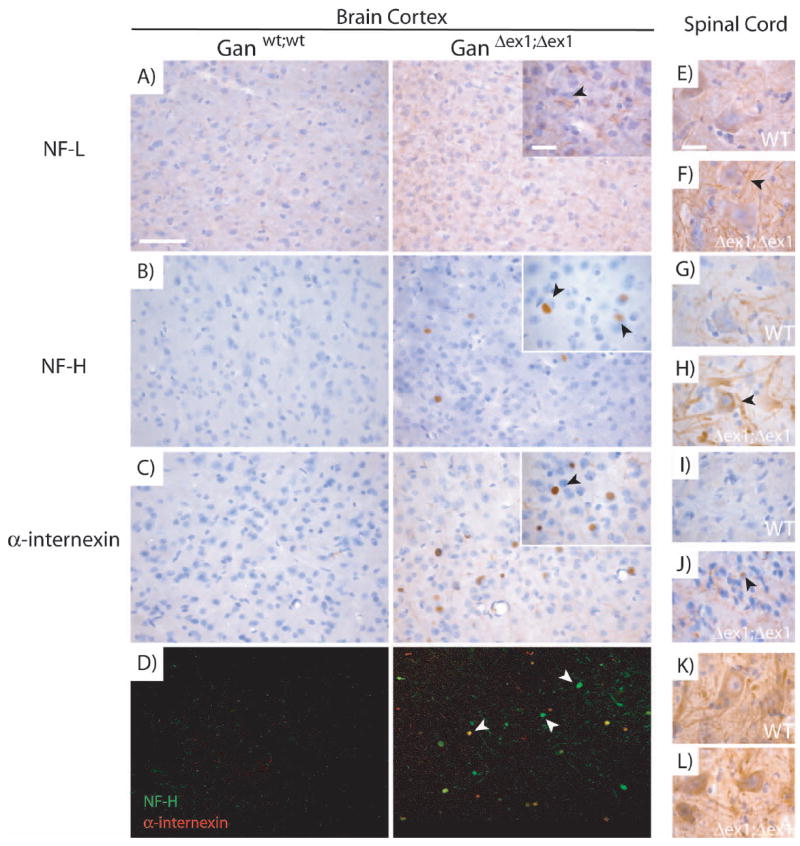
Accumulations of IFs in specific regions of the CNS in GanΔex1;Δex1 mice.
NF-L immunostaining was stronger in GanΔex1;Δex1 mice compared to normal mice (A). NF-H (B) and α-internexin (C) immunostaining revealed accumulations in the form of inclusion bodies in GanΔex1;Δex1 cerebral cortex. (D) Double labelling of NF-H and α-internexin in the cerebral cortex. NF-H and α-internexin do not always colocalize. NF-L (E-F) and NF-H (G-H) staining tend to be stronger in the lumbar spinal cord of motor neurons in GanΔex1;Δex1 mice unlike peripherin (K-L) which does not change. α-internexin (I-J) tends to form accumulations in the dorsal horn of the spinal cord as in the brain cortex. (Scale bar 100 and 10 μ.)
Absence of full length gigaxonin does not lead to significant motoneuron death
As expected, western blot analysis of both L4/L5 ventral (VR) and dorsal (DR) roots revealed a slight increase of NF content (Fig. 6A, 6D). In order to investigate if this increase in NF content was accompagned by axonal degeneration, L5 VR and DR of 6 months mice were dissected and cut into 1 semi thin sections. DR and VR axons number and caliber were then analyzed by stereomicroscopy. This allowed us to show that neither the number of sensory axons nor the axon caliber was altered in GanΔex1;Δex1 dorsal root compared to wild type (Fig. 6B, 6C). However, the number of motor axons was significantly diminished by 27% in GanΔex1;Δex1 ventral root compared to wild-type (Fig. 6E). A subset of axons from GanΔex1;Δex1 exhibited larger calibers than control mice (Fig. 6F).
Figure 6.

The number of axons is lower than normal in GanΔex1;Δex1 L5 ventral root but not in dorsal root.
