

Abstract
The forkhead box O subfamily has four members including FoxO1, FoxO3, FoxO4 and FoxO6. Unlike other three FoxO members, FoxO6 has garnered considerably less attention due to earlier reports that FoxO6 is limited to the brain. Recent data indicate that FoxO6 is produced in the liver of both rodent and human origins. Hepatic FoxO6 activity, which remains at low basal levels in fed states, is markedly induced in fasted mice. FoxO6 activity becomes abnormally higher in the liver of mice with dietary obesity or type 2 diabetes. Genetically engineered mice with elevated FoxO6 activity in the liver exhibit pre-diabetes, culminating in the development of glucose intolerance, fasting hyperglycemia and hyperinsulinemia. Conversely, inhibition of FoxO6 activity in insulin-resistant liver results in the reduction of fasting hyperglycemia, contributing to the amelioration of hyperinsulinemia in type 2 diabetic mice. These new data suggest that FoxO6 is an important regulator of hepatic glucose metabolism in response to insulin or physiological cues. Insulin inhibits FoxO6 activity by promoting its phosphorylation and disabling its activity in the nucleus without altering its subcellular distribution via a mechanism that is distinct from other members of the FoxO subfamily. In this article, we will provide a comprehensive review on the role of FoxO6 in glucose metabolism in health and disease. We will also address whether FoxO6 dysregulation is a contributing factor for the pathogenesis of fasting hyperglycemia and discuss whether FoxO6 is a potential therapeutic target for improving fasting hyperglycemia in type 2 diabetes.
The FoxO subfamily
The FoxO subfamily consists of four members including FoxO1, FoxO3, FoxO4 and FoxO6 (Fig. 1). This subfamily of nuclear transcription factors are characterized by a highly conserved DNA binding motif, known as forkhead box or winged helix domain [1]. They act as substrates of Akt/PKB to mediate the inhibitory effect of insulin (or IGF-1) on key functions in diverse pathways including cell survival, proliferation, differentiation, oxidative stress, and metabolism in mammals [1]. FoxO homologues, Daf16 in C. elegans and dFoxO in D. melanogaster, contribute to the induction of anti-oxidative function and prolongation of lifespan in response to environmental stress [2, 3].
Fig. 1. Phylogeny of the FoxO subfamily.

The phylogenetic tree for the FoxO subfamily was generated using the TreeFam program (http://www.treefam.org). The number in the phylogenic tree denotes the score that is linearly scaled from 1 to 100. The score measures how often the same ortholog (or paralog within-species) pair can be inferred from a resampled tree. The tree branches corresponding to the mammalian FoxO1 and FoxO6 are marked by red box.
It is well established that insulin or IGF-1 exerts its inhibitory effect on gene expression via a highly conserved sequence (TG/ATTTT/G), termed insulin response element (IRE) in target promoters [1] (Fig. 2). In the absence of insulin (or IGF-1), FoxO nuclear transcription factors reside in the nucleus and bind as a trans-activator to the IRE DNA motif, enhancing promoter activity. In response to insulin (or IGF-1), FoxO proteins are phosphorylated by Akt/PKB in the PI3K-dependent pathway. This effect promotes FoxO trafficking from the nucleus to cytoplasm, resulting in the inhibition of target gene expression [1]. Such phosphorylation-dependent nuclear export serves as an acute mechanism by which insulin (or IGF-1) effectively keeps FoxO transcriptional activity in check in cells. Failure in phosphorylation of FoxO results in its permanent nuclear localization and constitutive trans-activation of gene expression, an aberrant event that is often associated with pathogenesis of diseases [1, 4–9].
Fig. 2. FoxO mediates insulin action on target gene expression.
