

Abstract
The FOXL2C134W mutation has been identified in virtually all adult granulosa cell tumors (GCTs). Here we show that the exogenous FOXL2 expression is necessary for GDF-9 stimulation of follistatin transcription in the human GCT cell line, COV434 that lacks endogenous FOXL2 expression. Interestingly, in the presence of Smad3 co-expression, FOXL2C134W negated GDF-9 stimulation of follistatin transcription. However, mutation of the Smad binding element (SBE) located in the intronic enhancer elements in the follistatin gene restored normal FOXL2 activity to FOXL2C134W, thus the altered activity of FOXL2C134W is dependent on the ability of Smad3 to directly bind the SBE. Mutation of the FOXL2 binding element (FBE) or the FBE and SBE completely prevented GDF-9 activity, suggesting that the FBE is essential for GDF-9 stimulation in COV434. Overall, our study supports the view that altered interaction of FOXL2C134W with co-factors may underlie the pathogenesis of this mutation in GCTs.
Keywords: FOXL2, Follistatin, Granulosa cell tumor, Ovary, COV434, GDF-9, Smad
1. Introduction
Granulosa cell tumors (GCTs) represent around 5–10% of ovarian malignancies, with the majority (95%) of GCTs detected in adults [1]. Although most GCTs are diagnosed at an early stage that can be treated with surgery, there is a high rate of recurrence, particularly in patients diagnosed at a later stage [2]. Approximately 80% of patients with advanced stage or recurrent tumors succumb to their disease, mainly due to the characteristic slow, indolent pattern of GCT progression [3]. Since traditional chemotherapy is relatively ineffective in women with advanced-stage or recurrent GCTs a more effective therapeutic option for these patients is necessary. Unfortunately, the poorly understood etiology of GCTs has hampered the development of novel therapeutics. However, a recent breakthrough has surprisingly revealed that virtually all GCTs in adult women have a unique somatic FOXL2 mutation (FOXL2C134W) [4–9]. FOXL2, a member of the forkhead transcription factor family, is localized to GCs within the ovary and is essential for normal ovary development and function [10].
FOXL2 ablation in whole body knockout mouse models causes high perinatal mortality, which most likely reflects craniofacial defects [11,12]. However, surviving mice confirm a critical role of FOXL2 in ovarian function, with a block in follicle development at the primary stage associated with a failure of GCs to complete the squamous to cuboidal transition [11,12]. In contrast, GC-specific ablation of FOXL2 post-puberty in mice results in somatic sex reprogramming by induction of the transcription factor SOX9, with GCs becoming Sertoli cell-like and follicles changing into structures resembling seminiferous tubules [13].
There are findings suggesting that FOXL2C134W may promote GCT development at least in part by promoting cell cycle progression and helping cells evade apoptosis. In studies of human GCTs, a large proportion (58%) showed down-regulation of two members of the inhibitors of cyclin-dependent kinase 4 family (INK4A and INK4B), whose expression is altered in many different types of cancer [14]. Consistent with this, in the GCT cell line COV434 that has undetectable FOXL2 gene expression [6], overexpression of FOXL2wt but not FOXL2C134W induced transcriptional activity on INK4A [15]. Evidence for a difference in apoptotic activity between FOXL2wt and FOXL2C134W comes from another in vitro study using KGN cells, heterozygous for FOXL2C134W, which showed that FOXL2wt elicited significant apoptosis, while the activity of FOXL2C134W was impaired due to a failure to up-regulate the death receptors, TNF-R1 and Fas [16]. These findings provide us with valuable information, although rather limited, to help understand the mechanisms underlying how FOXL2C134W triggers GCT formation. Therefore, there is a need for further research on alternative cellular mechanisms that may be altered by FOXL2C134W.
Recent studies have shown that FOXL2 and Smad3 cooperatively regulate activin stimulation of follistatin, FSH β and GnRH receptor transcription in mouse pituitary cells [17–21]. As FOXL2 and Smad3 are similarly expressed in GCs [22–24], this led us to ask whether a similar cooperation between FOXL2 and Smad3 occurs in GCs. The mutated Cys134 residue is within the C-terminal region of the forkhead domain, required for both DNA-binding [25] and FOXL2 interaction with Smad3 [20]. Disruption of DNA- and/or Smad-binding capacity by this mutation would likely result in altered transcriptional activity and could ultimately result in tumor formation. As activation of Smad2/3 has been implicated in the pathogenesis of juvenile GCTs in humans [26], it is therefore plausible that known GC mitogens such as GDF-9, which signal through Smad2/3, may play a role in the pathogenesis of GCTs.
Follistatin is highly expressed in GCs of developing follicles [27–30] and binds activin [31–37] and some BMPs including BMP-7, and BMP-15 [38–40]. In so doing, follistatin inhibits the actions of each of these GC mitogens [41–43]. Thus, in doing so follistatin acts as an anti-proliferative factor. Our current data suggest that FOXL2 may act as a cell growth inhibitor by stimulating production of follistatin, an anti-proliferative factor. This hypothesis is additionally supported by the in vivo observation that follistatin expression was severely compromised in Foxl2 null mouse ovaries [11]. Dysregulation of follistatin by the FOXL2 GCT mutation could result in increased GC proliferation.
COV434 cells are a well-established immortalized human GC line derived from a 27-year-old patient with a metastatic GCT [44]. They possess certain morphologic and physiologic characteristics in common with normal GCs; in the presence of FSH and androstenedione COV434 cells secret estradiol and cAMP levels are increased indicating that the FSH receptor and P450 aromatase are present in these cells. In contrast to KGN cells used by other laboratories in recent studies [5,25] that heterozygously express the FOXL2C134W mutation, COV434 cells lack the FOXL2 mutation and FOXL2 mRNA and protein are undetectable[6]. Thus, COV434 cells provide a platform for direct comparison of FOXL2C134W and FOXL2wt.
