

Abstract
Background
Several recent studies implementing the standard “drinking-in-the-dark” (DID) model of short-term binge-like ethanol (EtOH) intake in C57BL/6J mice highlighted a role for the stress-related neuropeptide corticotropin-releasing factor (CRF) and its primary binding partner, the CRF type-1 receptor (CRF1).
Methods
We evaluated the selectivity of CRF1 involvement in binge-like EtOH intake by interrupting CRF1 function via pharmacological and genetic methods in a slightly modified two-bottle choice DID model that allowed calculation of an EtOH preference ratio. In addition to determining EtOH intake and preference, we also measured consumption of food and H2O during the DID period, both in the presence and absence of EtOH and sweet tastant solutions.
Results
Treatment with either of the CRF1-selective antagonists CP-376,395 (10–20 mg/kg, i.p.) or NBI-27914 (10–30 mg/kg, i.p.) decreased intake of 15% EtOH in male C57BL/6J mice, but did so in the absence of a concomitant decrease in EtOH preference. These findings were replicated genetically in a CRF1 knockout mouse model (also on a C57BL/6J background). In contrast to effects on EtOH intake, pharmacological blockade of CRF1 with CP-376,395 increased intake of 10% sucrose, consistent with previous findings in CRF1 knockout mice. Finally, pharmacological and genetic disruption of CRF1 activity significantly reduced feeding and/or total caloric intake in all experiments, confirming the existence of non-specific effects.
Conclusions
Our findings indicate that blockade of CRF1 receptors does not exert specific effects on EtOH intake in the DID paradigm, and that slight modifications to this procedure, as well as additional consummatory control experiments, may be useful when evaluating the selectivity of pharmacological and genetic manipulations on binge-like EtOH intake.
Keywords: corticotropin, binge, urocortin, alcohol, stress
INTRODUCTION
In 2004, the National Institute on Alcohol Abuse and Alcoholism defined “binge drinking” as a pattern of ethanol (EtOH) consumption leading to blood EtOH concentrations (BECs) of 80 mg/dL or above (NIH-NIAAA, 2004). Binge drinking is associated with numerous risk factors (Conover and Jones, 2012; Courtney and Polich, 2009; van de Wiel and de Lange, 2008), and is prevalent even in populations that do not otherwise meet the criteria for an alcohol use disorder (Deas and Brown, 2006; Lai et al., 2012). Given these observations, there has been substantial interest in unraveling the neurobiological contributions to binge-like EtOH consumption (Crabbe et al., 2011). To address this need for an animal model of binge drinking, several attempts were made to develop a procedure in which rodents would consume large quantities of EtOH during a brief time window in the circadian dark period, when consummatory behaviors are at their peak (Ryabinin et al., 2003; Sharpe et al., 2005). Ultimately, the “drinking-in-the-dark” (DID) method emerged as a favorable model for elucidating the neural and genetic substrates of binge-like drinking (Rhodes et al., 2005). A standard DID experiment lasts four days, during which C57BL/6J mice are given daily access to a single bottle of 20% EtOH for 2–4 hours in the dark cycle. Importantly, this procedure results in levels of EtOH intake that are sufficient to produce behavioral intoxication and binge-like BECs (Rhodes et al., 2007).
Stress-related neuropeptide circuits are thought to play an essential role in the adaptations that occur following exposure to drugs of abuse, including EtOH (Koob, 2008; Koob, 2009). Specifically, the corticotropin-releasing factor (CRF) system has been identified as a critical mediator of EtOH consumption under various conditions, including dependence-induced drinking and binge-like drinking (Heilig and Koob, 2007; Lowery and Thiele, 2010). Examination of the CRF system, which includes four endogenous ligands (CRF and the three urocortin peptides: Ucn1, Ucn2 Ucn3), the CRF binding protein (CRF-BP), and two G-protein-coupled receptors (CRF1, CRF2), has been complicated by the intricate pharmacological and anatomical relationships between these different components (Giardino and Ryabinin, 2012). Nevertheless, converging lines of evidence support the conclusion that binge-like EtOH intake in the standard DID procedure is dependent on activation of the CRF1 receptor by endogenous CRF (Kaur et al., 2012; Lowery et al., 2010).
A potential drawback of the standard DID model is that mice do not receive access to EtOH in the presence of an additional bottle containing H2O. Providing concurrent access to H2O during the EtOH consumption period would allow for the calculation of an EtOH preference ratio, which can be useful in determining whether manipulations selectively alter EtOH consumption vs. overall fluid consumption. Previous studies examining the role of the CRF1 receptor in DID EtOH consumption attempted to control for this possibility by replacing 20% EtOH with 3% or 10% sucrose, a highly-palatable solution that is also readily consumed in the dark period (Kaur et al., 2012; Lowery et al., 2010; Lowery-Gionta et al., 2012). In these experiments, EtOH intake was decreased by disruption of CRF1 signaling, whereas sucrose intake was either unaltered or increased by disruption of CRF1 signaling. Again, these studies implemented the single-bottle configuration that is standard for the DID procedure, preventing the calculation of a sucrose preference ratio. Therefore, the selectivity of CRF1 effects on binge-like EtOH consumption vs. overall fluid consumption remained largely unresolved.
We considered the possibility that the addition of a second bottle containing H2O during access to EtOH or other solutions might be helpful in determining the selectivity of CRF1 effects (via calculation of a preference ratio for each solution). Indeed, two-bottle choice configuations have long been implemented in tests for fluid preference. Therefore, we slightly modified the DID procedure (as others have in the past) so that mice received concurrent access to H2O while solutions of 15% EtOH, 10% sucrose, or 0.015% saccharin were available, and then examined the effects of interrupted CRF1 signaling under these conditions. In addition, we considered the possibility that sweet tastant solutions might not be ideal control fluids for examining whether CRF1 signaling modulates general consummatory behavior in the DID model. Therefore, we simply evaluated the impact of disrupted CRF1 signaling on food and H2O intake in the absence of additional fluids.