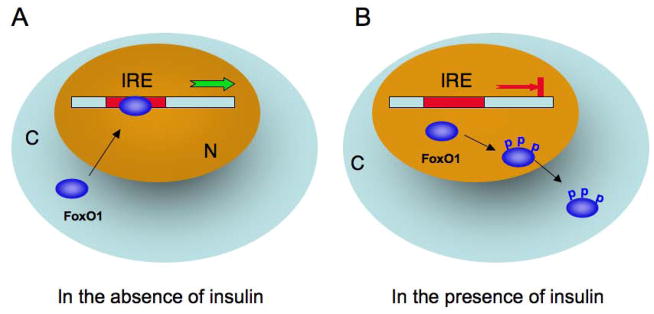
A. FoxO1 binds to the insulin response element (IRE) within the target promoter and promotes target gene expression in the absence of insulin. B. FoxO1 becomes phosphorylated in response to insulin. This effect promotes FoxO1 nuclear exclusion, resulting in the inhibition of target gene expression. N, nucleus. C, cytoplasm.
FoxO6 – a new member of the FoxO subfamily
FoxO6, as opposed to other FoxO members, has garnered the least attention in research. This derives in part from earlier reports that FoxO6 expression is limited to the brain [10, 11]. This finding is subsequently rectified by Kim et al. [12], who show that FoxO6 is also expressed in peripheral tissues including the liver, intestine, lung, kidney, muscle, and adipose tissues, a broad tissue distribution that is characteristic of other members of the FoxO subfamily. Although classified to the FoxO subfamily, FoxO6 differs from other FoxO members in fundamental ways (Fig. 3). First, FoxO6 has the lowest degree of homology (~ 30%) in amino acid sequence with the FoxO subfamily. Second, FoxO6 contains only two consesus AKT/PKB phosphorylation sites at Thr26 and Ser184 within the DNA binding domain. In contrast, other members of FoxO subfamily contains three highly conserved phosphorylation sites (Thr24, Ser253, Ser316 in FoxO1). Third, FoxO6 lacks the consensus motif that correponds to the nuclear export signal (NES) in other members of FoxO subfamily. Fourth, FoxO6 remains in the nucleus regardless of insulin action. In contrast, other FoxO members undergo insulin-dependent subcellular redistribution. It follows that FoxO6 mediates insulin action on target gene expression in a distinct mechanism. Below is our review on the role of FoxO6 in insulin action and carbohydrate metabolism. We will discuss the physiological significance of FoxO6 function in health and disease.
Fig. 3. Schematic depiction of mouse FoxO1 and FoxO6 proteins.

FoxO1 and FoxO6 are characterized by the amino DNA binding motif and carboxyl trans-activation domain. They share a conserved bipartite basic nuclear localization signal (NLS) within the DNA binding domain. FoxO1 contains a nuclear export signal (NES) within its carboxyl trans-activation domain. Such a characteristic motif is lacking in FoxO6. FoxO1 contains three Akt/PKB phosphorylation sites (T24, S253, and S316). In contrast, only two Akt/PKB phosphorylation sites (T26 and S184) are present in FoxO6.
FoxO6 in the liver
Our recent data show that FoxO6 is expressed in the liver of both rodent and human origins [12]. Preclinical studies indicate that FoxO6 is normally expressed at extremely low levels in fed states. However, hepatic FoxO6 production is markedly upregulated in fasted mice, indicating that hepatic FoxO6 activity is regulated in response to physiological cues [12]. Furthermore, hepatic FoxO6 expression at both mRNA and protein levels are significantly increased in mice with dietary obesity or type 2 diabetes. Likewise, hepatic FoxO6 mRNA and protein levels are significantly induced in streptozotocin-induced diabetic mice [12]. These data suggest that hepatic FoxO6 activity is subject to insulin inhibition. A lack of insulin inhibition, resulting from insulin deficiency or insulin resistance, contributes to FoxO6 overproduction in the liver in both type 1 and type 2 diabetes.