The ultimate purpose of our research is to illuminate the molecular and cellular mechanisms underlying altered GC function triggered by acquisition of the somatic FOXL2C134W mutation and gain a better understanding of what drives GCT formation. Towards this goal, the aim of the current study was to determine i) whether FOXL2 is necessary for follistatin transcription regulated by GDF-9 in COV434 cells, ii) whether FOXL2C134W has altered activities as compared with FOXL2wt and iii) whether FOXL2 and Smad3 coordinately regulate follistatin transcription in the ovary.
2. Materials and Methods
2.1. Plasmids and Reagents
Recombinant mouse GDF-9 was purchased from R & D systems (Minneapolis, MN). The mouse monoclonal anti-Flag M2 antibody and anti-Flag M2 antibody conjugated agarose beads were purchased from Sigma-Aldrich Co. (St. Louis, MO), the mouse monoclonal anti-Myc antibody was from BD Biosciences (San Jose, CA), and the α-tubulin antibody was from Santa Cruz Biotechnology (Santa Cruz, CA).
The rat follistatin luciferase rFS(0.3ex45)-luc reporter plasmid, N-terminal Flag-tagged human FOXL2wt and N-terminal Myc-tagged human Smad3 were kindly provided by Dr. Louise Bilezikjian of the Salk Institute [20]. The rFS(0.3ex45)-luc plasmid contains the +1784/+1912 section of intron 1 downstream of a −312/+136 minimal promoter. The +1784/+1912 section of intron 1 contains a forkhead-binding element (FBE) just downstream of a Smad-binding element (SBE). It is of note that the DNA sequences at and surrounding the SEB and FBE of the rat follistatin gene are identical to those of the human gene. Mutant versions of the rFS(0.3ex45)-luc reporter were generated with mutations in the SBE (mutant 1), FBE (mutant 2), or SBE and FBE (mutant 3) by site directed mutagenesis in a two-step PCR. DNA fragments containing the mutations were generated using the common primers 5′-AATCGCGCGGGCGGCCGGTGGCG-3′ and 5′-GGAATGCCAAGCTTAGTCCTAGG-3′ and the following specific primers to introduce the mutations: 5′-CAAGCTGCACGTGTTGTAATTGGGTCACTGGTAACTGACATTGATATGGCTAGGCGCAGCGGCTGCTGCTC-3′ and 5′-GAGCAGCAGCCGCTGCGCCTAGCCATATCAATGTCAGTTACCAGTGACCCA ATTACAACACGTGCAGCTTG-3′ for mutant 1; 5′-CAAGCTGCACGTGTTGTGTCTGGGTCACTGGTAACTGTCGAACTCTTGGCT AGGCGCAGCGGCTGCTGCTC-3′ and 5′-GAGCAGCAGCCGCTGCGCCTAGCCAAGAGTTCGACAGTTACCAGTGACCCAGACACAACAC GTGCAGCTTG-3′ for mutant 2; and 5′-CAAGCTGCACGTGTTGTAATTGGGTCACTGGTAACTGTCGAACTCTTGGCT AGGCGCAGCGGCTGCTGCTC-3′ and 5′-GAGCAGCAGCCGCTGCGCCTAGCCAAGAGTTCGACAGTTACCAGTGACCCAATTACAACAC GTGCAGCTTG-3′ for mutant 3. The resulting DNA fragments were cloned into to the rFS(0.3ex45)-luc plasmid using EagI and HindIII restriction sites. From the Flag-FOXL2wt expression plasmid we generated the Flag-FOXL2C134W mutant expression plasmid by site directed mutagenesis as explained above. Briefly, primers A (5′-TACGTGGCGCTCATCGCCATGGCGATC-3′) and D (5′-AGCGCCATGCTCTGCACGCGTGTGTAC-3′) contained Sty I and Mlu I restriction sites respectively, while primers B (5′-CTCGAACATGTCTTCCCAGGCCGGGTCCAG-3′) and C (5′-CTGGACCCGGCCTGGGAAGACATGTTCGAG-3′) included the 402 C to G mutation that corresponds to the C134W amino acid change. The resulting mutant FOXL2 was ligated to the pFlag-FOXL2/pCS2+ construct using standard procedures. A β-galactosidase reporter plasmid driven by the Herpes virus thymidine kinase promoter was used as internal control for transfection efficiency in all luciferase reporter experiments.
2.2. Harvest and culture of primary rat granulosa cells
Female Sprague Dawley rats (24 days old) were implanted with silastic implants (Dow Corning, Corp., Midland, MI) containing 10 mg diethylstilbestrol (DES) to stimulate GC proliferation. Four days after DES implantation GCs were harvested from the ovaries as previously described [45]. Briefly, ovarian follicles were punctured with needles to release GCs and oocytes, and oocytes were subsequently removed using a 40 μm cell strainer. GCs were cultured in serum-free McCoys 5A culture media containing antibiotics. The Institutional Animal Care and Use Committee at the University of California, San Diego, approved all animal protocols.
2.3. Transfections and luciferase reporter assays
For luciferase reporter assays, primary rat GCs were plated into serum-coated 12-well tissue culture plates at a density of 5 × 105 cells/well. The next day media were replaced with fresh media containing 10% fetal bovine serum (FBS) and transfected with indicated plasmids for 4 hr using Lipofectamine Ltx transfection reagent (Invitrogen, Carlsbad, CA) as per manufactures instructions. At the end of the transfection, the media containing transfection reagents was removed and replaced with fresh serum-free culture media and transfected cells cultured for a further 15 hr with vehicle or GDF-9 at described doses.