The content of NFs was reduced in GanΔex1;Δex1 L5 ventral root (D) but not in dorsal root (A). (B,E) show the average number of axons of wt;wt, Δex1;wt and Δex1; Δex1 mice in dorsal and ventral root, respectively. The number of axons was decreased by 27% in GanΔex1;Δex1 compared to WT littermates (p=0.01). (C,F) show the average caliber of the axons. Some ventral root axons are larger than normal in GanΔex1;Δex1(wt;wt vs Δex1; Δex1 ***p<0.001; **p<0.05). Scale bar 25μ.
As there was evidence of axonal degeneration in the L5 ventral root of GanΔex1;Δex1 mice, we carried out a motor neuron count in the lumbar spinal cord. Tissue sections from normal and GanΔex1;Δex1 mice at 1, 2, 3, 6, and 12 months of age were stained with thionine followed by neuronal count. There was a tendency for decreased number of motor neuron cell bodies at 6 months in GanΔex1;Δex1 as compared to normal mice but the changes were not significant (Fig. 7A). The GanΔex1;Δex1 mice did not exhibit motor dysfunction during aging. Grip strength analyses were performed and no differences could be observed between wild type and knockout mice (fig. 7B).
Figure 7.
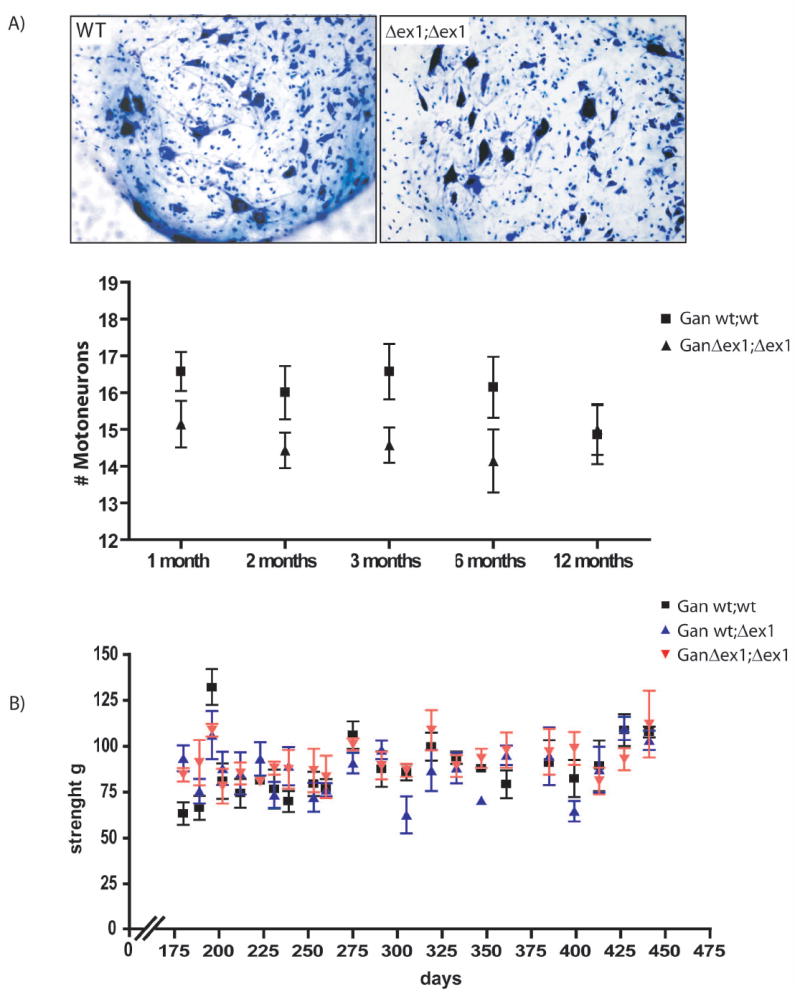
No significant reduction in the number of motor neurons in the lumbar region of the spinal cord.