FoxO6 in gluconeogenesis
Gluconeogenesis is a metabolic pathway that takes place mainly in the liver for converting non-carbohydrate metabolites (lactate, glycerol, and amino acids) to glucose [13, 14]. Hepatic gluconeogenesis accounts for up to 80% of total endogenous glucose production in healthy individuals during a prolonged fast [15]. Gluconeogenesis is tightly regulated by hormonal and nutritional cues. In response to postprandial insulin secretion, hepatic gluconeogenic activity is suppressed to limit glucose production. This effect serves two purposes; i) to prevent prolonged postprandial glucose excursion and ii) to replenish glycogen content in liver, as increased glucose influx into hepatocytes promotes the synthesis and storage of glycogen in the liver after meals. Conversely, in response to reduced insulin action and elevated glucagon secretion during fasting, hepatic gluconeogenesis is stimulated, resulting in increased glucose output from liver. Such a reciprocal mechanism, orchestrated by the two opposing hormones (insulin and glucagon), is crucial for rapid adaptation of the liver to metabolic shift between fed and fasting states for maintaining blood sugar levels within the physiological range [14].
FoxO6 is shown to stimulate the expression of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase), two key enzymes in hepatic gluconeogenesis. This effect contributes to increased hepatic gluconeogenesis, which is counteracted by insulin [12]. These findings are reproduced in cultured HepG2 cells and human primary hepatocytes. Furthermore, mice with adenovirus-mediated FoxO6 production in the liver exhibit fasting hyperglycemia due to increased hepatic glucose output [12]. Likewise, transgenic mice with FoxO6 gain-of-function in the liver develop pre-diabetes, culminating in the induction of glucose intolerance, fasting hyperglycemia and hyperinsulinemia, due to FoxO6-mediated induction of hepatic gluconeogenesis [12]. Conversely, hepatic FoxO6 depletion compromises the ability of the liver to undergo gluconeogenesis, resulting in fasting hypoglycemia in mice following an overnight fast [12]. Together these results indicate that FoxO6 plays an independent role in regulating hepatic gluconeogenesis in response to insulin (Fig. 4).
Fig. 4. FoxO6 mediates insulin action on gluconeogenesis.
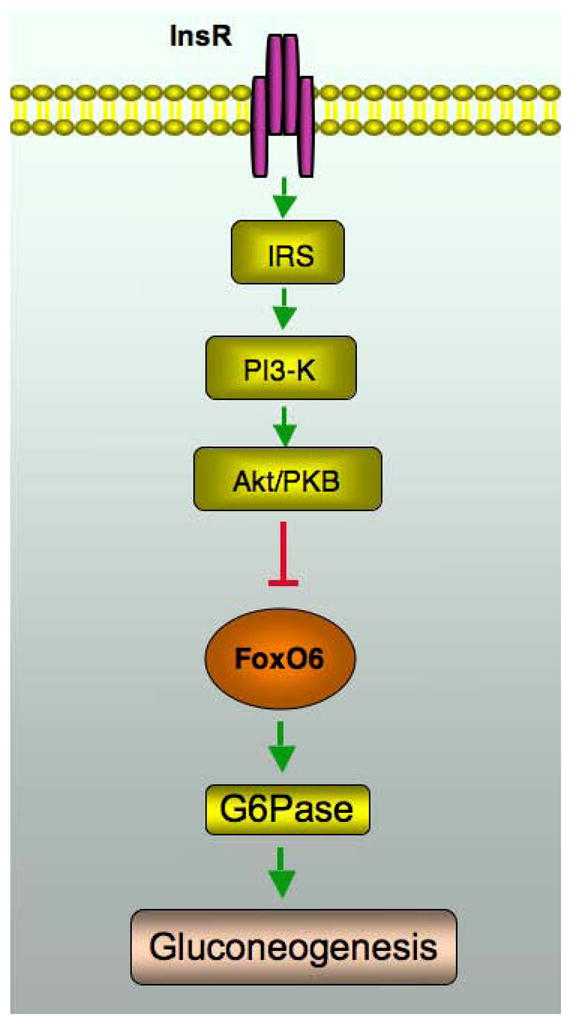
FoxO6 stimulates gluconeogenesis and this effect is counteracted by insulin. Insulin inhibits FoxO6 transcriptional activity by promoting FoxO6 phosphorylation, which in turn disables its cognate binding to the promoter of gluconeogenic genes (PEPCK and G6Pase) in liver. Insulin inhibition of FoxO6 activity in the liver is instrumental for limiting hepatic glucose production and preventing postprandial glucose excursion. In response to starvation, FoxO6 activity is upregulated, the resulting effect of which serves to prime the liver for augmented gluconeogenesis for maintaining fasting blood glucose levels within the physiological range. Unbridled FoxO6 activity, resulting from an impaired ability of insulin to keep FoxO6 action in check, promotes excessive gluconeogenesis in the liver and contributes to the episode of fasting hyperglycemia in diabetes. InsR, insulin receptor.