COV434 cells were routinely cultured in 10 % FBS in DMEM F-12 media (GIBCO-Invitrogen, Carlsbad, CA) with and antibiotics at 37 °C in a constant atmosphere of 5 % CO2. For transfections, COV434 cells were plated into 24-well plates at a density of 4 × 105 cells/well in 1 ml of culture medium. Cells were allowed to attach overnight. The next day the conditioned media was replaced with fresh culture media and cells transfected with indicated plasmids for 4 hr using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). At the end of transfection, the media containing the transfection reagent was discarded and fresh culture media added to the cells, and cells were incubated for 16 hr before an 8 hr treatment with vehicle, GDF-9 in serum-free culture media.
Cells were lysed for evaluation of luciferase reporter activity using a dual-light reporter assay (Applied Biosystems, Bedford, MA) on a Veritas Microplate luminometer. Luciferase activity was normalized to the β-gal internal control to correct for transfection efficiency. Data are represented as the ratio of the normalized luciferase activity to the untreated no-expression plasmid group.
2.3. Co-immuno precipitation assays
COV434 cells were plated in 12-well plates in a density of 2.5 × 105 cells/well in 2 ml of culture medium. Cells were allowed to attach overnight, then transfected as described above. Following the 20 hr culture after transfection, cells were lysed in 25mM Tris-HCl PH 7.5 containing 0.1 % triton x-100, 10 % glycerol, 1mM MgCl2, 0.5 mM EDTA, 100 mM NaCl, 5 mM NaF, 1 mM Na4P2O7, 1 mM DTT, and protease inhibitor cocktail (Roche) and sonicated at 50 % power for 5–10 seconds. Cells extracts were centrifuged at maximum speed for 10 minutes at 4 °C and the supernatant stored at −80 °C until use. Cell extracts were incubated with anti-Flag M2 antibody conjugated agarose beads previously equilibrated in lysis buffer by rotating for 2 hr at 4 °C. Beads were washed 3 times with lysis buffer, then immunocomplexes were analyzed by western blot.
2.4. Statistic analysis
All experiments contained triplicate samples, and all experiments were repeated 3 times. Data from the three replicate experiments were pooled for statistical analyses. Data analysis was performed using StatPlus (Chicago, IL) software using one-way ANOVA or two-way ANOVA (as required), supplemented with Tukey-Kramer post hoc tests. Significance was set at P < 0.05. Values or figures are all presented as the mean ± SEM.
3. Results
In this study, we used the rFS(0.3ex45)-luc reporter plasmid known to contain cis-elements for Smad3 (Smad binding element, SBE) and FOXL2 (forkhead binding element, FBE) necessary for an activin A response in the mouse pituitary gonadotrope cell line, α T3-1 [20]. Rat primary GCs and COV434 cells were transfected with the rFS(0.3ex45)-luc reporter plasmid followed by treatment with GDF-9, an important Smad3 signaling regulator of GC function [22–24]. Luciferase reporter activity in rat GCs was increased in a dose dependent manner by GDF-9, whereas that in COV434 cells was not changed (Fig. 1).
Figure 1. Effect of GDF-9 on the follistatin promoter activity in rat GCs and COV434 cells.
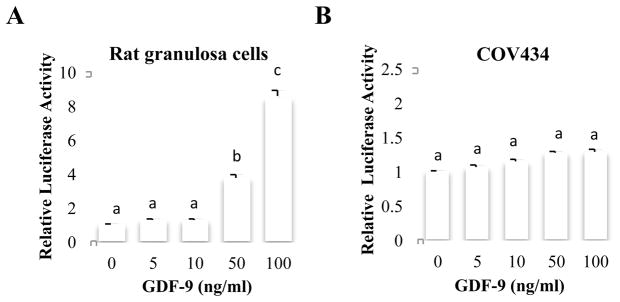
Primary rat GCs (A) were transfected with the rFS(0.3ex45)-luc reporter, then treated with indicated concentrations of GDF-9 for 15 hr, followed by measurement of luciferase activity. COV434 cells (B) were transfected with the rFS(0.3ex45)-luc reporter, then treated with indicated concentrations of GDF-9 for 8 hr, followed by measurement of luciferase activity. Each experiment contained triplicate wells and was repeated 3 times. Panel A shows the mean ± SEM of one representative experiment, Panel B values shown are the mean ± SEM of 3 replicate experiments. Different letters indicate a significant difference between treatments (p<0.05).
We then asked whether the difference observed in Fig. 1 was attributed to the presence or absence of endogenous FOXL2 expression. COV434 cells were transfected with FOXL2 and treated with GDF-9 (Fig. 2A). The luciferase activity was now induced by GDF-9, suggesting that FOXL2 is required for GDF-9 induction of follistatin in COV434 cells. In contrast, overexpression of Smad3 combined with GDF-9 treatment was not sufficient to induce follistatin transcription (Fig. 2B).
Figure 2. Effects of FOXL2wt and Smad3 expression in COV434 cells on GDF-9 regulation of follistatin promoter activity.