MATERIALS AND METHODS
Animals
For the pharmacological experiments, male C57BL/6J (B6) mice were used. Mice were delivered from The Jackson Laboratory (Sacramento, CA) at 8 weeks of age, housed 5 per cage, and spent one week acclimating to our colony room (12/12 schedule; lights on 0600h) before being single-housed and transferred to the experimental room (12/12 schedule; lights off at 0600h) for an additional ten-day acclimation period prior to the initiation of the experiment.
For the experiment using male and female CRF1 genetic knockout (KO) and wild-type (WT) littermate animals, we used single-gene mutant mice derived from embryonic stem cells that had undergone targeted gene deletion, as previously described in detail (Giardino et al., 2011b; Timpl et al., 1998). These mice have now been backcrossed onto the B6 strain for twelve generations. Mice were bred in our colony, weaned at 28–32 days of age, and isosexually housed 2–5 per cage. At 7–14 weeks of age, mice were single-housed and transferred to the experimental room (12/12 schedule; lights off at 0600h) for an additional ten-day acclimation period prior to the initiation of the experiment. Eleven separate litters of mice contributed to the KO and WT animals used in these experiments.
For all experiments, mice were housed in a temperature- and humidity-controlled environment with ad libitum access to food (LabDiet 5001; LabDiet, Richmond, IN, USA) and H2O. During the ten-day acclimation period, mice received 24h access to two 25 mL glass cylinder bottles with metal sipper tubes (both containing H2O) on either side of the cage, with food evenly distributed along the cage top. All protocols were approved by the Oregon Health & Science University animal care and use committee, and performed within the National Institutes for Health Guidelines for the Care and Use of Laboratory Animals, as well as the Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research.
Drugs and Solutions
For the pharmacological experiments, we used the brain-penetrable CRF1 antagonists CP-376,395 and NBI-27914 (CP and NBI; Tocris, Ellisville, MO, USA). CP was dissolved in 0.9% NaCl (saline) and administered intraperitoneally (i.p.) at a dose of either 0.0, 10.0, or 20.0 mg/kg (CP-0, CP-10, CP-20). NBI was dissolved in 10% Cremophor EL in saline and administered i.p. at a dose of either 0.0, 10.0, or 30.0 mg/kg (NBI-0, NBI-10, NBI-30). Vehicles, doses, and timepoint of injection (30 min prior to the start of the experiment) were chosen based on previous experiments from our laboratory (Giardino et al., 2012b) and others (Lowery-Gionta et al., 2012). All i.p. injections were given at a volume of 10 mL/kg body weight. Sucrose was dissolved in H2O at a concentration of 10% (weight/volume), saccharin was dissolved in H2O at a concentration of 0.015% (weight/volume), and 95% EtOH was diluted in H2O to reach a final concentration of 15% (volume/volume).
Procedure
Experiments were based on the “drinking-in-the-dark” (DID) paradigm, which has been described elsewhere in detail (Rhodes et al., 2005). This procedure resulted from efforts to identify a limited time window in the circadian dark period during which mice would consume quantities of EtOH capable of producing behavioral intoxication and pharmacologically-relevant BECs (Ryabinin et al., 2003; Sharpe et al., 2005). Indeed, naïve B6 mice in the standard DID paradigm display behavioral intoxication, and achieve BECs that surpass the 80 mg/dL criterion used by National Institute on Alcohol Abuse and Alcoholism to define an episode of binge-drinking (Rhodes et al., 2007). Instead of offering mice a single bottle of EtOH (as is standard for the DID procedure), we modeled our studies after earlier experiments that slightly modified this protocol by giving mice access to an additional bottle containing H2O (Kaur and Ryabinin, 2010; Rhodes et al., 2007). In addition, we adopted this two-bottle configuration for studies of sucrose-DID and saccharin-DID. Giving the mice concurrent access to H2O during the EtOH, sucrose, and saccharin drinking sessions allowed the calculation of preference ratios (based on the relative volumes of fluid consumed from each of the bottles).
Effects of CP-376,395 on food and fluid consumption
Male B6 mice (n=8–9 per group) were weighed at the beginning of each phase of the experiment (H2O phase, sucrose phase, EtOH phase). Each phase was separated by 1–3 days in which H2O was the only available solution.
In an effort to habituate mice to brief handling and i.p. drug administration, mice received injections of vehicle at 2.5h into the dark cycle during the final three days of the acclimation period. Over the next three days (Days 1–3 of the H2O phase), all mice continued to receive i.p. vehicle at 2.5h into the dark cycle. Thirty minutes after injection on Days 1–3, the two 25 mL H2O bottles on each cage top were replaced with two 10 mL plastic bottles containing H2O (for more precise measurement of small volumes). After 2h, the 10 mL bottles were removed and replaced with the original 25 mL H2O bottles. On Day 4, mice received i.p. injection of either 0.0, 10.0, or 20.0 mg/kg CP at 2.5h into the dark cycle, and food was collected and weighed from each subject’s cage. Thirty minutes later, 25 mL H2O bottles were replaced with 10 mL H2O bottles. After 4h, 10 mL bottles were replaced with 25 mL bottles, and food was again collected from cage tops and weighed. With each exchange of 25 mL and 10 mL bottles, volumes were recorded to the nearest 0.1 mL. In addition, volumes were read and recorded halfway through the 4h session on Day 4, in order to analyze the timecourse of CP’s effects on fluid intake. To avoid disturbing the mice halfway through the session, food consumption was only calculated for the entire four hour session.
During the next phase of the experiment (sucrose phase), CP dose groups were counterbalanced such that mice received a dose of CP distinct from that given during the H2O phase. Days 1–4 of the sucrose phase were identical to Days 1–4 of the H2O phase, with the exception that one of the two 10 mL bottles was filled with 10% sucrose instead of H2O.