FoxO6 in insulin action
Although FoxO6 is phosphorylated in response to insulin, phosphorylated FoxO6 does not undergo insulin-dependent subcellular redistribution [10, 12, 16]. - In contrast, FoxO1 is readily translocated from the nucleus to cytoplasm in response to insulin [17, 18]. This raises an intriguing question as to how insulin modulates FoxO6 activity. FoxO6 contains two consensus Akt/PKB phosphorylation sites, Thr26 and Ser184, within its DNA binding domain. One potential mechanism is that phosphorylation of Thr26 or Ser184 would alter its DNA binding activity and prevent FoxO6 from binding to its target promoter. Alternatively, phosphorylation of FoxO6 promotes its association with other factors such as the multifunctional factor 14-3-3, which masks FoxO6 DNA binding domain and precludes FoxO6 binding to target promoters.
FoxO6 bind to its cognate IRE site within the G6Pase promoter and stimulates G6Pase promoter activity. This effect is abolished in response to insulin. Converting Ser184 to non-phosphorylation residue Ala184 by site-directed mutagenesis results in a constitutively active FoxO6 allele, whose transcriptional activity is refractory to insulin inhibition. Furthermore, FoxO6 is able to complex with 14-3-3 in the nucleus of hepatocytes, coinciding with the conservation of a consensus 14-3-3 binding motif (23RSCTWP28) within FoxO6 DNA-binding domain. Together these results suggest that insulin inhibition of FoxO activity and FoxO nucleocytoplasmic trafficking are two distinct events. Consistent with our data, Tsai et al. [19] show that insulin inhibition of FoxO1 activity can be achieved without necessarily altering FoxO1 subcellular redistribution, but this inhibition depends on the ability of FoxO1 to undergo insulin-dependent phosphorylation.
FoxO trafficking
Nucleocytoplasmic shuttling constitutes a compartmental mechanism by which insulin inhibits FoxO activity in cells [1, 18]. However, such mechanism is lacking in FoxO6. Both FoxO1 and FoxO6 interacts with 14-3-3, precluding the possibility that 14-3-3 is liable for the idiosyncrasy in nucleocytoplasmic trafficking between FoxO1 and FoxO6 [12]. FoxO6 lacks NES, a consensus motif that is conserved in all other members of the FoxO subfamily. NES is capable of associating with the CRM-1, known as exportin-1 that is responsible for binding to the NES motif of a cargo protein and transporting the cargo protein from the nucleus to cytoplasm [20, 21]. Interestingly, FoxO1, but not FoxO6, is able to complex with CRM-1 in liver cells [12]. In response to insulin, FoxO1 (not FoxO6) in complex with CRM-1 is translocated from the nucleus to cytoplasm. This effect is abrogated by leptomycin B, an agent that binds specifically to CRM-1 and inhibits its cargo-trafficking activity [12]. These results underscore the significance of CRM-1 in facilitating insulin-dependent FoxO subcellular redistribution. It also implies that the inability of FoxO6 to associate with CRM-1 dictates the disability of FoxO6 to undergo insulin-stimulated nucleocytoplasmic trafficking (Fig. 5).
Fig. 5. A model of FoxO1 nucleocytoplasmic shuttling.