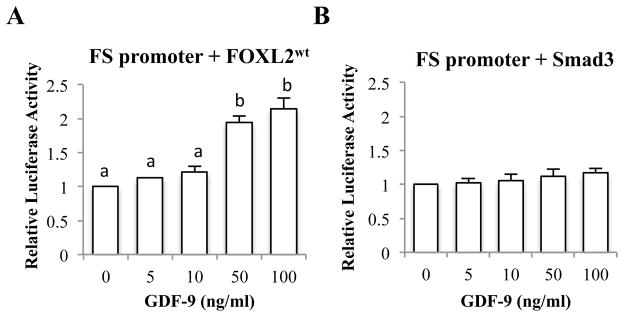
COV434 cells were co-transfected with the rFS(0.3ex45)-luc reporter and FOXL2wt (A) or Smad3 (B), then treated with indicated concentrations of GDF-9 for 8 hr, followed by measurement of luciferase activity. Each experiment contained triplicate wells and values shown are mean ± SEM of 3 replicate experiments. Different letters indicate a significant difference between treatments (p<0.05). FS: follistatin
We next proceeded to determine whether FOXL2C134W similarly regulates follistatin transcription in the absence or presence of GDF-9. To determine whether the C134W mutation alters FOXL2 protein expression or degradation, we first evaluated protein levels of FOXL2 and Smad3 in COV434 cells transfected with Smad3 along with FOXL2wt or FOXL2C134W. Importantly, the levels of FOXL2wt and FOXL2C134W were comparable as shown in Fig. 3A. The two bands detected with the flag antibody most likely reflect FOXL2 with different posttranslational modifications such as sumolyation, phosphorylation, acetylation, or ubiquitination as reported previously [46,47]. COV434 cells were then transfected with either Smad3, FOXL2wt or FOXL2C134W alone or in combination and cultured in the presence or absence of GDF-9 followed by the measurement of the rFS(0.3ex45)-luc activity (Fig. 3B). In untreated cells, expression of either Smad3 or FOXL2wt alone had no effect on the reporter activity. In contrast, expression of FOXL2C134W alone, Smad3 plus FOXL2wt, or Smad3 plus FOXL2C134W significantly increased follistatin luciferase activity. GDF-9 treatment had no effect on the reporter activity in the absence of FOXL2 overexpression, consistent with results in Fig. 1B. However, in cells transfected with FOXL2wt or FOXL2C134W, GDF-9 similarly enhanced reporter activity. In the presence of Smad3 overexpression, the ability of GDF-9 to induce follistatin reporter activity was markedly different depending on whether FOXL2wt or FOXL2C134W was co-expressed with Smad3. In stark contrast to the enhancement of GDF-9 induction of reporter activity in cells co-transfected with Smad3 plus FOXL2wt, GDF-9 had no activity in cells transfected with Smad3 plus FOXL2C134W. These results suggest differential regulation of follistatin transcription by FOXL2C134W, and show that the altered activity of FOXL2C134W is dependent on the presence of Smad3.
Figure 3. Effects of FOXL2wt and FOXL2C134W in the presence or absence of Smad3 overexpression in COV434 cells on GDF-9 regulation of follistatin promoter activity.

(A) COV434 cells were transfected with Myc-Smad3, Flag-FOXL2wt, Flag-FOXL2C134W or an empty vector as indicated, then protein expression analyzed by western blot. Anti-Myc and anti-Flag antibodies were used to detect Myc-Smad3 and Flag-FOXL2wt/Flag-FOXL2C134W, respectively.α-Tubulin was used as internal loading control. The experiment was performed in triplicate, with one representative experiment depicted. (B) COV434 cells were transfected with the rFS(0.3ex45)-luc reporter plasmid and Myc-Smad3, Flag- FOXL2wt, Flag-FOXL2C134W or an empty vector as indicated, then treated with vehicle, 100 ng/ml GDF-9 for 8 hr. Each experiment contained triplicate wells and values shown are mean ± SEM of 3 replicate experiments. Different letters indicate a significant difference between transfection conditions (p<0.05). # indicates a significant (p<0.05) effect of GDF-9 treatment compared with untreated cells within each transfection condition.
Given that Smad3 and FOXL2 can form a complex [20], it is reasonable to speculate that the C134W mutation in FOXL2 could modify the interaction between Smad3 and FOXL2. To address this possibility, we performed co-immunoprecipitation experiments using Myc-tagged Smad3 and Flag-tagged FOXL2wt or FOXL2C134W transiently expressed in COV434 cells. As shown in Fig. 4, FOXL2wt and FOXL2C134W co-immunoprecipitated Smad3 with equivalent efficiency, indicating that the C134W mutation does not alter its capacity to interact with Smad3. These data suggest that a change in the interaction between Smad3 and FOXL2 due to the C134W mutation may not be the main cause behind the loss of the GDF-9 responsiveness observed in the follistatin transcriptional activity.
Figure 4. Interaction of FOXL2 and Smad3.

COV434 cells were transfected with Myc-Smad3, Flag- FOXL2wt and Flag-FOXL2C134W for 24 hr. Cell lysates were incubated with anti-Flag antibody conjugatedbeads to immunoprecipitate Flag-FOXL2wt or Flag-FOXL2C134W. Bound proteins were subjected to SDS-PAGE and western blot analysis. Anti-Myc and anti-Flag antibodies were used to detect Myc-Smad3 and Flag-FOXL2wt/Flag-FOXL2C134W, respectively. The experiment was performed in triplicate, with one representative experiment depicted.
To determine how Smad3-FOXL2wt and Smad3-FOXL2C134W exhibit altered function on the intronic enhancer elements in the follistatin gene, we generated rFS(0.3ex45)-luc reporter plasmids with mutations in the SBE and FBE (Fig. 5A). The mutations were aimed to block Smad3 binding to SBE (mutant 1), FOXL2 binding to FBE (mutant 2), or both Smad3 and FOXL2 binding (mutant 3) as observed in the previous study [20]. Using these mutant reporter constructs, COV434 cells were transfected with Smad3, FOXL2wt or FOXL2C134W alone or in combination to examine GDF-9 effects on the mutant reporters. It was surprising that GDF-9 maintained activity in the absence of a functional SBE (Fig. 5B), suggesting that GDF-9 activity does not require Smad3 binding to DNA at the known SBE or that FOXL2 acts to tether Smad3 to the DNA. It is of particular note that mutation of the SBE abolished the suppressive effect of FOXL2C134W on GDF-9 activity previously observed on the wild-type luciferase reporter, which reinforces the concept that FOXL2C134W actions are dependent on its association with Smad3 and the ability of Smad3 to directly bind the SBE. Further, mutation of the FBE (mutant 2 and 3 reporters) completely prevented GDF-9 activity (Fig. 5C and D), suggesting that FBE in intron 1 of the follistatin gene is essential for GDF-9 stimulation in COV434 cells.
