

Abstract
Introduction
We have previously shown that the danger signal High Mobility Group Box 1 (HMGB1) promotes angiogenesis when administered to ischemic muscle. HMGB1 signals through Toll-like receptor 4 (TLR4) as well as the receptor for advanced glycation end-products (RAGE). However, the actions of these receptors in ischemic injury and muscle recovery are not known. We hypothesize that TLR4 mediates tissue recovery and angiogenesis in response to ischemia.
Methods
Femoral artery ligation was performed in control, TLR4 competent (C3H/HeOuJ), and incompetent (C3H/HeJ) mice, as well as RAGE knockout mice and their C57B6 control counterparts. In other experiments, control mice were pretreated with anti-HMGBI neutralizing antibody before femoral artery ligation. After two weeks, limb perfusion was evaluated using laser Doppler perfusion imaging (LDPI) and reported as the ratio of blood flow in the ischemic to nonischemic limb. Muscle necrosis, fat replacement, and vascular density in the anterior tibialis muscle were quantified histologically. In vitro, TLR4 and RAGE expression was evaluated in human dermal microvascular endothelial cells (HDMVECs) in response to hypoxia. HDMVECs treated with HMGB1 alone and in the presence of anti-TLR4 antibody were probed for phosphorylated ERK (p-ERK), a signaling molecule critical to EC angiogenic behavior.
Results
Both anti-HMGB1 antibody as well as defective TLR4 signaling in HeJ mice resulted in prominent muscle necrosis two weeks after femoral artery ligation. Control HeOuJ mice had less necrosis than TLR4 incompetent HeJ mice, but a greater amount of fat replacement. In contrast to control C3H mice, control C57B6 mice demonstrated prominent muscle regeneration with very little necrosis. Muscle regeneration was not dependent on RAGE. While vascular density did not differ between strains, mice with intact RAGE and TLR4 signaling had less blood flow in ischemic limbs compared to mutant strains. In vitro, EC TLR4 expression increased in response to hypoxia while TLR4 antagonism decreased HMGB1-induced activation of ERK.
Conclusion
Both HMGB1 and TLR4 protect against muscle necrosis after hindlimb ischemia. However, muscle regeneration does not appear to be tied to vascular density. HMGB1 likely activates angiogenic behavior in EC in vitro, and this activation may be modulated by TLR4. The improvement in blood flow seen in mice with absent TLR4 and RAGE signaling may suggest anti-angiogenic roles for both receptors, or vasoconstriction induced by TLR4 and RAGE mediated inflammatory pathways.
Introduction
Peripheral artery disease causes significant functional disability and can result in limb loss within six months of diagnosis in 25–40% of patients who present with non-reconstructable disease. 1 Responses to limb ischemia include arteriogenesis, angiogenesis and muscle regeneration. 2 Patients without either endovascular or surgical options for vascular reconstruction may benefit from medical therapies that promote perfusion and muscle recovery. The signals that promote angiogenesis, arteriogenesis and muscle regeneration are complex and not well characterized. Efforts to promote vessel growth with angiogenic agents have yielded little success with the development of inadequate or immature vascular networks. 3, 4 Thus, further study is required to characterize the signals that stimulate neovascularization and muscle regeneration to optimize current therapies for limb ischemia, and improve limb-salvage rates.
High Mobility Group Box-1 (HMGB1) is a ubiquitous nuclear protein that can be released by both necrotic and stressed cells in response to hypoxia and other insults. 5, 6 Once released, it signals through select Toll-like receptors (TLRs), including TLR2 and TLR4, as well as the Receptor for Advanced Glycation End-products (RAGE). HMGB1 has been shown to mediate lethality in sepsis and organ injury and in hemorrhagic shock.7 Recent studies suggest a role for HMGB1 and its receptors in angiogenesis and potentially muscle regeneration. 8–13 In our laboratory, we have demonstrated that HMGB1 is released by endothelial cells in response to hypoxia and promotes angiogenesis when administered to ischemic mouse hindlimbs.8 Based on this, we hypothesize that TLR4 mediates tissue recovery and angiogenesis in response to ischemia. Thus, we evaluated the roles of HMGB1, TLR4, and RAGE in promoting neovascularization and muscle regeneration after limb ischemia using a murine hindlimb ischemia model in this current study. Murine hindlimb ischemia is generally well tolerated due to compensatory arteriogenesis and angiogenesis and is thus a relevant model for these studies.14.
Methods
Endothelial Cells
Human dermal microvascular endothelial cells and (HDMVECs; VEC Technologies, Rensselaer, NY) were cultured in OptiMem with heparin and Endothelial Cell Growth Supplement (ECGS). Cells were used between passages 3–12. Preliminary experiments have demonstrated that early and late passages within this time frame behave similarly. Serum depletion was performed in DMEM with 1% FBS for 3–6 hours.
Reagents
Recombinant HMGB1 was derived from yeast using a modification of the vector YEpFLAG as described 15 and used at a dose of 1μg/ml which promotes endothelial tube formation.8 HMGB1 was stored in formulation buffer (25mM Tris chloride pH 8, 150mM KCl, 2mM dithiothreitol, 10% glycerol) which was used a control for rHMGB1 administration. Polyclonal HMGB1 blocking antibody, a generous gift from Dr. Kevin Tracey (Feinstein Institute for Medical Research, Manhasset NY), was developed in rabbit and affinity-purified as previously described 16. Control rabbit polyclonal IgG was obtained from Sigma (St. Louis, MO). Growth-factor reduced (GFR) Matrigel (BD Biosciences, San Jose, CA) was stored at 4°C, and allowed to solidify on either 24 well plates or glass coverslips for 30 min in a 37 °C standard incubator. Neutralizing antibodies against TLR2 and TLR4 were obtained from Biolegend (San Diego, CA). Antibodies for immunohistochemistry were obtained from the following companies: Abcam, Cambridge, MA (anti-HMGB1, anti-RAGE); eBiosciences, San Diego, CA (anti-TLR 4); Dako Carpinteria, CA (von Willebrand Factor); and Vector Laboratories, Burlingame, CA (Goat-anti-rabbit secondary antibody). Alexa 488 was obtained from Molecular Probes (Eugene, OR), Rhodamine-labeled phalloidin from Invitrogen (Carlsbad, CA), DRAQ 5 from Biostatus Limited (Bath, UK), and DAPI from Sigma (St. Louis, MO).