During the final phase of the experiment (EtOH phase), CP dose groups were again counterbalanced such that mice received a dose of CP distinct from that given during the H2O and sucrose phases. Days 1–4 of the EtOH phase were identical to Days 1–4 of the H2O and sucrose phases, with the exception that one of the 10 mL bottles was instead filled with 15% EtOH. Following the end of the 4h session on Day 4 of the EtOH phase, mice were euthanized by CO2 asphyxiation, and trunk blood was collected for later analysis of BECs, as previously reported by our laboratory (Kaur and Ryabinin, 2010).
Effects of NBI-27914 on food and fluid consumption
Male B6 mice (n=12 per group) were weighed at the beginning of each phase of each experiment (EtOH phase, saccharin phase). The general protocol was identical to the CP-376,395 experiment, with the exceptions that there was not a H2O phase, that an EtOH phase preceded a saccharin phase (rather than a sucrose phase preceding an EtOH phase), and that NBI-27914 was administered rather than CP-376,395. Dose groups (NBI-0, NBI-10, NBI-30) were counterbalanced between the phases such that each animal received a different dose of NBI during each phase.
Effects of genetic deletion of CRF1 on food and fluid consumption
Male and female CRF1 KO and WT mice (n=9–18 per genotype) were weighed at the beginning of each phase of the experiment (H2O phase, EtOH phase). The general procedure was nearly identical to that of the pharmacological experiments, with the exceptions that mice did not receive i.p. injections of vehicle or drug, that fluid consumption was not measured at the halfway point during the 4h session, and that there were only two phases: a H2O phase followed by an EtOH phase.
Calculations of Food, Fluid, and Caloric Intake
Intakes of H2O, sucrose, and saccharin are expressed as mL consumed per kg body weight (mL/kg). EtOH intakes were calculated by multiplying the volume of EtOH consumed (mL) by the density of 15% EtOH (0.11835 g/mL), and are expressed as g consumed per kg body weight (g/kg). Food intakes are also expressed as g/kg. Total caloric intakes for the sucrose and EtOH phases were calculated using the caloric contents of sucrose (3.87 kcal/g; 10% sucrose density = 0.1587 g/mL), EtOH (7.1 kcal/g; 15% EtOH density = 0.11835 g/mL), and the 5001 LabDiet mouse chow (4.07 kcal/g). Total kcal from combined food and sucrose or food and EtOH intakes were expressed relative to mouse body weights (kcal/kg). Sucrose, saccharin, and EtOH preference ratios were calculated by dividing the mL of sucrose, saccharin, or EtOH consumed by the total mL of fluid consumed during the given timeperiod. All intake values were normalized to body weight, and no significant differences in body weight were identified between treatment groups or genotypes in any of the phases of any of the experiments.
Statistical Analyses
For experiments in male B6 mice receiving the CRF1 antagonists, we used repeated measures analysis of variance (RM-ANOVA) with CRF1 antagonist treatment (vehicle, low dose, high dose) as the between-subjects factor, and Time (First two hours, Last two hours) as the repeated measure. We also used one-way ANOVA where appropriate to analyze effects of CRF1 antagonist treatment. Significant main or interacting effects of CRF1 antagonist treatment were followed up by Bonferroni-corrected (RM-ANOVA) or Newman-Keuls (one-way ANOVA) post-hoc comparisons.
For experiments performed in male and female CRF1 genetic KO and WT littermate mice, preliminary analyses uncovered no statistically significant interactions with sex. Therefore, sexes were combined for all analyses. We used RM-ANOVA with genotype (KO, WT) as the between-subjects factor and Day (Days 1, 2, 3) as the repeated measure. We also performed t-tests on data from Day 4 in order to make direct comparisons between genotypes. Analyses for each phase were performed separately. All data are presented as mean ± SEM, and effects were considered statistically significant at p < 0.05.
RESULTS
CP-376,395 attenuates H2O intake
On Day 4 of the H2O phase, administration of the CRF1 antagonist CP-376,395 (CP) reduced H2O intake (F2,22 = 5.62; p < .05) and food intake (F2,22 = 15.19; p < .0001) during the 4h period corresponding to the DID time window. Post-hoc comparisons confirmed that CP dose-dependently attenuated intake of H2O (Figure 1A) and food (Figure 1B), with H2O intake affected specifically during the first half of the session.
Figure 1. CP-376,395 attenuates H2O and food intake.
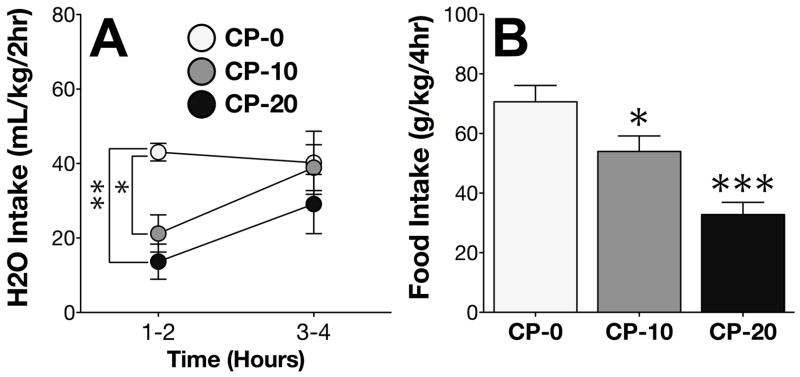
(A) Administration of CP on Day 4 dose-dependently decreased H2O intake during the first half of the 4h DID time window. (B) CP also dose-dependently decreased food intake. *p < .05 vs. CP-0; **p < .005 vs. CP-0; ***p < .0005 vs. CP-0.
CP-376,395 increases sucrose intake
On Day 4 of the sucrose phase, CP differentially altered intake of 10% sucrose over the course of the session (time x CP treatment interaction; F2,22 = 7.84; p < .005). Post-hoc comparisons revealed that CP administration significantly increased sucrose intake during the second half of the session (Figure 2A). Analysis of the sucrose preference ratio identified a main effect of CP treatment (F2,22 = 8.16; p < .005), and post-hoc comparisons showed that high-dose CP administration slightly decreased sucrose preference during the first half of the session (Figure 2B). CP treatment on Day 4 also significantly decreased food consumption (F2,22 = 10.73; p < .001; Figure 2C), and decreased total caloric intake from food and sucrose combined (F2,22 = 8.93; p < .005; Figure 2D).