Figure 5. Effects of mutations at the SBE and/or FBE within the follistatin intronic enhancer.

(A) Partial DNA sequences of the intron 1 of the human and rat follistatin genes with a mutation in the SBE (mutant 1), FBE (mutant 2), or both the SBE and FBE (mutant 3) of the rFS (0.3ex45)-luc reporter plasmid used for this study. The wild type sequences at and surrounding the SEB and FBE of the human and rat genes are identical. (B–D) COV434 cells were transfected with mutant 1 (B), 2 (C) or 3 (D) of the rFS(0.3ex45)-luc reporter in combination with Myc-Smad3, Flag-FOXL2wt, Flag-FOXL2C134W or an empty vector as indicated. Following transfection, cells were treated with vehicle or 100 ng/ml GDF-9 for 8 hr. Each experiment contained triplicate wells and values shown are mean ± SEM of 3 replicate experiments. Different letters indicate a significant difference between transfection conditions (p<0.05). # indicates a significant (p<0.05) effect of GDF-9 treatment compared with untreated cells within each transfection condition.
Collectively, these data show that ectopic FOXL2C134W expression in COV434 cells prevents GDF-9 enhancement of follistatin transcription only when it was co-expressed with Smad3. Moreover, mutation of the SBE alone restored the follistatin promoter response to GDF-9 in cells co-expressing FOXL2C134W and Smad3.
4. Discussion
Understanding how a single somatic FOXL2 mutation (FOXL2C134W) that is present in almost all GCTs contributes to altered GC function and ultimately promotes tumor development is of central importance to advance the development of improved detection methods and novel therapeutic options. It has been shown that in the pituitary exogenously expressed FOXL2 and Smad3 associate and co-operate to promote activin A induction of follistatin transcription [20]. Transactivation studies in KGN and HeLa cells on known FOXL2 target reporters showed that FOXL2C134W and FOXL2wt behaved similarly except for the artificial 3xGRAS-luc reporter on which FOXL2C134W was slightly hyperactive [25]. The 3xGRAS-luc contains three repeats of a composite regulatory element with overlapping Smad3, FOXL2 and AP1 binding sites that are present in the murine GnRH receptor promoter. The FOXL2-Smad3 complex binds this synthetic reporter [17], therefore it seemed possible that FOXL2C134W could also affect other common targets for Smad3 and FOXL2 in the ovary that may contribute to GCT formation. In the current study, we showed that in COV434 cells GDF-9 did not regulate follistatin transcription, even in the presence of Smad3 overexpression. The inability of Smad3 to elicit a GDF-9 response in COV434 cells is likely explained by the fact that COV434 cells do not express endogenous FOXL2. We showed that restoring FOXL2 through expression of FOXL2wt or FOXL2C134W facilitated GDF-9 stimulation of follistatin promoter activity (Fig. 2A and 3B). Interestingly, co-expression of FOXL2wt and Smad3 in COV434 cells enhanced the response to GDF-9 above that in cells expressing only FOXL2wt, suggesting that Smad3 supported FOXL2 actions. We attribute the ability of FOXL2, without exogenous Smad3, to facilitate GDF-9 induction of follistatin promoter activity to the presence of endogenous Smad3 in COV434 cells. Overall, our results showed that both transcription factors were necessary to mediate GDF-9 induction of the follistatin promoter in COV434 cells.
Comparison of the activity of the GCT mutant FOXL2C134W with wild-type FOXL2wt yielded interesting results. We demonstrated that FOXL2C134W, but not FOXL2wt, abruptly prevented the follistatin promoter activity in response to GDF-9 in the presence of exogenous Smad3 (Fig. 3B). Interestingly, in the absence of exogenous Smad3, FOXL2C134W had equivalent activity to FOXL2wt, promoting GDF-9 induction of follistatin luciferase activity. Mutational studies supported these findings, with mutation of the SBE reversing the negative effects of FOXL2C134W on the follistatin promoter (Fig. 5B). Surprisingly, mutating the SBE conferred Smad3 activity on the follistatin promoter. It is possible that mutating the SBE alone could make a small change in the promoter activity in a way that allows another unidentified factor(s) to confer Smad3 activity in the absence of direct Smad3 binding to the DNA. It is worth noting this effect of Smad3 is specific to the SBE mutation, as mutation of the FBE in the presence or absence of the mutated SBE does not exhibit Smad3 activity. Overall, we showed that mutation of either the SBE or FBE or both prevented the altered suppressive FoxL2C134W activity. Thus, we propose that the negative effect of FOXL2C134W on GDF-9-induced follistatin promoter involves a change in the interaction between Smad3 and FOXL2C134W. We speculate that SBE-bound Smad3 associated with FBE-bound FOXL2C134W exhibits an altered conformation of the complex, compared with the case of Smad3 and -FOXL2wt, leading to the loss of GDF-9 signaling activity. Further, we propose that when the SBE is mutated, Smad3 is similarly tethered to FOXL2wt and FOXL2C134W, which facilitate normal GDF-9-induced activity.
5. Conclusions
In summary, our study shows that i) FOXL2 expression is required for GDF-9 stimulation of follistatin transcription in COV434 cells, ii) FOXL2wt and FOXL2C134W associate with Smad3 with equivalent efficiency in COV434 cells, iii) ectopic FOXL2C134W expression in COV434 cells prevents GDF-9 enhancement of follistatin transcription when co-expressed with Smad3 and iv) mutation of the SBE restores normal FOXL2 activity to FOXL2C134W, thus the altered activity of FOXL2C134W is dependent on the ability of Smad3 to directly bind the SBE. These findings suggest that there is a critical conformational change in the structure of the complex between FOXL2wt/Smad3 and FOXL2C134W/Smad3 only when they occupy both the SBE and FBE in the follistatin intronic enhancer. Such a structural change may cause an altered interaction with other co-regulatory factors, leading to altered GDF-9 activity on the follistatin transcription. Understanding how FOXL2C134W alters GC function may ultimately drive the development of novel therapeutic options for GCT patients.