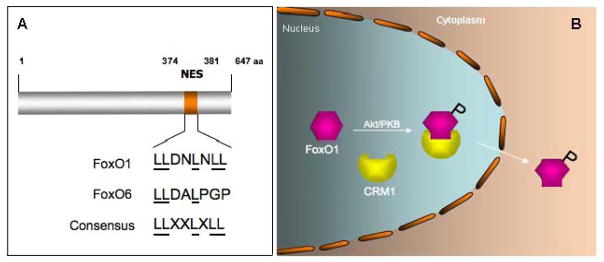
A. FoxO1 contains a conserved NES within the trans-activation domain. In contrast, FoxO6 contains a semi-conserved NES. B. FoxO1 upon phosphorylation is associated with CRM-1, an exportin that is responsible for transporting FoxO1 from the nucleus to the cytoplasm.
FoxO6 – a deviant from the FoxO subfamily
Why does FoxO1, but not FoxO6, evolve such a NES-dependent nucleocytoplasmic trafficking mechanism for mediating insulin inhibition on gluconeogenesis? An intuitive explanation is that FoxO6 serves as a basal mechanism for monitoring hepatic insulin action and fine-tuning gluconeogenesis in the liver between fasting and fed states, such that after FoxO1 is translocated to the cytoplasm in response to postprandial insulin release, constitutively nuclear FoxO6 can prime the liver for augmented gluconeogenesis in the ensuing post-absorption phase.
Alternatively, FoxO1 upon its nuclear exit has a secondary function in the cytoplasm. Implicit in this explanation is a recent study showing that cytosolic FoxO1 is essential for the induction of autophagy, an adaptive response of cells to survive in the face of serum starvation or oxidative stress, as this action is independent of FoxO1 transcriptional activity [22]. Altered autophagy in peripheral tissues is linked to the pathophysiology of insulin resistance in both human subjects and animal models with type 2 diabetes [23–26]. Yang et al. [27] show that defective autophagy in the liver directly induces endoplasmic reticulum stress and instigates insulin resistance in dietary obese mice. Interestingly, this effect correlates with the diminution of cytosolic FoxO1 proteins in insulin resistant liver, as FoxO1 is predominantly nuclear, due to its inability to undergo insulin-dependent nuclear export in obesity and type 2 diabetes [6, 17]. These data provide important physiological underpinning for cytosolic FoxO1 in regulating hepatic autophagy, in keeping with the idea that FoxO6 is functionally divergent from the FoxO subfamily [28] (Fig. 1).
FoxO6 – a non-redundant pathway of gluconeogenesis
Although FoxO1 is shown to play a key role in integrating insulin signaling to hepatic gluconeogenesis, FoxO1 depletion in the liver does not result in abolition of hormonal regulation of gluconeogenesis in mice [29]. Mice with FoxO1 loss-of-function in the liver are associated with diminished gluconeogenic activities (by ~50%) and impaired abilities to maintain fasting blood glucose levels within the normal range [17]. Furthermore, FoxO1 loss-of-function reduces, but does not abrogate the responsiveness of the liver to insulin or glucagon (via cAMP) signaling. These data suggest that there are additional factors or compensatory mechanisms in mediating hormonal regulation of hepatic gluconeogenesis [17, 29]. One such factor is FoxO6 that is shown to independently mediate the inhibitory effect of insulin on hepatic gluconeogenesis. Hepatic FoxO6 depletion impairs the ability of the liver to undergo gluconeogenesis and contributes to fasting hyperglycemia in mice. This effect is independent of other FoxO members, as hepatic FoxO6 ablation does not result in altered expression of FoxO1, FoxO3 and FoxO4, three redundant pathways in regulating gluconeogenesis in the liver [30]. Together these results suggest that FoxO6 plays a non-redundant role in adjusting the rate of hepatic glucose production in response to nutritional states under different physiological conditions (Fig. 4).