Figure 2. CP-376,395 increases sucrose intake.

(A) Administration of CP on Day 4 increased sucrose intake during the second half of the 4h DID session. (B) High-dose CP administration on Day 4 decreased sucrose preference during the first half of the 4hr DID session. (C) CP decreased both food intake and (D) total caloric intake. *p < .05 vs. CP-0; **p < .005 vs. CP-0; ***p < .0005 vs. CP-0.
CP-376,395 attenuates EtOH intake but not EtOH preference
On Day 4 of the EtOH phase, CP reduced intake of 15% EtOH (main effect of CP treatment; F2,22 = 5.33; p < .05). Post-hoc comparisons confirmed that high-dose CP administration significantly decreased EtOH intake during the first half of the session (Figure 3A). CP treatment also altered total fluid consumption on Day 4 (F1,22 = 11.00; p = .0005), specifically during the first half of the session (Figure 3B).
Figure 3. CP-376,395 attenuates EtOH intake but not EtOH preference.

(A) High-dose CP administration on Day 4 decreased EtOH intake during the first half of the 4h DID session. (B) High-dose CP also decreased total fluid intake during the first half of the session. (C) Administration of CP on Day 4 did not alter EtOH preference. (D) High-dose CP administration decreased BECs. (E) CP decreased both food intake and (F) total caloric intake. *p < .05 vs. CP-0.
Analysis of the EtOH preference ratio revealed a significant main effect of time (F1,22 = 9.03; p < .01), but no significant main or interacting effects of CP treatment (Figure 3C). Similar to the effects on EtOH intake, mice that received CP-20 displayed significantly reduced blood ethanol concentrations (BECs) at the end of the EtOH drinking session (Figure 3D).
Consistent with our findings from the H2O and sucrose phases, CP treatment on Day 4 significantly decreased food consumption (F2,22 = 4.23; p < .05; Figure 3E), and decreased the total number of calories consumed from food and EtOH combined (F2,22 = 5.09; p < .05; Figure 3F).
NBI-27914 attenuates EtOH intake but not EtOH preference
On Day 4 of the EtOH phase, the CRF1 antagonist NBI-27914 (NBI) differentially altered intake of 15% EtOH over the course of the 4hr DID session (NBI x time interaction; F2,33 = 3.77; p < .05). Post-hoc comparisons confirmed that high-dose NBI significantly decreased EtOH intake during the first half of the session (Figure 4A). NBI also altered total fluid consumption on Day 4, specifically during the first half of the session (NBI x time interaction; F2,33 = 4.00; p < .05; Figure 4B).
Figure 4. NBI-27914 attenuates EtOH intake but not EtOH preference.

(A) Administration of high-dose NBI on Day 4 decreased EtOH intake during the first half of the 4h DID session. (B) High-dose NBI also decreased total fluid intake during the first half of the session. (C) Administration of NBI did not alter EtOH preference. (D) High-dose NBI administration decreased both food intake and (E) total caloric intake. ***p < .0005 vs. NBI-0, *p < .05 vs. NBI-0.
Analysis of the EtOH preference ratio revealed no significant main or interacting effects (Figure 4C). Consistent with our findings from the CP experiments, NBI significantly reduced food intake (F2,33 = 5.52; p < .005; Figure 4D), and total caloric intake from food and EtOH combined (F2,33 = 6.75; p < .005; Figure 4E).
NBI-27914 attenuates saccharin intake and preference
On Day 4 of the saccharin phase, NBI differentially altered intake of 0.015% saccharin over the course of the 4hr DID session (NBI x time interaction; F2,33 = 6.95; p < .005). Post-hoc comparisons confirmed that high-dose NBI significantly decreased saccharin intake during the first half of the session (Figure 5A). NBI treatment also altered total fluid consumption on Day 4 (F2,33 = 3.62; p < .05), specifically during the first half of the session (Figure 5B).
Figure 5. NBI-27914 attenuates saccharin intake and preference.

(A) High-dose NBI administration on Day 4 decreased saccharin intake during the first half of the 4h DID session. (B) High-dose NBI also decreased total fluid intake during the first half of the session. (C) Administration of NBI decreased saccharin preference. (D) High-dose NBI administration decreased food intake. **p < .005 vs. NBI-0, ## main effect of NBI treatment, p < .005.
CRF1 blockade with NBI reduced the saccharin preference ratio (main effect of NBI treatment; F2,33 = 5.85; p < .005), although post-hoc comparisons did not quite reach significance (Figure 5C). Consistent with our findings from the previous experiments, NBI also significantly decreased food consumption (F2,33 = 5.41; p < .005; Figure 5D).
Genetic deletion of CRF1 attenuates H2O intake
Data collected during the final two days of habituation to the reverse dark/light cycle revealed that daily baseline H2O intakes were lower among CRF1 KO mice, relative to WT littermate mice (main effect of genotype; F1,25 = 9.75; p < .005; Figure 6A). In order to determine whether these reduced H2O intakes were also present during the 2hr time window used for DID experiments, we measured H2O consumption on Days 1–3 of the H2O phase, revealing that CRF1 KO mice indeed drank less H2O than WT littermate mice (main effect of genotype; F1,25 = 5.44; p < .05; Figure 6B).
Figure 6. Genetic deletion of CRF1 attenuates H2O intake.

(A) Genetic deletion of CRF1 resulted in decreased H2O intake during two baseline days prior to the start of the experiment. (B) CRF1 KO mice also showed lower H2O intakes during the 2hr DID time window on Days 1–3. (C) H2O intake during the 4hr DID time window on Day 4 was also decreased in KO mice, relative to WT littermates. (D) Food intake on Day 4 was not different between genotypes. ## main effect of genotype p < .005; # main effect of genotype p < .05. * p < .05 vs. CRF1 WT.