Highlights.
FOXL2 is required for GDF-9 activation of follistatin transcription in COV434 cells
FOXL2wt and FOXL2C134W similarly bind Smad3
In co-ordination with Smad3, FoxL2C134W prevents GDF-9 activity
FOXL2C134W altered activity requires intact Smad binding cis-element
Acknowledgments
The authors thank Dr. Louise Bilezikjian for providing us with the follistatin luciferase reporters and Flag-tagged human FOXL2 and Myc-tagged human Smad3 expression plasmids. We also thank Dr. Rik Derynck for the original human Smad3 cDNA clone. This work was supported by National Institutes of Health (NIH) grant R01 HD41494 and the Eunice Kennedy Shriver National Institute of Child Health and Human Development/NIH through cooperative agreement (U54 HD012303) as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research.
Footnotes
Disclosure summary: The authors have nothing to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kalfa N, Veitia RA, Benayoun BA, Boizet-Bonhoure B, Sultan C. The new molecular biology of granulosa cell tumors of the ovary. Genome Med. 2009;1:81. doi: 10.1186/gm81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colombo N, Parma G, Zanagnolo V, Insinga A. Management of ovarian stromal cell tumors. J Clin Oncol. 2007;25:2944–51. doi: 10.1200/JCO.2007.11.1005. [DOI] [PubMed] [Google Scholar]
- 3.Amsterdam A, Selvaraj N. Control of differentiation, transformation, and apoptosis in granulosa cells by oncogenes, oncoviruses, and tumor suppressor genes. Endocrine Reviews. 1997;18:435–461. doi: 10.1210/edrv.18.4.0306. [DOI] [PubMed] [Google Scholar]
- 4.Shah SP, Kobel M, Senz J, Morin RD, Clarke BA, Wiegand KC, Leung G, Zayed A, Mehl E, Kalloger SE, Sun M, Giuliany R, Yorida E, Jones S, Varhol R, Swenerton KD, Miller D, Clement PB, Crane C, Madore J, Provencher D, Leung P, DeFazio A, Khattra J, Turashvili G, Zhao Y, Zeng T, Glover JN, Vanderhyden B, Zhao C, Parkinson CA, Jimenez-Linan M, Bowtell DD, Mes-Masson AM, Brenton JD, Aparicio SA, Boyd N, Hirst M, Gilks CB, Marra M, Huntsman DG. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med. 2009;360:2719–29. doi: 10.1056/NEJMoa0902542. [DOI] [PubMed] [Google Scholar]
- 5.Schrader KA, Gorbatcheva B, Senz J, Heravi-Moussavi A, Melnyk N, Salamanca C, Maines-Bandiera S, Cooke SL, Leung P, Brenton JD, Gilks CB, Monahan J, Huntsman DG. The Specificity of the FOXL2 c.402C>G Somatic Mutation: A Survey of Solid Tumors. PLoS One. 2009;4:e7988. doi: 10.1371/journal.pone.0007988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jamieson S, Butzow R, Andersson N, Alexiadis M, Unkila-Kallio L, Heikinheimo M, Fuller PJ, Anttonen M. The FOXL2 C134W mutation is characteristic of adult granulosa cell tumors of the ovary. Mod Pathol. 2010;23:1477–85. doi: 10.1038/modpathol.2010.145. [DOI] [PubMed] [Google Scholar]
- 7.Kim T, Sung CO, Song SY, Bae DS, Choi YL. FOXL2 mutation in granulosa-cell tumours of the ovary. Histopathology. 2010;56:408–10. doi: 10.1111/j.1365-2559.2010.03487.x. [DOI] [PubMed] [Google Scholar]
- 8.Kim MS, Hur SY, Yoo NJ, Lee SH. Mutational analysis of FOXL2 codon 134 in granulosa cell tumour of ovary and other human cancers. J Pathol. 2010;221:147–52. doi: 10.1002/path.2688. [DOI] [PubMed] [Google Scholar]
- 9.Al-Agha OM, Huwait HF, Chow C, Yang W, Senz J, Kalloger SE, Huntsman DG, Young RH, Gilks CB. FOXL2 is a sensitive and specific marker for sex cord-stromal tumors of the ovary. Am J Surg Pathol. 2011;35:484–94. doi: 10.1097/PAS.0b013e31820a406c. [DOI] [PubMed] [Google Scholar]
- 10.Pisarska MD, Barlow G, Kuo FT. Minireview: Roles of the Forkhead Transcription Factor FOXL2 in Granulosa Cell Biology and Pathology. Endocrinology. 2011 doi: 10.1210/en.2010-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt D, Ovitt CE, Anlag K, Fehsenfeld S, Gredsted L, Treier AC, Treier M. The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development. 2004;131:933–42. doi: 10.1242/dev.00969. [DOI] [PubMed] [Google Scholar]
- 12.Uda M, Ottolenghi C, Crisponi L, Garcia JE, Deiana M, Kimber W, Forabosco A, Cao A, Schlessinger D, Pilia G. Foxl2 disruption causes mouse ovarian failure by pervasive blockage of follicle development. Hum Mol Genet. 2004;13:1171–81. doi: 10.1093/hmg/ddh124. [DOI] [PubMed] [Google Scholar]
- 13.Uhlenhaut NH, Jakob S, Anlag K, Eisenberger T, Sekido R, Kress J, Treier AC, Klugmann C, Klasen C, Holter NI, Riethmacher D, Schutz G, Cooney AJ, Lovell-Badge R, Treier M. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell. 2009;139:1130–42. doi: 10.1016/j.cell.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 14.Arcellana-Panlilio MY, Egeler RM, Ujack E, Magliocco A, Stuart GC, Robbins SM, Coppes MJ. Evidence of a role for the INK4 family of cyclin-dependent kinase inhibitors in ovarian granulosa cell tumors. Genes Chromosomes Cancer. 2002;35:176–81. doi: 10.1002/gcc.10108. [DOI] [PubMed] [Google Scholar]