Animals
Male C3H/HeJ (TLR4 incompetent; Stock # 000659; N=9) and C3H/HeOuJ (TLR4 competent; Stock # 000635) and C57BL/6J mice (Stock # 000664) were purchased from Jackson Laboratories (Bar Harbor, ME). The C3H/HeJ mice express a mutant TLR4 receptor and are not responsive to the TLR4 ligands LPS or HMGB1. The C3H/HeOuJ mice are the wild-type controls for this strain. RAGE KO (C57Bl/6J background) mice were generous gifts from Tim Oury, PhD (University of Pittsburgh, Pittsburgh, PA). All animal housing and procedures were approved by the Institutional Animal Use and Care Committee of the University of Pittsburgh (protocol #0911093B-7). Mice were used at 12 wks of age and weighed 20–40gm.
In vitro immunohistochemistry and confocal microscopy
HDMVECs were seeded onto gelatin-coated glass coverslips and incubated in either normoxia or 1% hypoxia for 6 or 24 hrs. The hypoxic conditions were achieved in a hypoxia chamber (COY Laboratory Products, Inc, Grass Lake, MI). Desired hypoxia was regulated by replacing ambient air in the chamber with a mixture of humidified nitrogen (95%) and CO2 (5%) until the set oxygen content is reached. The cells were undisturbed for the duration of the hypoxia treatment to maintain a consistent level of hypoxia during the experiments. Cells were fixed with 2% paraformaldehyde, permeabilized with 0.1% Triton X for 10 min, and blocked with 2% BSA for 1 hr. Coverslips were incubated with primary antibody to TLR4 or RAGE, as well as rhodamine phalloidin and DRAQ-5 to evaluate the cytoskeleton (phalloidin) and cell nuclei (DRAQ 5), respectively. Alexa 488-conjugated secondary antibody was used to detect antigen staining. After mounting onto glass slides, 4–5 images per coverslip were obtained at 400x using an Olympus Provis II microscope with Magnafire image acquisition software.
Western blot analysis
HDMVECs were treated with HMGB1 or formulation buffer and lysed in phenylmethanesulfonylfluoride (PMSF) at 30 minutes. Protein content was quantified using BCA. Cell proteins were separated on SDS-PAGE, transferred to nitrocellulose membranes and immunoblotted for phospho-ERK (p-ERK), and total ERK (t-ERK) as a loading control. Quantification of relative band density was obtained using Image J (NIH) analysis programming.
Skeletal muscle obtained from ischemic and nonischemic hindlimbs after femoral artery ligation (see below) were snap frozen in liquid nitrogen and stored at −80°C until use. For immunoblotting, muscles were homogenized and lysed in PMSF. Nuclear and cytoplasmic fractions were separated as described 5 on 14% SDS-PAGE. After transfer to nitrocellulose, Western blot was performed on nuclear and cytoplasmic fractions for HMGB1 and β-actin.
Murine hind-limb ischemia model
Femoral artery ligation promotes robust angiogenic and arteriogenic responses in mice,14 and was used to evaluate ischemic muscle injury and neovascularization in numerous studies. We have chosen a 2 week time point for the maximum duration of the ischemia because other studies have demonstrated that mice achieve near maximal recovery of perfusion at this time. Plateau occurs at about 21 days.14 Also, we have observed that muscle recovery is complete by 2 weeks. Mice were anesthetized with pentobarbital (0.1cc/gm IP). Bilateral groins were shaved and prepped with iodine prep solution. Transverse incisions were made in each groin and the femoral structures, were identified. On the right, the external iliac and femoral veins and arteries, as well as all visible branches were ligated with 6-0 silk 17, avoiding the femoral nerve. On the left, the femoral vessels were exposed but not ligated. Mice were sacrificed by overdose of inhaled isoflurane and cervical dislocation either 24 hrs or 14 days later. In separate experiments, HeOuJ mice were injected with 600μg of either functional anti-HMGB-1 antibody or control IgG before the initiation of hind-limb ischemia. This dose has previously been shown to attenuate hepatic damage in a model of ischemic-reperfusion injury.5
Laser Doppler perfusion imaging (LDPI)
Mice were anesthetized with inhaled isoflurane which was maintained throughout the procedure. Heart rate and respiratory rate were visually monitored to assess depth of anesthesia, and animals were kept warm with a heating lamp. Excess hairs were removed from the limb. The blood flow to both hind-limbs was measured using a Laser Doppler blood-flow meter (PERIMED III, Stockholm, Sweden). Three sequential images were obtained, and the one demonstrating the greatest perfusion to both extremities was chosen for analysis. The entire hind limb starting from the incision site proximally to the paw distally was incorporated into the region of interest. Perfusion was expressed as a ratio of the ischemic to non-ischemic leg.