During the 4h session on Day 4, H2O intakes were also lower among KO mice, as compared to WT mice (t25 = 2.56; p < .05; Figure 6C). Despite a 29% decrease in food intake among KO mice relative to WT mice, this genotypic difference did not reach significance (p > 0.2; Figure 6D).
Genetic deletion of CRF1 dampens EtOH intake but not EtOH preference
During the 2h DID time window on Days 1–3 of the EtOH phase, CRF1 KO mice consumed less EtOH (F1,25 = 6.90; p < .05; Figure 7A), and also consumed less fluid overall (F1,25 = 14.22; p < .001; Figure 7B). Surprisingly, the reduction in EtOH intake among CRF1 KO mice on Days 1–3 was accompanied by an overall enhancement in EtOH preference (F1,25 = 6.75; p < .05; Figure 7C), indicating that the effects of CRF1 deletion on H2O intake were more pronounced than those on EtOH intake.
Figure 7. Genetic deletion of CRF1 attenuates EtOH intake but not EtOH preference.

During the 2hr sessions on Days 1–3, CRF1 KO mice displayed (A) reduced EtOH intakes, (B) reduced total fluid intakes, and (C) increased EtOH preference, relative to WT mice. On Day 4, EtOH (D) and total fluid (E) intakes were also reduced in KO mice, but EtOH preference did not significantly differ between the genotypes (F). Deletion of CRF1 had no effect on (G) food intake, but significantly decreased (H) total caloric intake from food and ethanol combined (**p < .005, t-test).
During the 4h session on Day 4, KO mice consumed less EtOH (t25 = 2.29; p < .05; Figure 7D). Total fluid consumption on Day 4 was also decreased in CRF1 KO mice (t25 = 2.55; p < .05; Figure 7E), but the difference in EtOH preference did not reach significance (p > 0.1; Figure 7F). Although the genotypic difference in food intake also failed to reach significance (p > 0.2; Figure 7G), the total caloric intake from EtOH and food combined was significantly lower among KO mice, relative to WT mice (t25 = 3.20; p < .005; Figure 7H).
DISCUSSION
In this study, we provide evidence against a selective role for CRF1 signaling in short-term, binge-like EtOH drinking in mice on a B6 genetic background. Although pharmacological blockade of CRF1 with two chemically distinct antagonists (and constitutive genetic knockout of CRF1) reduced the g/kg dose of EtOH consumed in all experiments, the two bottle-choice modification of the standard DID paradigm revealed that these manipulations did so in the absence of a concomitant decrease in the EtOH preference ratio. Moreover, these treatments also decreased overall consumption of fluid and calories (both in the presence and absence of EtOH), and had complex effects on consumption of sweet tastant control solutions.
Clearly, our findings hold immediate relevance to several publications that aimed to identify the CRF-related neural substrates of short-term binge-like drinking (Kaur et al., 2012; Lowery et al., 2010; Lowery-Gionta et al., 2012; Ryabinin et al., 2008; Sparta et al., 2008). However, we are aware that multiple lines of converging evidence strongly implicate a selective role for the CRF system in a number of alcohol-related phenotypes (Giardino et al., 2011a; Heilig et al., 2011; Heilig and Koob, 2007), and our data do not refute the well-established role for CRF and CRF-related urocortin peptides in the neural processes underlying addiction (Koob, 2010; Ryabinin et al., 2012; Schank et al., 2012). Rather, they suggest that the ability of some manipulations of the CRF system to affect intake of addictive drugs may rest on the innate contribution of the CRF system to regulation of food and/or H2O consumption. It is important to note that components of the CRF system are differentially expressed between mouse genotypes that differ in EtOH-related phenotypes (Giardino et al., 2012a; Ryabinin and Weitemier, 2006), and our findings in B6 mice await replication in other genetic backgrounds.
Within the greater context of the CRF literature on feeding and homeostatic processes, our data may at first appear contradictory. Several early reports found that food and fluid consumption were decreased by activation of CRF1 via intracranial infusions of its two endogenous ligands, CRF and urocortin-1 (Ucn1) (Gosnell et al., 1983; Negri et al., 1985; Spina et al., 1996). Yet in the present experiments, a state of dampened or absent CRF1 signaling also reduced consummatory behavior. This apparent inconsistency can be explained by the fact that CRF and Ucn1 also bind to the CRF type-2 receptor (CRF2) (Fekete and Zorrilla, 2007), which has well-documented anorectic properties (Inoue et al., 2003; Pelleymounter et al., 2000; Smagin et al., 1998). Indeed, two separate research groups demonstrated that the anorectic properties of endogenous CRF1-binding ligands likely occur via their actions at CRF2 rather than CRF1 (Bradbury et al., 2000; Contarino et al., 2000). Thus, our series of findings using both pharmacological and genetic methods suggest that, unlike CRF2, activation of CRF1 may in fact promote overall consumptive processes under conditions of low or moderate stress (i.e., isolate housing). Indeed, we are aware of earlier studies in which two different CRF1 antagonists also decreased food intake in other mammalian species (Hope et al., 2000; Sabino et al., 2006).
One alternative possibility is that blockade of CRF1 resulted in a non-selective decrease in behavior, and that reduced consumption was an indirect effect of reduced locomotor activity levels. However, the doses that reduced intake of food, H2O, and EtOH also increased intake of sucrose, suggesting that these outcomes are unlikely to be mediated via drug-induced sedation. Furthermore, we previously demonstrated that CRF1 KO and WT mice did not differ in baseline locomotor activity levels, and that the 10 mg/kg dose of CP did not produce locomotor depression in drug-naïve B6 mice (Giardino et al., 2012b). Thus, our findings cannot be explained by effects of drug treatments or genetic manipulation on general activity levels.