- 15.Benayoun BA, Georges AB, L’Hote D, Andersson N, Dipietromaria A, Todeschini AL, Caburet S, Bazin C, Anttonen M, Veitia RA. Transcription factor FOXL2 protects granulosa cells from stress and delays cell cycle: role of its regulation by the SIRT1 deacetylase. Hum Mol Genet. 2011;20:1673–86. doi: 10.1093/hmg/ddr042. [DOI] [PubMed] [Google Scholar]
- 16.Kim JH, Yoon S, Park M, Park HO, Ko JJ, Lee K, Bae J. Differential apoptotic activities of wild-type FOXL2 and the adult-type granulosa cell tumor-associated mutant FOXL2 (C134W) Oncogene. 2010 doi: 10.1038/onc.2010.541. [DOI] [PubMed] [Google Scholar]
- 17.Ellsworth BS, Burns AT, Escudero KW, Duval DL, Nelson SE, Clay CM. The gonadotropin releasing hormone (GnRH) receptor activating sequence (GRAS) is a composite regulatory element that interacts with multiple classes of transcription factors including Smads, AP-1 and a forkhead DNA binding protein. Mol Cell Endocrinol. 2003;206:93–111. doi: 10.1016/s0303-7207(03)00235-1. [DOI] [PubMed] [Google Scholar]
- 18.Ellsworth BS, Egashira N, Haller JL, Butts DL, Cocquet J, Clay CM, Osamura RY, Camper SA. FOXL2 in the pituitary: molecular, genetic, and developmental analysis. Mol Endocrinol. 2006;20:2796–805. doi: 10.1210/me.2005-0303. [DOI] [PubMed] [Google Scholar]
- 19.Lamba P, Fortin J, Tran S, Wang Y, Bernard DJ. A novel role for the forkhead transcription factor FOXL2 in activin A-regulated follicle-stimulating hormone beta subunit transcription. Mol Endocrinol. 2009;23:1001–13. doi: 10.1210/me.2008-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blount AL, Schmidt K, Justice NJ, Vale WW, Fischer WH, Bilezikjian LM. FoxL2 and Smad3 coordinately regulate follistatin gene transcription. J Biol Chem. 2009;284:7631–45. doi: 10.1074/jbc.M806676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corpuz PS, Lindaman LL, Mellon PL, Coss D. FoxL2 Is required for activin induction of the mouse and human follicle-stimulating hormone beta-subunit genes. Molecular Endocrinology. 2010;24:1037–51. doi: 10.1210/me.2009-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pangas SA, Matzuk MM. Genetic models for transforming growth factor beta superfamily signaling in ovarian follicle development. Molecular and Cellular Endocrinology. 2004;225:83–91. doi: 10.1016/j.mce.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Shimasaki S, Moore RK, Otsuka F, Erickson GF. The bone morphogenetic protein system in mammalian reproduction. Endocrine Reviews. 2004;25:72–101. doi: 10.1210/er.2003-0007. [DOI] [PubMed] [Google Scholar]
- 24.Knight PG, Glister C. TGF-beta superfamily members and ovarian follicle development. Reproduction. 2006;132:191–206. doi: 10.1530/rep.1.01074. [DOI] [PubMed] [Google Scholar]
- 25.Benayoun BA, Caburet S, Dipietromaria A, Georges A, D’Haene B, Pandaranayaka PJ, L’Hote D, Todeschini AL, Krishnaswamy S, Fellous M, De Baere E, Veitia RA. Functional exploration of the adult ovarian granulosa cell tumor-associated somatic FOXL2 mutation p. Cys134Trp (c.402C>G) PLoS One. 2010;5:e8789. doi: 10.1371/journal.pone.0008789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Middlebrook BS, Eldin K, Li X, Shivasankaran S, Pangas SA. Smad1-Smad5 ovarian conditional knockout mice develop a disease profile similar to the juvenile form of human granulosa cell tumors. Endocrinology. 2009;150:5208–17. doi: 10.1210/en.2009-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimasaki S, Koga M, Esch F, Cooksey F, Mercado M, Koba A, Ueno N, Ying SY, Ling N. Primary structure of the human follistatin precursor and its genomic organization. Proceedings of the National Academy of Sciences, USA. 1988;85:4218–4222. doi: 10.1073/pnas.85.12.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimasaki S, Koga M, Buscaglia ML, Simmons DM, Bicsak TA, Ling N. Follistatin gene expression in the ovary and extragonadal tissues. Molecular Endocrinology. 1989;3:651–659. doi: 10.1210/mend-3-4-651. [DOI] [PubMed] [Google Scholar]
- 29.Nakatani A, Shimasaki S, DePaolo LV, Erickson GF, Ling N. Cyclic changes in follistatin messenger ribonucleic acid and its protein in the rat ovary during the estrous cycle. Endocrinology. 1991;129:603–611. doi: 10.1210/endo-129-2-603. [DOI] [PubMed] [Google Scholar]
- 30.Roberts VJ, Barth S, El-Roeiy A, Yen SS. Expression of inhibin/activin subunits and follistatin messenger ribonucleic acids and proteins in ovarian follicles and the corpus luteum during the human menstrual cycle. Journal of Clinical Endocrinology and Metabolism. 1993;77:1402–1410. doi: 10.1210/jcem.77.5.8077341. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura T, Takio K, Eto Y, Shibai H, Titani K, Sugino H. Activin-binding protein from rat ovary is follistatin. Science. 1990;247:836–838. doi: 10.1126/science.2106159. [DOI] [PubMed] [Google Scholar]