Histologic Analysis
Tibialis anterior muscle was removed at the time of sacrifice, fixed in formalin, paraffin-embedded and sectioned (8μm thickness). Sections were stained with hematoxylin and eosin (H&E) for morphologic evaluation. Three H&E sections 60μm apart were digitally captured at 20x and evaluated for muscle necrosis, muscle regeneration and fat replacement using Metamorph image analysis program (Molecular Devices, Downington, PA) – all quantifications were performed in a blinded fashion. Muscle necrosis was identified by loss of muscle archictecture as well as inflammatory infiltrate. Regenerating muscle was characterized by rounded shape and centrally located nuclei.18 Corresponding sections were also stained with primary antibody to HMGB1 in samples obtained at 4 hours, and vWF to quantify vascular density in samples obtained after 14 days. After washing, sections were incubated with biotinylated goat-anti-rat secondary antibody. ABC horseradish peroxidase reagent was added after washing. Antigen detection was performed by adding AEC chromogen substrate, and sections were counterstained with hematoxylin. Sections were photographed at 200x magnification. Vascular density was determined using Metamorph and expressed as a value per high powered field (HPF).
Statistical analysis
SigmaStat (San Jose CA) analysis program was used to evaluate statistical significance (P<.05). Analysis was performed using analysis of variance (ANOVA) or t-test.
Results
Ischemia results in a sustained reduction in nuclear HMGB1 in skeletal muscle: In our previous study 8, HMGB1 had prominent effects on endothelial cells when given exogenously, and nuclear HMGB1 was reduced in HeOuJ mice after as little as 4 hours of muscle ischemia. In this experiment, muscle from both ischemic and nonischemic hindlimbs was examined for HMGB1 release 24 hours after arterial ligation to evaluate whether release persists for longer than 4 hours. Similar to our previous findings at 4 hours, ischemic muscle from both the TLR4 mutant and the control mice had significantly reduced nuclear HMGB1 content when compared to the nonischemic limb (Figure 1A–C). Western blot confirmed the reduction in nuclear HMGB1 in ischemic muscle compared to nonischemic muscle (Figure 1D). Beta-actin, which is involved in chromatin remodeling complexes within the nucleus 19 and was used as a loading control and remained unchanged regardless of ischemia. However, cytoplasmic fractions from ischemic muscle did not demonstrate an increase in HMGB1, suggesting that it was released extracellularly in the setting of ischemia (Figure 1E).
Figure 1.

C57BL6 (A) and C3H/HeOuJ (B) mice underwent femoral artery ligation on the right hindlimb, and exposure of the femoral vessels without ligation on the left. Anterior tibialis muscles were harvested from both sides after sacrifice on 24 hours after femoral ligation. Paraffin sections were stained for HMGB1 (arrow). (C) Quantification of HMGB1 staining nuclei (arrow) was expressed as % total nuclei (*P<.001;¶ P<.05; N=4–5; Scale bar = 50μm). Western blots for HMGB1 were performed on nuclear (D) and cytoplasmic (E) fractions of skeletal muscle from C3H/HeOuJ mice. Animal number refers to individual mice within the experiment.
HMGB1 and TLR4 protect against muscle necrosis: We hypothesized that HMGB1 is mobilized and extracellularly released to mediate muscle regeneration and angiogenesis. We have previously reported that administration of exogenous HMGB1 into ischemic skeletal muscle improved perfusion recovery and led to greater vascular density. Additionally, we have demonstrated loss of nuclear HMGB1 from both ischemic EC and ischemic myocytes. 8 In the following experiments, we evaluated the role of endogenous HMGB1 and its receptor TLR4 on tissue responses 2 weeks after femoral artery ligation. HeOuJ control mice treated with anti-HMGB1 blocking antibody (N=9) developed significantly more muscle necrosis than mice treated with nonspecific IgG (N=6) (25.5% ± 5.6 vs. 5.2% ± 3.6, respectively, P = .02; Figure 2A–B). Vessel density, as determined by vWF staining, did not differ between the treatment groups (anti-HMGB1 (N=6) 5.6 ± 1.5/HPF; IgG (N=4) 5.4 ± 1.1). Similarly, TLR4 incompetent mice (HeJ) also developed greater areas of muscle necrosis in response to limb ischemia. In contrast, the control mice (HeOuJ) developed larger areas of fat replacement (Figure 3). Vascular density was similar between HeJ and HeOuJ mice.
Figure 2.