(A) Motor neurons were stained (Nissl) and counted at different ages. No change in number of motor neurons was detected in GanΔex1;Δex1 mice when compared to wild-type littermates. (B) Grip strength test was carried out at 6 months old mice. No weakness of the hind limbs was detected in GanΔex1;Δex1 mice when compared to heterozygous and wild-type littermate (n=5).
Discussion
Here we report the characterization of mice with targeted disruption the Gan gene by insertion of a Neo cassette in exon 1. This method succeeded in eliminating expression of the full length form of gigaxonin (Fig. 2B). The GanΔex1;Δex1 mice exhibited enhanced levels of several IF proteins, a histological pathological feature in GAN patients (Fig.3-4). Increase levels of IF proteins in nervous tissue were detected for NF proteins, α-internexin, peripherin as well as vimentin. Microscopy of peripheral nerve revealed that some of the motor axons in GanΔex1;Δex1 mice at 6 months of age were larger than normal and that there was a significant axonal loss of 26% (Fig. 6). However, the GanΔex1;Δex1 mice failed to develop giant axons typical of the human GAN disease. The modest but significant loss of motor axons was not associated with reduced number of spinal motor neurons and with motor dysfunction (Fig. 7). A most intriguing result came from histological analyses of brain sections that revealed accumulations of NF-H and of α-internexin specifically in the cortex (Fig. 5). These accumulations formed neuronal intracytoplasmic inclusions that are highly reminiscent of α-internexin inclusions found in human neuronal filament inclusion disease (NFID) (Cairns et al., 2004a; Cairns et al., 2004a; Joseph 2005). Like in human NFID, the inclusions in GanΔex1;Δex1 mice are mainly composed of α-internexin and they occur predominantly in the cerebral cortex. These inclusions were positive for NF-H and α-internexin but negative for other NF subunits, tau and α-synuclein like accumulations found in Alzheimer’s disease or Parkinson’s disease. No genetic mutations have been linked to NFID so far (Momeni et al., 2006).
Giant axonal neuropathy is a progressive and fatal sensory motor neuropathy that affects both the CNS and PNS (Asbury et al., 1972; Berg et al., 1972; Ouvrier et al., 1974; Igisu et al., 1975). Hence, mutations in GAN often lead to severe phenotypes in humans. Despite a disorganization of IF network, formation of neuronal filament inclusions in cerebral cortex and slight increase in caliber of motor axons, the GanΔex1;Δex1 mice exhibited only mild phenotypes when compared to human GAN disease. Here, the GanΔex1;Δex1 mice did not show limb weakness despite a significant loss of motor axons. The mice lacking exon 3-5 of Gan described in a previous study exhibited heterogenous phenotypes with some of them having observable neurological phenotypes (Ding et al., 2006; Yang et al., 2007). However, Ding et al. (2006) did not report the extent of axonal degeneration in their Gan null mouse. One plausible explanation for the differences in phenotypes between the two Gan null mouse models is that the presence of a shorter form of gigaxonin (<50 kDa) in the spinal cord of GanΔex1;Δex1 mice may compensate in part for the absence of full length Gan protein (Fig. 2B). This smaller Gan protein (s-Gig) would lack the first 80 amino acids corresponding to exon 1 and part of exon 2 (Fig. 2D-E).
It has been shown that vimentin forms aggregates in fibroblasts from patients with GAN (Pena, 1981; Bomont and Koenig, 2003). However, fibroblasts from patients bearing the GAN R15S mutation in the N-terminal domain do not develop vimentin aggregates. This result suggests that a perfectly intact BTB domain is not necessary for gigaxonin activity even thought its function would not be optimal (Bomont and Koenig, 2003). This would be in line with the view that even if s-Gig lacks part of the BTB-domain, it may still remain partly functional. It is noteworthy that we detected increased IF protein levels and IF inclusions in the cortex where there was complete absence of Gigaxonin (Fig. 3 and 5). No IF inclusions occurred in spinal cord where s-Gig is detected (Fig. 3A and 5). So, the presence of s-Gig may explain the lack of neurological phenotypes in the GanΔex1;Δex1 mice.