An additional alternative explanation is that CP-376,395 failed to reduce EtOH preference because of carryover effects from drug treatment or sweet tastant experience during the prior experimental phases. However, we carefully counterbalanced the dose groups in each phase of the pharmacological experiments, and kept at least five days between treatments, minimizing the potential of residual drug effects. Moreover, although mice treated with CP had experienced sucrose drinking prior to EtOH drinking, our experiment with NBI reversed the order of the EtOH and sweet tastant phases, illustrating that CRF1 blockade also failed to reduce EtOH preference in tastant- and antagonist-naïve mice. Furthermore, in relation to their WT counterparts, CRF1 KO mice displayed reduced consumption of food and fluids in the absence of any pharmacological manipulations or experience with sweet tastant solutions. Therefore, our data are highly unlikely to be confounded by experiential drug or tastant effects, and more likely reflect the direct outcome of CRF1 blockade or deletion on homeostatic or consummatory processes.
In this regard, our studies highlight how simple measures of consummatory behavior can be used to calculate combined total caloric intakes from food and EtOH, or food and sucrose. These additional control variables may be of use to investigators attempting to parse the effects of their manipulations on motivation for the pharmacological vs. caloric properties of EtOH (especially those examining stress- and feeding-related neuropeptide systems). Although a series of experiments from the Thiele group showed that B6 mice do not consume binge-like doses of EtOH in the DID paradigm primarily for caloric reasons (Lyons et al., 2008), this conclusion may vary depending on the neural substrates and manipulations of interest, and should be reevaluated on a case-by-case basis. Aside from issues of caloric need, our decision to provide mice with access to H2O during all drinking sessions highlights the advantage of using preference ratios and total fluid intake values as additional variables for evaluating selectivity.
Although the CP-induced increase in sucrose consumption appears to contrast with the effects of CRF1 blockade on consumption of food and other solutions, our lab previously reported elevated consumption of 10% sucrose in CRF1 KO mice (Kaur et al., 2012), consistent with the present findings. CRF produces behavioral avoidance (Land et al., 2008), and has pro-depressant effects that are associated with decreased sucrose consumption, a proxy of anhedonia (Chen et al., 2012). Therefore, it is reasonable to hypothesize that blockade of CRF1 signaling potentiated the hedonic properties of a 10% sucrose solution, leading to the increased intake observed in our study (Fig. 2A). Although we reported in the same experiment that CP-20 reduced sucrose preference during the first half of the session (1.000 ± 0.000 vs. 0.980 ± 0.008), this minute difference is unlikely to be biologically meaningful, and more likely reflects the large variability in exceptionally low volumes of H2O that were consumed during this time period (0.02 ± 0.01 mL/2hr). The opposite effects of CRF1 blockade on sucrose intake vs. food intake may be explained by the bi-phasic, time-dependent effects of these manipulations, as we observed effects on sucrose intake specifically during the second half of the 4hr drinking session. Unfortunately, because measuring food consumption in the midst of the session would have been a major disturbance to the mice, we could not directly compare the timecourse of CP-376,395 effects on intake of sucrose vs. food.
It is important to note that the rates of sucrose drinking in our study were substantially higher than rates of intake of EtOH, water or saccharin. CRF1 blockade decreased intake of these fluids, but increased intake of sucrose. One cannot exclude the possibility that CRF1 blockade differentially affects self-administration of reinforcers with different motivational valence. While testing this idea is beyond the scope of the current study, it underscores our finding that 10% sucrose is not an appropriate control for specificity of EtOH self-administration. Interestingly, this idea also suggests that the effect of CRF1 on intake of a reinforcer can change direction depending on the motivational valence of a reinforcer. Thus, while CRF1 blockade non-specifically decreased EtOH intake in the present study, this manipulation could increase intake of EtOH if the motivational valence of EtOH were increased. In agreement with this possibility, increased self-administration of EtOH has been observed in CRF1 KO mice following prolonged exposure to EtOH and repeated stress (Molander et al., 2012; Sillaber et al., 2002).
Taken together, our findings indicate that CRF1 blockade produces non-specific effects on consummatory behaviors, and argue against a specific involvement of CRF1 signaling in regulation of binge alcohol consumption. Finally, we acknowledge that CRF/CRF1 adaptations occurring in the central nucleus of the amygdala are critical for the binge-like drinking phenotype (Lowery-Gionta et al., 2012), and our systemic manipulations cannot address CRF1 actions at the site-specific level within the brain. Nevertheless, human alcoholics enrolled in clinical trials are currently undergoing systemic treatments with CRF1 antagonists, exemplifying the relevance of our current findings to pharmacotherapeutic treatment of addiction-related disorders.
Acknowledgments
This research was supported by NIH grants to William J. Giardino (F31 AA021023) and Andrey E. Ryabinin (AA013738, AA016647, AA010760). We thank Dr. John C. Crabbe and Amanda M. Barkley-Levenson for helpful discussion, Dr. Mary Stenzel-Poore for donation of CRF1 heterozygous knockout breeder mice, Dr. Deborah A. Finn for generous donation of NBI-27914, Ju Li and Eduardo D. Rodriguez for excellent technical assistance, and Christopher Snelling for logistical assistance.
Footnotes
The authors have no conflicts of interest.