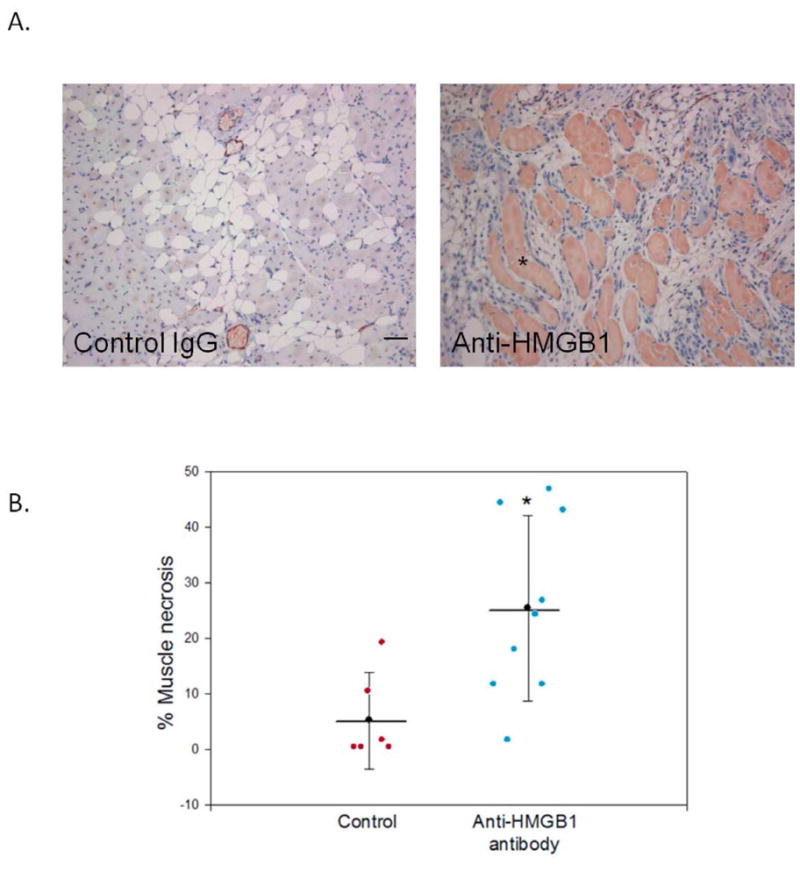
A. C3H/HeOuJ mice were treated with IP injection of polyclonal anti-HMGB1 antibody (600mg, N= 9) or IgG (N=6) 30 minutes before femoral artery ligation. The anterior muscle compartments of both limbs were harvested at day 14 for histologic analysis (asterisk = necrotic muscle, scale bar = 50μm. B. Scatter plot of the % muscle necrosis identified in each mouse (error bars = SEM, * P = .02).
Figure 3.

A. Calf muscles from ischemic hind-limbs of HeJ and HeOuJ mice obtained at day 14 were paraffin embedded and stained for vonWillebrand (vWF) factor (red). Note areas of fat replacement (arrow), and muscle necrosis (asterisk). Scale bar = 50μm. B. Quantification of vascular density (vessels per high power field; HPF), % area muscle regeneration, necrosis, and fat replacement in HeOuJ and HeJ mice. Values are obtained from 4–5 images per section, 3 sections per animal; N=3–4 animals/group. (* P = .03, ¶ P = .04).
Loss of RAGE and TLR4 signaling in limb ischemia improved limb perfusion but not muscle recovery
The role of RAGE, another HMGB1 receptor, was also examined. In contrast to the C3H strain of mice, prominent muscle regeneration was observed in the RAGE KO mice as well as their C57B6 controls. Architecturally, there was no difference between WT mice and those lacking RAGE with neither group of mice developing necrosis. Both had comparable amounts of fat replacement, muscle regeneration, and vascular density 14 days after femoral artery ligation (Figure 4). Thus, absence of RAGE or TLR4 signaling did not improve muscle regeneration or protect against necrosis. Paradoxically, mice lacking either RAGE or TLR4 signaling had better return of blood flow in the ischemic hindlimbs than control mice as measured by LDPI. (Figure 5).
Figure 4.
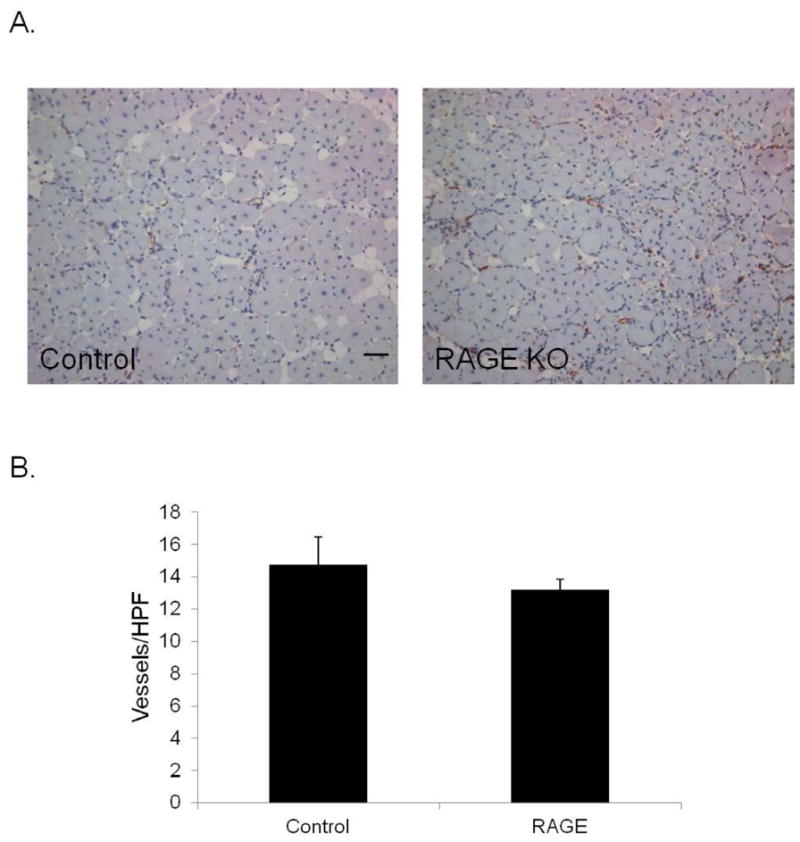
(A) Calf muscle from control C57B6 and RAGE KO mice were harvested 14 days after femoral ligation and stained for vWF (red) to evaluate vascular density (scale bar = 50 μm; N=4 each), (B) Quantification of number of vWF positive structures per HPF. Values are obtained from 4–5 images per section, 3 sections per animal.
Figure 5.