FoxO6 in diabetes
FoxO6 becomes deregulated in insulin deficient or insulin resistant liver, culminating in its increased production in type 1 or type 2 diabetic mice [12]. Likewise, abnormally higher FoxO6 production is detectable in the liver of high fat-induced obese mice [12]. Unchecked FoxO6 activity, resulting from impaired insulin action, is a causative factor for unrestrained gluconeogenesis in the liver, contributing to fasting hyperglycemia in morbid obesity and type 2 diabetes. Diabetic db/db mice with selective FoxO6 knockdown in the liver display improved fasting glycemia with a concomitant reduction in fasting hyperinsulinemia. This effect translates into a significant improvement in whole-body insulin sensitivity in db/db mice [12].
Excessive glucose production, resulting from impaired insulin suppression of gluconeogenesis in liver, exerts its deleterious effect on whole-body metabolism in two fundamental ways: 1) It prolongs postprandial blood glucose excursion, 2) It contributes to the pathogenesis of fasting hyperglycemia in diabetes. Indeed, the gluconeogenic pathway is being explored as a major therapeutic target for improving glycemic control in diabetes. This is exemplified by metformin, an oral anti-hyperglycemia agent that is widely prescribed for lowering blood sugar levels in patients with diabetes [13, 31, 32]. Our data characterize FoxO6 as a potential therapeutic target, presaging that selective inhibition of FoxO6 activity in insulin resistant liver by small molecule antagonists would suppress hepatic gluconeogenesis and ameliorate fasting hyperglycemia in diabetes.
FoxO6 in Aging
Although FoxO6 is expressed in the brain, its central function remains elusive. Zemva et al. [33] report that FoxO6 mRNA expression is markedly upregulated in the brain in aged mice, implicating that the insulin/IGF-1-FoxO6 signaling pathway is involved in aging. However, clinical studies reveal no association between FoxO6 and life expectancy in humans [34]. Instead, a close association between genetic FoxO3 variants and human longevity is consistently reproduced in a variety of ethnic groups including Chinese, Danish, German, Italian, Japanese, Jewish and US-American [34–40]
It is noted that central FoxO6 expression is significantly down-regulated in the brain of obese mice [33]. This effect has been implicated in the pathogenesis of obesity-associated dementia. The underlying mechanism remains unknown. Nor is it clear whether the altered FoxO6 expression is a cause or consequence of neurodegeneration in the brain. A recent study indicates that FoxO6 activity in the hippocampus is required for memory consolidation in mice [41]. This finding implicates that altered FoxO6 expression may contribute to pathological age-dependent decline in memory. Further studies are warranted to delineate the FoxO6 pathway in the brain to understand the cause and mechanism of the cognitive impairment in obesity and type 2 diabetes.
Conclusion
FoxO6 constitutes a distinct route by which the liver orchestrates insulin-dependent regulation of gluconeogenesis. Hepatic FoxO6 activity is increased and this effect primes the liver for promoting glucose production in response to fasting. Conversely, hepatic FoxO6 activity is inhibited by insulin and this action curbs glucose production in the liver and limits postprandial glucose excursion after meals. Unlike other members of the FoxO family, FoxO6 mediates insulin action in a distinct mechanism without altering its subcellular redistribution. FoxO6 activity becomes unchecked in insulin resistant liver and this effect contributes to hepatic glucose overproduction – the underlying cause of fasting hyperglycemia in diabetes. Hepatic FoxO6 inhibition curbs glucose overproduction in the liver and ameliorates fasting hyperglycemia in diabetic mice. Thus, FoxO6 has emerged as an important signaling molecule for fine-tuning hepatic glucose output and regulating whole-body glucose metabolism.
Acknowledgments
This study was supported by National Institute of Health Grant DK087764.