Our results confirm the importance of Gigaxonin in modulating the levels and organization of IF proteins. It supports the notion that a Gigaxonin deficiency can provoke formation of neuronal IF inclusions. As neuronal IF proteins have very long half live (Millecamps et al., 2007), IFs are prone to form abnormal accumulations following microtubule-based transport defects such as those caused by GAN gene mutations. The GanΔex1;Δex1 mouse model presented here exhibit some features of the human GAN disease including the presence of neuronal IFs inclusions. In future, these mice should provide a useful tool for testing potential therapeutic approaches for this disease.
Acknowledgments
We thank Roxanne Larivière, Mélanie Lalancette-Hébert, Geneviève Soucy and Makoto Urushitani for their advices and technical assistance. Jean-Pierre Julien holds a Canada Research Chair in Neurodegeneration. Florence Dequen is a recipient of a FRSQ Studentship. Pascale Bomont was supported in part by a Fellowship from the Fondation pour la Recherche Medicale. Geneviève Gowing is recipient of a CIHR Doctoral Research Award. This work was supported by grants from the Canadian Institutes of Health and Research to J.-P.J and from National Institutes of Health (USA) to DWC.
References
- Asbury AK, Gale MK, Cox SC, Baringer JR, Berg BO. Giant axonal neuropathy--a unique case with segmental neurofilamentous masses. Acta Neuropathol (Berl) 1972;20:237–247. doi: 10.1007/BF00686905. [DOI] [PubMed] [Google Scholar]
- Ben Hamida C, Cavalier L, Belal S, Sanhaji H, Nadal N, Barhoumi C, M’Rissa N, Marzouki N, Mandel JL, Ben Hamida M, Koenig M, Hentati F. Homozygosity mapping of giant axonal neuropathy gene to chromosome 16q24.1. Neurogenetics. 1997;1:129–133. doi: 10.1007/s100480050019. [DOI] [PubMed] [Google Scholar]
- Berg BO, Rosenberg SH, Asbury AK. Giant axonal neuropathy. Pediatrics. 1972;49:894–899. [PubMed] [Google Scholar]
- Bomont P, Koenig M. Intermediate filament aggregation in fibroblasts of giant axonal neuropathy patients is aggravated in non dividing cells and by microtubule destabilization. Hum Mol Genet. 2003;12:813–822. doi: 10.1093/hmg/ddg092. [DOI] [PubMed] [Google Scholar]
- Bomont P, Ioos C, Yalcinkaya C, Korinthenberg R, Vallat JM, Assami S, Munnich A, Chabrol B, Kurlemann G, Tazir M, Koenig M. Identification of seven novel mutations in the GAN gene. Hum Mutat. 2003;21:446. doi: 10.1002/humu.9122. [DOI] [PubMed] [Google Scholar]
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tuysuz B, Landrieu P, Hentati F, Koenig M. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26:370–374. doi: 10.1038/81701. [DOI] [PubMed] [Google Scholar]
- Bruno C, Bertini E, Federico A, Tonoli E, Lispi ML, Cassandrini D, Pedemonte M, Santorelli FM, Filocamo M, Dotti MT, Schenone A, Malandrini A, Minetti C. Clinical and molecular findings in patients with giant axonal neuropathy (GAN) Neurology. 2004;62:13–16. doi: 10.1212/01.wnl.0000101676.41505.a7. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Uryu K, Bigio EH, Mackenzie IR, Gearing M, Duyckaerts C, Yokoo H, Nakazato Y, Jaros E, Perry RH, Arnold SE, Lee VM, Trojanowski JQ. alpha-Internexin aggregates are abundant in neuronal intermediate filament inclusion disease (NIFID) but rare in other neurodegenerative diseases. Acta Neuropathol (Berl) 2004a;108:213–223. doi: 10.1007/s00401-004-0882-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Zhukareva V, Uryu K, Zhang B, Bigio E, Mackenzie IR, Gearing M, Duyckaerts C, Yokoo H, Nakazato Y, Jaros E, Perry RH, Lee VM, Trojanowski JQ. alpha-internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease. Am J Pathol. 2004b;164:2153–2161. doi: 10.1016/s0002-9440(10)63773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Goldman RD. Intermediate filaments mediate cytoskeletal crosstalk. Nat Rev Mol Cell Biol. 2004;5:601–613. doi: 10.1038/nrm1438. [DOI] [PubMed] [Google Scholar]