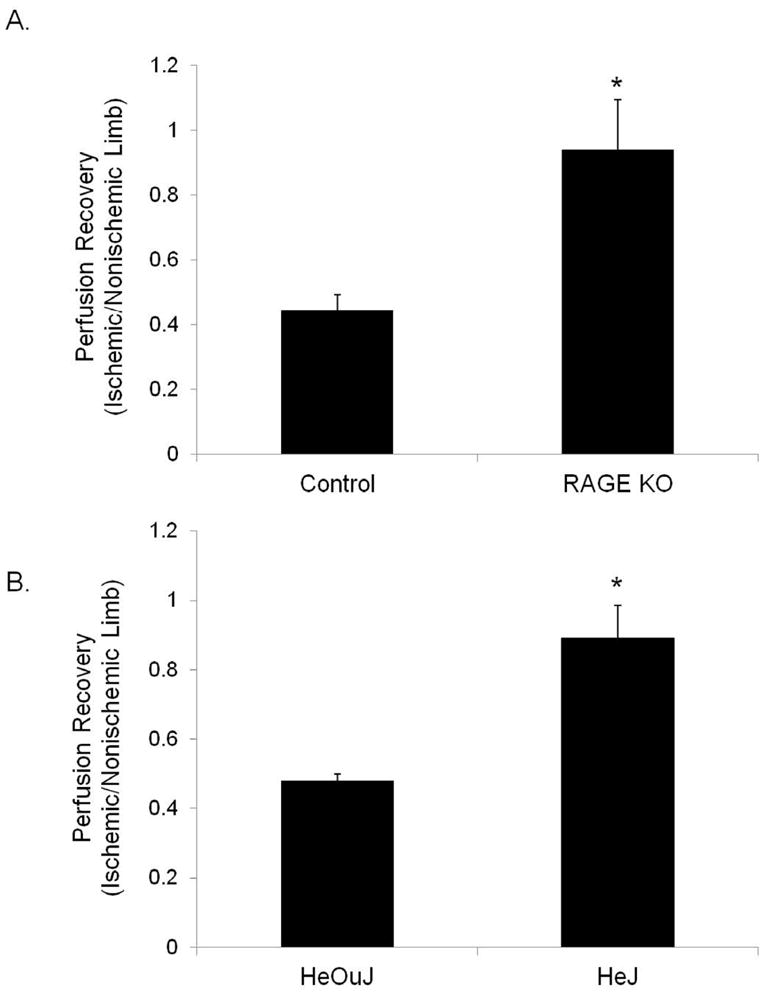
Laser Doppler perfusion imaging was performed 14 days after femoral artery ligation in (A) control and RAGE KO mice (N=4 each), as well as (B) HeOuJ and HeJ mice (N=6 each). Perfusion recovery is shown as ratio between ischemic and nonischemic limb. *P<.05.
Hypoxia results in increased expression of TLR4 but not RAGE in ECs: We have previously shown that hypoxic endothelial cells mobilize HMGB1 from their nuclei, similar to findings in hypoxic myoctes and the administration of exogenous HMGB1 increased EC tubing.8 In this experiment, we evaluated the expression of relevant HMGB1 receptors in ECs in response to normoxic and hypoxic conditions. TLR4 and RAGE expression in HDMVECs exposed to normal oxygen concentration or 1% hypoxia was examined by immunohistochemistry. While RAGE expression remained fairly stable under all the conditions, TLR4 expression increased significantly following 6 hours of hypoxia (Figure 6). Additionally, important differences existed in the distribution of TLR4 and RAGE within the cells. TLR4 appeared to be located in a perinuclear fashion during normoxia but became more diffuse in hypoxia. In contrast, RAGE staining was throughout the cytoplasm under both conditions.
Figure 6.

(A) HDMVECs were exposed to 1% hypoxia for 6 or 24 hours, and probed for expression of either TLR4 or (B) RAGE. Protein expression was determined using mean fluorescence intensity normalized to actin. Percent change in protein expression with hypoxia compared to air is depicted (C) (*P<.03, N=4/group)
HMGB1 stimulates ERK activation in HDMVECs: ERK is an important signal transduction pathway in EC angiogenic behavior. 20 Treatment of HDMVECs with rHMGB1 resulted in sustained ERK phosphorylation for over one hour (Figure 7A). In the presence of HMGB1 and the TLR4 neutralizing antibody, ERK phosphorylation was diminished. In contrast, treatment with control IgG or anti-TLR2 antibody, did not significantly affect ERK activation in HDMVECs (Figure 7B–C). This effect was seen 30 minutes after incubation with HMGB1 and the antibodies.
Figure 7.

(A) Phospho (P)-ERK and total (T)-ERK expression in HDMVECs exposed to HMGB1 over one hour. (B–C) Percent change in P-ERK/T-ERK ratio with exposure to IgG, Anti-TLR2 and Anti-TLR4 after thirty minutes; *P<.03, N=3
Conclusion
HMGB1, a danger associated molecular pattern that has been shown to be released from cells during stress and necrosis, has important cytokine-like functions including the activation of the innate immune system through TLRs and RAGE. 7 The goal of this study was to identify the role of HMGB1 and its receptors TLR4 and RAGE in mediating recovery after muscle ischemia. Interactions between HMGB1 and RAGE are critical to cell migration in the central nervous system. 21–24 In smooth muscle cells, HMGB1 promotes cell migration through actin cytoskeletal rearrangement.25 Systemically, extracellular HMGB1 induced the migration and proliferation of mesangioblasts.26 HMGB1 release may be through cell injury/death and it may also be actively secreted by inflammatory cells. The secreted HMGB1 has been shown to function in a cytokine type fashion, initiating a variety of downstream inflammatory pathways. The release of HMGB1 during cell damage may mediate regenerative processes such as angiogenesis and muscle recovery after an ischemic insult which may have important implications for patients with severe peripheral arterial disease who cannot undergo revascularization. These effects may be mediated by direct effects on skeletal muscle, endothelial cells, progenitor cells and/or immune cells. The role of TLRs and RAGE on resident and bone-marrow derived cells in ischemic muscle is an area of ongoing investigation in our laboratory.