Abbreviations
- FoxO1
Forkhead box O1
- FoxO3
Forkhead box O3
- FoxO4
Forkhead box O4
- FoxO6
Forkhead box O6
- Daf-16
Dauer abnormal formation 16
- IGF-1
Insulin-like growth factor 1
- PKB
Protein kinase B
- G6Pase
Glucose-6-phosphatase
- PEPCK
Phosphoenolpyruvate carboxykinase
- CRM-1
Chromosome region maintenance 1, also known as Exportin 1
- NLS
Nuclear localization signal
- NES
Nuclear export signal
Footnotes
Disclosure:
None of the authors has a conflict of interest to declare in this manuscript
References
- 1.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 2.Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429:562–566. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- 3.Lee SS, Kennedy S, Tolonen AC, Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- 4.Barthel A, Schmoll D, Unterman TG. FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab. 2005;16:183–189. doi: 10.1016/j.tem.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 5.Sparks DJ, Dong HH. FoxO1 and hepatic lipid metabolism. Current Opinion in Lipidology. 2009;20:217–226. doi: 10.1097/MOL.0b013e32832b3f4c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altomonte J, Cong L, Harbaran S, Richter A, Xu J, Meseck M, Dong HH. Foxo1 Mediates Insulin Action on ApoC-III and Triglyceride Metabolism. J Clin Invest. 2004;114:1493–1503. doi: 10.1172/JCI19992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qu S, Altomonte J, Perdomo G, He J, Fan Y, Kamagate A, Meseck M, Dong HH. Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology. 2006;147:5641–5652. doi: 10.1210/en.2006-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cifarelli V, et al. FOXO1 Mediates the Autocrine Effect of Endothelin-1 on Endothelial Cell Survival. Mol Endocrinol. 2012;26:1213–1224. doi: 10.1210/me.2011-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464–2472. doi: 10.1172/JCI27047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003;278:35959–35967. doi: 10.1074/jbc.M302804200. [DOI] [PubMed] [Google Scholar]
- 11.Hoekman MF, Jacobs FM, Smidt MP, Burbach JP. Spatial and temporal expression of FoxO transcription factors in the developing and adult murine brain. Gene Expr Patterns. 2006;6:134–140. doi: 10.1016/j.modgep.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Kim DH, et al. FoxO6 Integrates Insulin Signaling With Gluconeogenesis in the Liver. Diabetes. 2011;60:2763–2774. doi: 10.2337/db11-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgerton DS, Johnson KM, Cherrington AD. Current strategies for the inhibition of hepatic glucose production in type 2 diabetes. Front Biosci. 2009;14:1169–1181. doi: 10.2741/3301. [DOI] [PubMed] [Google Scholar]
- 14.Wahren J, Ekberg K. Splanchnic regulation of glucose production. Annu Rev Nutr. 2007;27:329–345. doi: 10.1146/annurev.nutr.27.061406.093806. [DOI] [PubMed] [Google Scholar]
- 15.Ekberg K, Landau BR, Wajngot A, Chandramouli V, Efendic S, Brunerngraber H, Wahren J. Contributions by kidney and liver to glucose production in the postabsorptive state and after 60 h of fasting. Diab. 1999:48. doi: 10.2337/diabetes.48.2.292. [DOI] [PubMed] [Google Scholar]
- 16.van der Heide LP, Jacobs FM, Burbach JP, Hoekman MM, Smidt MP. FoxO6 transcriptional activity is regulated by Thr26 and Ser184, independent of nucleo-cytoplasmic shuttling. Biochem J. 2005;391:623–629. doi: 10.1042/BJ20050525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamagate A, Qu S, Perdomo G, Su D, Kim DH, Slusher S, Meseck M, Dong HH. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J Clin Invest. 2008;118:2347–2364. doi: 10.1172/JCI32914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamagate A, Dong HH. FoxO1 integrates insulin signaling to VLDL production. Cell Cycle. 2008;7:3162–3170. doi: 10.4161/cc.7.20.6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai WC, Bhattacharyya N, Han LY, Hanover JA, Rechler MM. Insulin inhibition of transcription stimulated by the forkhead protein Foxo1 is not solely due to nuclear exclusion. Endocrinology. 2003;144:5615–5622. doi: 10.1210/en.2003-0481. [DOI] [PubMed] [Google Scholar]