- Couillard-Despres S, Zhu Q, Wong PC, Price DL, Cleveland DW, Julien JP. Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proc Natl Acad Sci U S A. 1998;95:9626–9630. doi: 10.1073/pnas.95.16.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir E, Bomont P, Erdem S, Cavalier L, Demirci M, Kose G, Muftuoglu S, Cakar AN, Tan E, Aysun S, Topcu M, Guicheney P, Koenig M, Topaloglu H. Giant axonal neuropathy: clinical and genetic study in six cases. J Neurol Neurosurg Psychiatry. 2005;76:825–832. doi: 10.1136/jnnp.2003.035162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Liu JJ, Kowal AS, Nardine T, Bhattacharya P, Lee A, Yang Y. Microtubule-associated protein 1B: a neuronal binding partner for gigaxonin. J Cell Biol. 2002;158:427–433. doi: 10.1083/jcb.200202055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Allen E, Wang W, Valle A, Wu C, Nardine T, Cui B, Yi J, Taylor A, Jeon NL, Chu S, So Y, Vogel H, Tolwani R, Mobley W, Yang Y. Gene targeting of GAN in mouse causes a toxic accumulation of microtubule-associated protein 8 and impaired retrograde axonal transport. Hum Mol Genet. 2006;15:1451–1463. doi: 10.1093/hmg/ddl069. [DOI] [PubMed] [Google Scholar]
- Donaghy M, King RH, Thomas PK, Workman JM. Abnormalities of the axonal cytoskeleton in giant axonal neuropathy. J Neurocytol. 1988;17:197–208. doi: 10.1007/BF01674207. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Cleveland DW. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279:514–519. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- Furukawa M, He YJ, Borchers C, Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol. 2003;5:1001–1007. doi: 10.1038/ncb1056. [DOI] [PubMed] [Google Scholar]
- Georgiou DM, Zidar J, Korosec M, Middleton LT, Kyriakides T, Christodoulou K. A novel NF-L mutation Pro22Ser is associated with CMT2 in a large Slovenian family. Neurogenetics. 2002;4:93–96. doi: 10.1007/s10048-002-0138-4. [DOI] [PubMed] [Google Scholar]
- Houlden H, Groves M, Miedzybrodzka Z, Roper H, Willis T, Winer J, Cole G, Reilly MM. New mutations, genotype phenotype studies and manifesting carriers in giant axonal neuropathy. J Neurol Neurosurg Psychiatry. 2007 doi: 10.1136/jnnp.2007.118968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igisu H, Ohta M, Tabira T, Hosokawa S, Goto I. Giant axonal neuropathy. A clinical entity affecting the central as well as the peripheral nervous system. Neurology. 1975;25:717–721. doi: 10.1212/wnl.25.8.717. [DOI] [PubMed] [Google Scholar]
- Kerr DA, Llado J, Shamblott MJ, Maragakis NJ, Irani DN, Crawford TO, Krishnan C, Dike S, Gearhart JD, Rothstein JD. Human embryonic germ cell derivatives facilitate motor recovery of rats with diffuse motor neuron injury. J Neurosci. 2003;23:5131–5140. doi: 10.1523/JNEUROSCI.23-12-05131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koop O, Schirmacher A, Nelis E, Timmerman V, De Jonghe P, Ringelstein B, Rasic VM, Evrard P, Gartner J, Claeys KG, Appenzeller S, Rautenstrauss B, Huhne K, Ramos-Arroyo MA, Worle H, Moilanen JS, Hammans S, Kuhlenbaumer G. Genotype-phenotype analysis in patients with giant axonal neuropathy (GAN) Neuromuscul Disord. 2007;17:624–630. doi: 10.1016/j.nmd.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Kruger R, Fischer C, Schulte T, Strauss KM, Muller T, Woitalla D, Berg D, Hungs M, Gobbele R, Berger K, Epplen JT, Riess O, Schols L. Mutation analysis of the neurofilament M gene in Parkinson’s disease. Neurosci Lett. 2003;351:125–129. doi: 10.1016/s0304-3940(03)00903-0. [DOI] [PubMed] [Google Scholar]