In vivo, our results suggest that for up to 24 hours after the induction of ischemia, the loss of nuclear HMGB1 from muscle cells is prominent. HMGB1 does not appear to be present in the sarcoplasm during this time, suggesting that it is being released into the extracellular milieu. We have not been able to detect HMGB1 in the serum by Western blot in the setting of acute limb ischemia (data not shown). This may be due to relatively low levels of HMGB1 arising from the damaged skeletal muscle that is sufficient for local signaling in the local muscle bed. The 3 mechanisms governing HMGB1 release from skeletal muscle are not clear but are likely due to direct cellular injury or necrosis7.
HIF-1 alpha, a cellular sensor of low oxygen concentration which is stabilized by hypoxia and promotes the transcription of angiogenic proteins, promotes upregulation of RAGE in tumor cells. 27 The role of HIF-1 alpha in HMGB1 release was not investigated in our current study and there is no evidence in the literature to link these two molecules. Instead, there is evidence that HIF-1alpha does have some interplay with TLR4 signaling with reports indicating that LPS signaling through TLR4 requires HIF-1alpha to manifest signs of sepsis. 28 In addition, HIF-1alpha appears to be downstream of TLR4 activation in mediating dendritic cell maturation.29 These studies favor a role for HIF-1alpha in downstream TLR4 signaling which needs to be examined in the setting of hindlimb ischemia.
Interestingly, neutralization of HMGB1 as well as impaired TLR4 signaling both resulted in marked muscle necrosis that was still prominent two weeks after femoral artery ligation. These data suggest that HMGB1 released in the setting of ischemia is critical in promoting muscle recovery, potentially through TLR4 mediated pathways, at either a local or systemic level. The severity of muscle necrosis appeared to be independent of vascular density which did not differ between control or TLR4 mutant mice. The release of local HMGB1 following ischemia may recruit regenerative mesenchymal stem cells or circulating endothelial progenitor cells (EPCs) that can contribute to muscle regeneration and angiogenesis. This hypothesis is supported by the finding that HMGB1 enhanced homing of mesangioblasts to dystrophic muscle 30. HMGB1 has also been shown to promote homing of EPCs to ischemic muscle and tumors 31.
Both HMGB1 and TLR4 have been shown to promote cardiac remodeling after ischemia, and have been associated with decreased fibrosis and improved angiogenesis at the ischemic border. However, in cardiac ischemia-reperfusion, HMGB1 and TLR4 both mediate worse outcomes, presumably by driving an exaggerated inflammatory response. It has been postulated that in ischemia without reperfusion, HMGB1 and TLR4 drive recovery, which may be important therapeutically. 32 Interestingly, TLR4 polymorphisms have been associated with differing levels of cardiac functional recovery in patients with dilated cardiomyopathy, which may have implications for patients with critical limb ischemia.33 This has not been extensively investigated in the literature to date.
While HMGB1-RAGE interactions are known to be important in the recovery of the nervous system following injury, RAGE does not appear to play a significant role in muscle regeneration after ischemia. RAGE KO mice exhibited muscle histology that was indistinguishable from control animals following hind limb ischemia. However, it is difficult to compare the findings between the RAGE KO and the TLR4 mutant mice because the parental strain responses differ greatly in their responses to ischemia. The parent strain for the RAGE KO mice tolerated muscle ischemia well with robust muscle regeneration and little necrosis. Strain differences in muscle regeneration after ischemia are well described in mice and may be related to regenerative capacity as well as to an arteriogenic potential that can restore earlier tissue perfusion and limit ischemic injury. 34
While absent RAGE and impaired TLR4 signaling offered no advantage over controls in terms of muscle regeneration, both groups demonstrated improvements in perfusion recovery compared to their controls, without differences in vessel density. These findings would suggest that angiogenesis following ischemia is not regulated by RAGE or TLR4 signaling. Instead, TLR4 and RAGE signaling may negatively regulate arteriogenesis which has a greater ability to restore distal perfusion than angiogenesis. 35 The regulation of arteriogenesis by TLR4 and/or RAGE will be the focus of future investigations. Alternative explanations for the paradoxical LDPI findings exist. Because LDPI is a measurement of cutaneous perfusion, it is also possible that activation of inflammatory pathways through RAGE and TLR4 may result in cutaneous vasoconstriction which abates in the absence of these inflammatory signaling pathways. Thus, the relationship between cutaneous perfusion and vascular density in muscle may not be linear, particularly when evaluating inflammatory mediators.
The ability of hypoxia to upregulate TLR4 expression on ECs in vitro supports the potential for TLR4 to play an important role in mediating vascular responses. While RAGE expression was not altered in hypoxia, it is abundantly expressed in ECs and may still be an important receptor. In conjunction with our finding that ECs also release HMGB1 in the setting of ischemia, these data suggest a potential autocrine or paracrine mechanism in which HMGB1 released by ECs can mediate vascular responses to ischemia or flow changes derived from vascular occlusion, possibly through TLR4, in ECs or vascular smooth muscle cells. In support of this hypothesis, exogenous HMGB1 promotes ERK activation, which is critical for endothelial tube formation. In the setting of TLR4 antagonism, HMGB1 results in an initial peak of ERK phosphorylation followed by a significant nadir. Thus, effects of HMGB1 on EC that are mediated by TLR4 are likely to be temporal and context specific.