- 20.Monecke T, Guttler T, Neumann P, Dickmanns A, Gorlich D, Ficner R. Crystal Structure of the Nuclear Export Receptor CRM1 in Complex with Snurportin1 and RanGTP. Science. 2009;324:1087–1091. doi: 10.1126/science.1173388. [DOI] [PubMed] [Google Scholar]
- 21.Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, Chook YM. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458:1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12:665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez CD, Lee MS, Marchetti P, Pietropaolo M, Towns R, Vaccaro MI, Watada H, Wiley JW. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011;7:2–11. doi: 10.4161/auto.7.1.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovsan J, et al. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab. 2011;96:E268–277. doi: 10.1210/jc.2010-1681. [DOI] [PubMed] [Google Scholar]
- 25.Zhou L, Liu F. Autophagy: roles in obesity-induced ER stress and adiponectin downregulation in adipocytes. Autophagy. 2010;6:1196–1197. doi: 10.4161/auto.6.8.13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, Liu Z, Cao W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284:31484–31492. doi: 10.1074/jbc.M109.033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang M, Zhang X, Zhao H, Wang Q, Pan Y. FoxO gene family evolution in vertebrates. BMC Evol Biol. 2009;9:222. doi: 10.1186/1471-2148-9-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor foxo1 in liver. Cell Metab. 2007;6:208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Haeusler RA, Kaestner KH, Accili D. FoxOs function synergistically to promote glucose production. J Biol Chem. 2010;285:35245–35248. doi: 10.1074/jbc.C110.175851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garber AJ. Metformin. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus: A Fundamental and Clinical Text. 2. Lippincott Williams & Wilkins Press; Philadelphia, PA. USA: 2000. pp. 778–788. [Google Scholar]
- 32.Hundal RS, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–2069. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zemva J, Schilbach K, Stohr O, Moll L, Franko A, Krone W, Wiesner RJ, Schubert M. Central FoxO3a and FoxO6 Expression is Down-regulated in Obesity Induced Diabetes but not in Aging. Exp Clin Endocrinol Diabetes. 2012;120:340–350. doi: 10.1055/s-0031-1297970. [DOI] [PubMed] [Google Scholar]
- 34.Kleindorp R, Flachsbart F, Puca AA, Malovini A, Schreiber S, Nebel A. Candidate gene study of FOXO1, FOXO4, and FOXO6 reveals no association with human longevity in Germans. Aging Cell. 2011;10:622–628. doi: 10.1111/j.1474-9726.2011.00698.x. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, et al. Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum Mol Genet. 2009;18:4897–4904. doi: 10.1093/hmg/ddp459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flachsbart F, Caliebe A, Kleindorp R, Blanche H, von Eller-Eberstein H, Nikolaus S, Schreiber S, Nebel A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci U S A. 2009;106:2700–2705. doi: 10.1073/pnas.0809594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anselmi CV, Malovini A, Roncarati R, Novelli V, Villa F, Condorelli G, Bellazzi R, Puca AA. Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res. 2009;12:95–104. doi: 10.1089/rej.2008.0827. [DOI] [PubMed] [Google Scholar]
- 38.Willcox BJ, et al. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A. 2008;105:13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soerensen M, Dato S, Christensen K, McGue M, Stevnsner T, Bohr VA, Christiansen L. Replication of an association of variation in the FOXO3A gene with human longevity using both case-control and longitudinal data. Aging Cell. 2010;9:1010–1017. doi: 10.1111/j.1474-9726.2010.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pawlikowska L, et al. Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging Cell. 2009;8:460–472. doi: 10.1111/j.1474-9726.2009.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salih DA, et al. FoxO6 regulates memory consolidation and synaptic function. Genes Dev. 2012;26:2780–2801. doi: 10.1101/gad.208926.112. [DOI] [PMC free article] [PubMed] [Google Scholar]