- Kuhlenbaumer G, Young P, Oberwittler C, Hunermund G, Schirmacher A, Domschke K, Ringelstein B, Stogbauer F. Giant axonal neuropathy (GAN): case report and two novel mutations in the gigaxonin gene. Neurology. 2002;58:1273–1276. doi: 10.1212/wnl.58.8.1273. [DOI] [PubMed] [Google Scholar]
- Lariviere RC, Julien JP. Functions of intermediate filaments in neuronal development and disease. J Neurobiol. 2004;58:131–148. doi: 10.1002/neu.10270. [DOI] [PubMed] [Google Scholar]
- Leung CL, Pang Y, Shu C, Goryunov D, Liem RK. Alterations in lipid metabolism gene expression and abnormal lipid accumulation in fibroblast explants from giant axonal neuropathy patients. BMC Genet. 2007;8:6. doi: 10.1186/1471-2156-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Gowing G, Corti O, Mallet J, Julien JP. Conditional NF-L transgene expression in mice for in vivo analysis of turnover and transport rate of neurofilaments. J Neurosci. 2007;27:4947–4956. doi: 10.1523/JNEUROSCI.5299-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momeni P, Cairns NJ, Perry RH, Bigio EH, Gearing M, Singleton AB, Hardy J. Mutation analysis of patients with neuronal intermediate filament inclusion disease (NIFID) Neurobiol Aging. 2006;27:778 e771–778 e776. doi: 10.1016/j.neurobiolaging.2005.03.030. [DOI] [PubMed] [Google Scholar]
- Ouvrier RA, Prineas J, Walsh JC, Reye RD, McLeod JG. Giant axonal neuropathy -- a third case. Proc Aust Assoc Neurol. 1974;11:137–144. [PubMed] [Google Scholar]
- Pena SD. Giant axonal neuropathy: intermediate filament aggregates in cultured skin fibroblasts. Neurology. 1981;31:1470–1473. doi: 10.1212/wnl.31.11.1470. [DOI] [PubMed] [Google Scholar]
- Pintard L, Willis JH, Willems A, Johnson JL, Srayko M, Kurz T, Glaser S, Mains PE, Tyers M, Bowerman B, Peter M. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature. 2003;425:311–316. doi: 10.1038/nature01959. [DOI] [PubMed] [Google Scholar]
- Tomkins J, Usher P, Slade JY, Ince PG, Curtis A, Bushby K, Shaw PJ. Novel insertion in the KSP region of the neurofilament heavy gene in amyotrophic lateral sclerosis (ALS) Neuroreport. 1998;9:3967–3970. doi: 10.1097/00001756-199812010-00036. [DOI] [PubMed] [Google Scholar]
- Wang W, Ding J, Allen E, Zhu P, Zhang L, Vogel H, Yang Y. Gigaxonin interacts with tubulin folding cofactor B and controls its degradation through the ubiquitin-proteasome pathway. Curr Biol. 2005;15:2050–2055. doi: 10.1016/j.cub.2005.10.052. [DOI] [PubMed] [Google Scholar]
- Xia CH, Roberts EA, Her LS, Liu X, Williams DS, Cleveland DW, Goldstein LS. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol. 2003;161:55–66. doi: 10.1083/jcb.200301026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, McLean J, Robertson J. Neuronal intermediate filaments and ALS: a new look at an old question. Biochim Biophys Acta. 2006;1762:1001–1012. doi: 10.1016/j.bbadis.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, Elledge SJ, Harper JW. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature. 2003;425:316–321. doi: 10.1038/nature01985. [DOI] [PubMed] [Google Scholar]
- Yang Y, Allen E, Ding J, Wang W. Giant axonal neuropathy. Cell Mol Life Sci. 2007 doi: 10.1007/s00018-007-6396-4. [DOI] [PMC free article] [PubMed] [Google Scholar]