In summary, TLR4 and HMGB1 appear to play a role in muscle protection and regeneration after muscle ischemia. Our in vivo data also suggest that muscle regeneration is not dependent on angiogenesis. A plausible hypothesis is that HMGB1 mobilizes stem cells in a TLR4 dependent manner. The ability of HMGB1 to recruit mesenchymal stem cells has been previously described. 30 Alternatively, HMGB1-TLR4 interactions may be important in arteriogenesis or collateral formation which was also not specifically addressed in this study. Of interest, lack of TLR4 or RAGE resulted in improved perfusion recovery after ischemia. Thus, RAGE and TLR4 may mediate cutaneous vasoconstriction after injury which may, in part, be a consequence of actions on endothelial and perivascular cells. Further studies are required to determine how HMGB1’s interaction with both RAGE and TLR4 may play a role in mediating muscle recovery and angiogenesis after ischemic injury.
Clinical Relevance.
Nonreconstructable peripheral artery disease causes significant functional disability, and is associated with a high risk of limb loss. The mechanisms that govern muscle recovery and angiogenesis after ischemia are important to understand in order to improve medical therapy for patients who can not have an intervention. This paper evaluates the role of High Mobility Group Box-1 (HMGB1) and the innate immune receptor TLR4 in mediating muscle recovery after ischemia. While HMGB1 has been shown to mediate end-organ damage in other clinical scenarios, it may also play an important role in regenerative processes such as myocyte regeneration and angiogenesis.
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the VA Pittsburgh Healthcare System (ET). The work was supported by funding through an American Heart Association Established Investigator Award (ET), Foundation for Accelerated Vascular Research through the Wylie Award (US) and the UPMC Competitive Medical Research Foundation Award (US). The contents of this publication do not represent the views of the Department of Veterans Affairs or the United States Government. The project described was additionally supported by Grant Number 1UL1RR024153 (US) from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and NIH Roadmap for Medical Research, and its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. Information on NCRR is available at http://www.ncrr.nih.gov.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marston WA, Davies SW, Armstrong B, Farber MA, Mendes RC, Fulton JJ, Keagy BA. Natural history of limbs with arterial insufficiency and chronic ulceration treated without revascularization. J Vasc Surg. 2006;44(1):108–114. doi: 10.1016/j.jvs.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 2.Buschmann I, Schaper W. The pathophysiology of the collateral circulation (arteriogenesis) J Pathol. 2000;190(3):338–342. doi: 10.1002/(SICI)1096-9896(200002)190:3<338::AID-PATH594>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 3.Masaki I, Yonemitsu Y, Yamashita A, Sata S, Tanii M, Komori K, Nakagawa K, Hou X, Nagai Y, Hasegawa M, Sugimachi K, Sueishi K. Angiogenic gene therapy for experimental critical limb ischemia: acceleration of limb loss by overexpression of vascular endothelial growth factor 165 but not of fibroblast growth factor-2. Circ Res. 2002;90(9):966–973. doi: 10.1161/01.res.0000019540.41697.60. [DOI] [PubMed] [Google Scholar]
- 4.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9(6):685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 5.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7):1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu J, Nishimura M, Wang Y, Sims JR, Qiu S, Savitz SI, Salomone S, Moskowitz MA. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab. 2008;28(5):927–938. doi: 10.1038/sj.jcbfm.9600582. [DOI] [PubMed] [Google Scholar]
- 7.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5(4):331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 8.Sachdev U, Cui X, Hong G, Namkoong S, Karlsson JM, Baty CJ, Tzeng E. High mobility group box 1 promotes endothelial cell angiogenic behavior in vitro and improves muscle perfusion in vivo in response to ischemic injury. Journal of Vascular Surgery. 55(1):180–191. doi: 10.1016/j.jvs.2011.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Mori R, Straino S, Di Carlo A, Mangoni A, Pompilio G, Palumbo R, Bianchi ME, Capogrossi MC, Germani A. Multiple effects of high mobility group box protein 1 in skeletal muscle regeneration. Arterioscler Thromb Vasc Biol. 2007;27(11):2377–2383. doi: 10.1161/ATVBAHA.107.153429. [DOI] [PubMed] [Google Scholar]
- 10.Biscetti F, Straface G, De Cristofaro R, Lancellotti S, Rizzo P, Arena V, Stigliano E, Pecorini G, Egashira K, De Angelis G, Ghirlanda G, Flex A. High-Mobility Group Box 1 Protein Promotes Angiogenesis after Peripheral Ischemia in Diabetic Mice through a VEGF-dependent Mechanism. Diabetes. doi: 10.2337/db09-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlueter C, Weber H, Meyer B, Rogalla P, Roser K, Hauke S, Bullerdiek J. Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol. 2005;166(4):1259–1263. doi: 10.1016/S0002-9440(10)62344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitola S, Belleri M, Urbinati C, Coltrini D, Sparatore B, Pedrazzi M, Melloni E, Presta M. Cutting edge: extracellular high mobility group box-1 protein is a proangiogenic cytokine. J Immunol. 2006;176(1):12–15. doi: 10.4049/jimmunol.176.1.12. [DOI] [PubMed] [Google Scholar]