- 32.DePaolo LV, Bicsak TA, Erickson GF, Shimasaki S, Ling N. Follistatin and activin: A potential intrinsic regulatory system within diverse tissues. Proceedings of the Society for Experimental Biology and Medicine. 1991;198:500–512. doi: 10.3181/00379727-198-43286a. [DOI] [PubMed] [Google Scholar]
- 33.Xiao S, Findlay JK. Interaction between activin and follicle-stimulating hormone-suppressing protein and their mechanisms of action on cultured rat granulosa cells. Molecular and Cellular Endocrinology. 1991;79:99–107. doi: 10.1016/0303-7207(91)90100-7. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura T, Hasegawa Y, Sugino K, Kogawa K, Titani K, Sugino H. Follistatin inhibits activin-induced differentiation of rat follicular granulosa cells in vitro. Biochim Biophys Acta. 1992;1135:103–109. doi: 10.1016/0167-4889(92)90173-9. [DOI] [PubMed] [Google Scholar]
- 35.Xiao S, Robertson DM, Findlay JK. Effects of activin and follicle-stimulating hormone (FSH)-suppressing protein/follistatin on FSH receptors and differentiation of cultured rat granulosa cells. Endocrinology. 1992;131:1009–1016. doi: 10.1210/endo.131.3.1505447. [DOI] [PubMed] [Google Scholar]
- 36.Cataldo NA, Rabinovici J, Fujimoto VY, Jaffe RB. Follistatin antagonizes the effects of activin-A on steroidogenesis in human luteinizing granulosa cells. Journal of Clinical Endocrinology and Metabolism. 1994;79:272–277. doi: 10.1210/jcem.79.1.7517947. [DOI] [PubMed] [Google Scholar]
- 37.Erämaa M, Hildén K, Tuuri T, Ritvos O. Regulation of inhibin/activin subunit messenger ribonucleic acids (mRNAs) by activin A and expression of activin receptor mRNAs in cultured human granulosa-luteal cells. Endocrinology. 1995;136:4382–4389. doi: 10.1210/endo.136.10.7664658. [DOI] [PubMed] [Google Scholar]
- 38.Yamashita H, ten Dijke P, Huylebroeck D, Sampath TK, Andries M, Smith JC, Heldin CH, Miyazono K. Osteogenic protein-1 binds to activin type II receptors and induces certain activin-like effects. Journal of Cell Biology. 1995;130:217–226. doi: 10.1083/jcb.130.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iemura S, Yamamoto TS, Takagi C, Uchiyama H, Natsume T, Shimasaki S, Sugino H, Ueno N. Direct binding of follistatin to a complex of bone morphogenetic protein and its receptor inhibits ventral and epidermal cell fates in early Xenopus embryo. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:9337–9342. doi: 10.1073/pnas.95.16.9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otsuka F, Moore RK, Iemura SI, Ueno N, Shimasaki S. Follistatin inhibits the function of the oocyte-derived factor BMP-15. Biochemical and Biophysical Research Communication. 2001;289:961–966. doi: 10.1006/bbrc.2001.6103. [DOI] [PubMed] [Google Scholar]
- 41.Miró F, Hillier SG. Modulation of granulosa cell deoxyribonucleic acid synthesis and differentiation by activin. Endocrinology. 1996;137:464–468. doi: 10.1210/endo.137.2.8593790. [DOI] [PubMed] [Google Scholar]
- 42.Otsuka F, Yao Z, Lee TH, Yamamoto S, Erickson GF, Shimasaki S. Bone morphogenetic protein-15: Identification of target cells and biological functions. Journal of Biological Chemistry. 2000;275:39523–39528. doi: 10.1074/jbc.M007428200. [DOI] [PubMed] [Google Scholar]
- 43.Lee MJ, Yang CW, Jin DC, Chang YS, Bang BK, Kim YS. Bone morphogenetic protein-7 inhibits constitutive and interleukin-1b-induced monocyte chemoattractant protein-1 expression in human mesangial cells: Role for JNK/AP-1 pathway. Journal of Immunology. 2003;170:2557–2563. doi: 10.4049/jimmunol.170.5.2557. [DOI] [PubMed] [Google Scholar]
- 44.van den Berg-Bakker CA, Hagemeijer A, Franken-Postma EM, Smit VT, Kuppen PJ, van Ravenswaay Claasen HH, Cornelisse CJ, Schrier PI. Establishment and characterization of 7 ovarian carcinoma cell lines and one granulosa tumor cell line: growth features and cytogenetics. Int J Cancer. 1993;53:613–20. doi: 10.1002/ijc.2910530415. [DOI] [PubMed] [Google Scholar]
- 45.Otsuka F, Shimasaki S. A negative feedback system between oocyte bone morphogenetic protein 15 and granulosa cell kit ligand: its role in regulating granulosa cell mitosis. Proceedings of the National Academy of Sciences U S A. 2002;99:8060–8065. doi: 10.1073/pnas.122066899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benayoun BA, Auer J, Caburet S, Veitia RA. The post-translational modification profile of the forkhead transcription factor FOXL2 suggests the existence of parallel processive/concerted modification pathways. Proteomics. 2008;8:3118–23. doi: 10.1002/pmic.200800084. [DOI] [PubMed] [Google Scholar]
- 47.Georges A, Benayoun BA, Marongiu M, Dipietromaria A, L’Hote D, Todeschini AL, Auer J, Crisponi L, Veitia RA. SUMOylation of the Forkhead Transcription Factor FOXL2 Promotes Its Stabilization/Activation through Transient Recruitment to PML Bodies. PLoS One. 2011;6:e25463. doi: 10.1371/journal.pone.0025463. [DOI] [PMC free article] [PubMed] [Google Scholar]