References
- Bradbury MJ, McBurnie MI, Denton DA, Lee KF, Vale WW. Modulation of urocortin-induced hypophagia and weight loss by corticotropin-releasing factor receptor 1 deficiency in mice. Endocrinology. 2000;141(8):2715–24. doi: 10.1210/endo.141.8.7606. [DOI] [PubMed] [Google Scholar]
- Chen YW, Rada PV, Butzler BP, Leibowitz SF, Hoebel BG. Corticotropin-releasing factor in the nucleus accumbens shell induces swim depression, anxiety, and anhedonia along with changes in local dopamine/acetylcholine balance. Neuroscience. 2012;206:155–66. doi: 10.1016/j.neuroscience.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Conover EA, Jones KL. Safety concerns regarding binge drinking in pregnancy: A review. Birth Defects Res A Clin Mol Teratol. 2012;94(8):570–5. doi: 10.1002/bdra.23034. [DOI] [PubMed] [Google Scholar]
- Contarino A, Dellu F, Koob GF, Smith GW, Lee KF, Vale WW, Gold LH. Dissociation of locomotor activation and suppression of food intake induced by CRF in CRFR1-deficient mice. Endocrinology. 2000;141(7):2698–702. doi: 10.1210/endo.141.7.7653. [DOI] [PubMed] [Google Scholar]
- Courtney KE, Polich J. Binge drinking in young adults: Data, definitions, and determinants. Psychol Bull. 2009;135(1):142–56. doi: 10.1037/a0014414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Harris RA, Koob GF. Preclinical studies of alcohol binge drinking. Ann N Y Acad Sci. 2011;1216:24–40. doi: 10.1111/j.1749-6632.2010.05895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas D, Brown ES. Adolescent substance abuse and psychiatric comorbidities. J Clin Psychiatry. 2006;67(7):e02. doi: 10.4088/jcp.0706e02. [DOI] [PubMed] [Google Scholar]
- Fekete EM, Zorrilla EP. Physiology, pharmacology, and therapeutic relevance of urocortins in mammals: ancient CRF paralogs. Front Neuroendocrinol. 2007;28(1):1–27. doi: 10.1016/j.yfrne.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardino WJ, Cocking DL, Kaur S, Cunningham CL, Ryabinin AE. Urocortin-1 within the centrally-projecting Edinger-Westphal nucleus is critical for ethanol preference. PLoS One. 2011a;6(10):e26997. doi: 10.1371/journal.pone.0026997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardino WJ, Cote DM, Li J, Ryabinin AE. Characterization of Genetic Differences within the Centrally Projecting Edinger-Westphal Nucleus of C57BL/6J and DBA/2J Mice by Expression Profiling. Front Neuroanat. 2012a;6:5. doi: 10.3389/fnana.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardino WJ, Mark GP, Stenzel-Poore MP, Ryabinin AE. Dissociation of corticotropin-releasing factor receptor subtype involvement in sensitivity to locomotor effects of methamphetamine and cocaine. Psychopharmacology (Berl) 2012b;219(4):1055–63. doi: 10.1007/s00213-011-2433-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardino WJ, Pastor R, Anacker AM, Spangler E, Cote DM, Li J, Stenzel-Poore MP, Phillips TJ, Ryabinin AE. Dissection of corticotropin-releasing factor system involvement in locomotor sensitivity to methamphetamine. Genes Brain Behav. 2011b;10(1):78–89. doi: 10.1111/j.1601-183X.2010.00641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardino WJ, Ryabinin AE. orticotropin-releasing factor: innocent until proven guilty. Nat Rev Neurosci. 2012;13(1):70. doi: 10.1038/nrn3110-c1. author reply 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosnell BA, Morley JE, Levine AS. A comparison of the effects of corticotropin releasing factor and sauvagine on food intake. Pharmacol Biochem Behav. 1983;19(5):771–5. doi: 10.1016/0091-3057(83)90078-3. [DOI] [PubMed] [Google Scholar]
- Heilig M, Goldman D, Berrettini W, O’Brien CP. Pharmacogenetic approaches to the treatment of alcohol addiction. Nat Rev Neurosci. 2011;12(11):670–84. doi: 10.1038/nrn3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007;30(8):399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope PJ, Turnbull H, Farr S, Morley JE, Rice KC, Chrousos GP, Torpy DJ, Wittert GA. Peripheral administration of CRF and urocortin: effects on food intake and the HPA axis in the marsupial Sminthopsis crassicaudata. Peptides. 2000;21(5):669–77. doi: 10.1016/s0196-9781(00)00196-0. [DOI] [PubMed] [Google Scholar]
- Inoue K, Valdez GR, Reyes TM, Reinhardt LE, Tabarin A, Rivier J, Vale WW, Sawchenko PE, Koob GF, Zorrilla EP. Human urocortin II, a selective agonist for the type 2 corticotropin-releasing factor receptor, decreases feeding and drinking in the rat. J Pharmacol Exp Ther. 2003;305(1):385–93. doi: 10.1124/jpet.102.047712. [DOI] [PubMed] [Google Scholar]
- Kaur S, Li J, Stenzel-Poore M, Ryabinin AE. Corticotropin-releasing factor acting on corticotropin-releasing factor receptor type 1 is critical for binge alcohol drinking in mice. Alcohol Clin Exp Res. 2012;36(2):369–76. doi: 10.1111/j.1530-0277.2011.01610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Ryabinin AE. Ghrelin receptor antagonism decreases alcohol consumption and activation of perioculomotor urocortin-containing neurons. Alcohol Clin Exp Res. 2010;34(9):1525–34. doi: 10.1111/j.1530-0277.2010.01237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59(1):11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Brain stress systems in the amygdala and addiction. Brain Res. 2009;1293:61–75. doi: 10.1016/j.brainres.2009.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. The role of CRF and CRF-related peptides in the dark side of addiction. Brain Res. 2010;1314:3–14. doi: 10.1016/j.brainres.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CL, Liu MT, Yin SJ, Lee JT, Lu CC, Peng GS. Heavy Binge Drinking May Increase Risk of Stroke in Nonalcoholic Hypertensives Carrying Variant ALDH2*2 Gene Allele. Acta Neurol Taiwan. 2012;21(1):39–43. [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28(2):407–14. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery EG, Spanos M, Navarro M, Lyons AM, Hodge CW, Thiele TE. CRF-1 antagonist and CRF-2 agonist decrease binge-like ethanol drinking in C57BL/6J mice independent of the HPA axis. Neuropsychopharmacology. 2010;35(6):1241–52. doi: 10.1038/npp.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery EG, Thiele TE. Pre-clinical evidence that corticotropin-releasing factor (CRF) receptor antagonists are promising targets for pharmacological treatment of alcoholism. CNS Neurol Disord Drug Targets. 2010;9(1):77–86. doi: 10.2174/187152710790966605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery-Gionta EG, Navarro M, Li C, Pleil KE, Rinker JA, Cox BR, Sprow GM, Kash TL, Thiele TE. Corticotropin Releasing Factor Signaling in the Central Amygdala is Recruited during Binge-Like Ethanol Consumption in C57BL/6J Mice. J Neurosci. 2012;32(10):3405–13. doi: 10.1523/JNEUROSCI.6256-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons AM, Lowery EG, Sparta DR, Thiele TE. Effects of food availability and administration of orexigenic and anorectic agents on elevated ethanol drinking associated with drinking in the dark procedures. Alcohol Clin Exp Res. 2008;32(11):1962–8. doi: 10.1111/j.1530-0277.2008.00784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander A, Vengeliene V, Heilig M, Wurst W, Deussing JM, Spanagel R. Brain-specific inactivation of the Crhr1 gene inhibits post-dependent and stress-induced alcohol intake, but does not affect relapse-like drinking. Neuropsychopharmacology. 2012;37(4):1047–56. doi: 10.1038/npp.2011.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri L, Noviello L, Noviello V. Effects of sauvagine, urotensin I and CRF on food intake in rats. Peptides. 1985;6(Suppl 3):53–7. doi: 10.1016/0196-9781(85)90350-x. [DOI] [PubMed] [Google Scholar]
- NIH-NIAAA. NIAAA council approves definition of binge drinking. NIAAA Newsletter. 2004;3(3) [Google Scholar]
- Pelleymounter MA, Joppa M, Carmouche M, Cullen MJ, Brown B, Murphy B, Grigoriadis DE, Ling N, Foster AC. Role of corticotropin-releasing factor (CRF) receptors in the anorexic syndrome induced by CRF. J Pharmacol Exp Ther. 2000;293(3):799–806. [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84(1):53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Ford MM, Yu CH, Brown LL, Finn DA, Garland T, Jr, Crabbe JC. Mouse inbred strain differences in ethanol drinking to intoxication. Genes Brain Behav. 2007;6(1):1–18. doi: 10.1111/j.1601-183X.2006.00210.x. [DOI] [PubMed] [Google Scholar]
- Ryabinin AE, Galvan-Rosas A, Bachtell RK, Risinger FO. High alcohol/sucrose consumption during dark circadian phase in C57BL/6J mice: involvement of hippocampus, lateral septum and urocortin-positive cells of the Edinger-Westphal nucleus. Psychopharmacology (Berl) 2003;165(3):296–305. doi: 10.1007/s00213-002-1284-y. [DOI] [PubMed] [Google Scholar]
- Ryabinin AE, Tsoory MM, Kozicz T, Thiele TE, Neufeld-Cohen A, Chen A, Lowery-Gionta EG, Giardino WJ, Kaur S. Urocortins: CRF’s siblings and their potential role in anxiety, depression and alcohol drinking behavior. Alcohol. 2012;46(4):349–57. doi: 10.1016/j.alcohol.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryabinin AE, Weitemier AZ. The urocortin 1 neurocircuit: ethanol-sensitivity and potential involvement in alcohol consumption. Brain Res Rev. 2006;52(2):368–80. doi: 10.1016/j.brainresrev.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Ryabinin AE, Yoneyama N, Tanchuck MA, Mark GP, Finn DA. Urocortin 1 microinjection into the mouse lateral septum regulates the acquisition and expression of alcohol consumption. Neuroscience. 2008;151(3):780–90. doi: 10.1016/j.neuroscience.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabino V, Cottone P, Koob GF, Steardo L, Lee MJ, Rice KC, Zorrilla EP. Dissociation between opioid and CRF1 antagonist sensitive drinking in Sardinian alcohol-preferring rats. Psychopharmacology (Berl) 2006;189(2):175–86. doi: 10.1007/s00213-006-0546-5. [DOI] [PubMed] [Google Scholar]
- Schank JR, Heilig M, Ryabinin AE, Giardino WJ, Ciccocioppo R. Stress related neuropeptides and addictive behaviors: beyond the usual suspects. Neuron. 2012;76(1):192–208. doi: 10.1016/j.neuron.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe AL, Tsivkovskaia NO, Ryabinin AE. Ataxia and c-Fos expression in mice drinking ethanol in a limited access session. Alcohol Clin Exp Res. 2005;29(8):1419–26. doi: 10.1097/01.alc.0000174746.64499.83. [DOI] [PubMed] [Google Scholar]
- Sillaber I, Rammes G, Zimmermann S, Mahal B, Zieglgansberger W, Wurst W, Holsboer F, Spanagel R. Enhanced and delayed stress-induced alcohol drinking in mice lacking functional CRH1 receptors. Science. 2002;296(5569):931–3. doi: 10.1126/science.1069836. [DOI] [PubMed] [Google Scholar]
- Smagin GN, Howell LA, Ryan DH, De Souza EB, Harris RB. The role of CRF2 receptors in corticotropin-releasing factor- and urocortin-induced anorexia. Neuroreport. 1998;9(7):1601–6. doi: 10.1097/00001756-199805110-00063. [DOI] [PubMed] [Google Scholar]
- Sparta DR, Sparrow AM, Lowery EG, Fee JR, Knapp DJ, Thiele TE. Blockade of the corticotropin releasing factor type 1 receptor attenuates elevated ethanol drinking associated with drinking in the dark procedures. Alcohol Clin Exp Res. 2008;32(2):259–65. doi: 10.1111/j.1530-0277.2007.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina M, Merlo-Pich E, Chan RK, Basso AM, Rivier J, Vale W, Koob GF. Appetite-suppressing effects of urocortin, a CRF-related neuropeptide. Science. 1996;273(5281):1561–4. doi: 10.1126/science.273.5281.1561. [DOI] [PubMed] [Google Scholar]
- Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19(2):162–6. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- van de Wiel A, de Lange DW. Cardiovascular risk is more related to drinking pattern than to the type of alcoholic drinks. Neth J Med. 2008;66(11):467–73. [PubMed] [Google Scholar]