- 13.van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1) Angiogenesis. 2008;11(1):91–99. doi: 10.1007/s10456-008-9093-5. [DOI] [PubMed] [Google Scholar]
- 14.Couffinhal T, Silver M, Zheng LP, Kearney M, Witzenbichler B, Isner JM. Mouse model of angiogenesis. Am J Pathol. 1998;152(6):1667–1679. [PMC free article] [PubMed] [Google Scholar]
- 15.Ngamkitidechakul C, Twining SS. Buffered non-fermenter system for lab-scale production of secreted recombinant His-tagged proteins in Saccharomyces cerevisiae. Biotechniques. 2002;33(6):1296–1300. doi: 10.2144/02336pt02. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 17.Messina LM, Brevetti LS, Chang DS, Paek R, Sarkar R. Therapeutic angiogenesis for critical limb ischemia: invited commentary. J Control Release. 2002;78(1–3):285–294. doi: 10.1016/s0168-3659(01)00501-6. [DOI] [PubMed] [Google Scholar]
- 18.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84(1):209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 19.McDonald D, Carrero G, Andrin C, de Vries G, Hendzel MJ. Nucleoplasmic beta-actin exists in a dynamic equilibrium between low-mobility polymeric species and rapidly diffusing populations. J Cell Biol. 2006;172(4):541–552. doi: 10.1083/jcb.200507101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim YM, Namkoong S, Yun YG, Hong HD, Lee YC, Ha KS, Lee H, Kwon HJ, Kwon YG. Water extract of Korean red ginseng stimulates angiogenesis by activating the PI3K/Akt-dependent ERK1/2 and eNOS pathways in human umbilical vein endothelial cells. Biol Pharm Bull. 2007;30(9):1674–1679. doi: 10.1248/bpb.30.1674. [DOI] [PubMed] [Google Scholar]
- 21.Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000;275(51):40096–40105. doi: 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- 22.Rauvala H, Pihlaskari R. Isolation and some characteristics of an adhesive factor of brain that enhances neurite outgrowth in central neurons. J Biol Chem. 1987;262(34):16625–16635. [PubMed] [Google Scholar]
- 23.Rong LL, Yan SF, Wendt T, Hans D, Pachydaki S, Bucciarelli LG, Adebayo A, Qu W, Lu Y, Kostov K, Lalla E, Yan SD, Gooch C, Szabolcs M, Trojaborg W, Hays AP, Schmidt AM. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 2004;18(15):1818–1825. doi: 10.1096/fj.04-1900com. [DOI] [PubMed] [Google Scholar]
- 24.Rong LL, Trojaborg W, Qu W, Kostov K, Yan SD, Gooch C, Szabolcs M, Hays AP, Schmidt AM. Antagonism of RAGE suppresses peripheral nerve regeneration. FASEB J. 2004;18(15):1812–1817. doi: 10.1096/fj.04-1899com. [DOI] [PubMed] [Google Scholar]
- 25.Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, Arrigoni G, Bianchi ME. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152(6):1197–1206. doi: 10.1083/jcb.152.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palumbo R, Sampaolesi M, De Marchis F, Tonlorenzi R, Colombetti S, Mondino A, Cossu G, Bianchi ME. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol. 2004;164(3):441–449. doi: 10.1083/jcb.200304135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tafani M, Schito L, Pellegrini L, Villanova L, Marfe G, Anwar T, Rosa R, Indelicato M, Fini M, Pucci B, Russo MA. Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-κB. Carcinogenesis. 32(8):1167–1175. doi: 10.1093/carcin/bgr101. [DOI] [PubMed] [Google Scholar]
- 28.Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting Edge: Essential Role of Hypoxia Inducible Factor-1α in Development of Lipopolysaccharide-Induced Sepsis. The Journal of Immunology. 2007;178(12):7516–7519. doi: 10.4049/jimmunol.178.12.7516. [DOI] [PubMed] [Google Scholar]
- 29.Spirig R, Djafarzadeh S, Regueira T, Shaw SG, von Garnier C, Takala J, Jakob SM, Rieben R, Lepper PM. Effects of TLR Agonists on the Hypoxia-Regulated Transcription Factor HIF-1α and Dendritic Cell Maturation under Normoxic Conditions. PLoS ONE. 5(6):e10983. doi: 10.1371/journal.pone.0010983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D’Antona G, Pellegrino MA, Barresi R, Bresolin N, De Angelis MG, Campbell KP, Bottinelli R, Cossu G. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. 2003;301(5632):487–492. doi: 10.1126/science.1082254. [DOI] [PubMed] [Google Scholar]
- 31.Chavakis E, Hain A, Vinci M, Carmona G, Bianchi ME, Vajkoczy P, Zeiher AM, Chavakis T, Dimmeler S. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ Res. 2007;100(2):204–212. doi: 10.1161/01.RES.0000257774.55970.f4. [DOI] [PubMed] [Google Scholar]
- 32.Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 8(5):292–300. doi: 10.1038/nrcardio.2011.38. [DOI] [PubMed] [Google Scholar]
- 33.Riad A, Meyer zu Schwabedissen H, Weitmann K, Herda LR, Dörr M, Empen K, Kieback A, Hummel A, Reinthaler M, Grube M, Klingel K, Nauck M, Kandolf R, Hoffmann W, Kroemer HK, Felix SB. Variants of Toll-like Receptor 4 Predict Cardiac Recovery in Patients with Dilated Cardiomyopathy. Journal of Biological Chemistry. 287(32):27236–27243. doi: 10.1074/jbc.M112.369728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shireman PK, Quinones MP. Differential necrosis despite similar perfusion in mouse strains after ischemia. J Surg Res. 2005;129(2):242–250. doi: 10.1016/j.jss.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 35.Baumgartner I, Pieczek A, Manor O, Blair R, Kearney M, Walsh K, Isner JM. Constitutive expression of phVEGF165 after intramuscular gene transfer promotes collateral vessel development in patients with critical limb ischemia. Circulation. 1998;97(12):1114–1123. doi: 10.1161/01.cir.97.12.1114. [DOI] [PubMed] [Google Scholar]