

Abstract
Due to growing environmental concerns and diminishing petroleum reserves, a wide range of oxygenated species has been proposed as possible substitutes to fossil fuels: alcohols, methyl esters, acyclic and cyclic ethers. After a short review the major detailed kinetic models already proposed in the literature for the combustion of these molecules, the specific classes of reactions considered for modeling the oxidation of acyclic and cyclic oxygenated molecules respectively, are detailed.
Keywords: Biofuel, combustion, gas-phase oxidation, detailed kinetic models, alcohol, ester, ether, furan
I. INTRODUCTION
As traditional fossil fuels are considered to be largely responsible for important atmospheric degradations, there is an increasing interest to shift from hydrocarbon fossil fuels to bio-fuels. It has been shown that the presence of additional oxygenated compounds could reduce the formation of soot in diesel engines [1], but may also promote the formation of some toxic pollutants, such as aldehydes [2][3]. To better assess the effect of the presence of these new molecules in fuels on the engine efficiency and the pollutants formation, it is necessary to developed well validated detailed kinetic models for the combustion and the oxidation of the oxygenated components of biofuels. In order to be used in a predictive way, these detailed kinetic models need to include a large number of elementary reactions [4]. Before writing such detailed kinetic models, the types of involved reacting species have to be defined. The first part of this paper focuses then on the identification of the main types of oxygenated molecules present in the fuels which can be obtained from biomass. A second part reviews the major detailed kinetic models already proposed in the literature for the combustion of potential oxygenated components of biofuels and of molecules representative of them. Finally, a third and a fourth part describe the specific classes of reactions considered in detailed kinetic models for the combustion of acyclic and cyclic oxygenated molecules, respectively.
II. SUMMARY OF THE MAIN COMPONENTS OF THE FUELS DERIVED FROM BIOMASS AND WAYS TO OBTAIN THEM
The major difficulty in developing biofuels is to transform the very large and very oxygenated molecules present in biomass (e.g. carbohydrates, such as cellulose and starch, are large polymer chains assembled from thousand of sugar units which each contains 6 carbon atoms and a similar number of oxygen atoms) into molecules suitable to be used as fuels for engines, (e.g. small molecules including from 5 to 20 carbon atoms and very few oxygen atoms) [5]. A review of the ways to obtain transportation fuel from biomass has been recently published [6]. A method widely proposed is to partially oxidize biomass to produce a ‘syngas’ (a mixture of mainly carbon monoxide and hydrogen) [7] which can thereafter feed a catalytic process (Fisher-Tropsch synthesis) to produce a liquid fuel. This latter, made of linear saturated hydrocarbons, can easily be used in diesel engines [8]. However, as shown in figure 1, many types of oxygenated molecules (see Table 2 for their main chemical and physical properties) considered as potential additives to gasoline or diesel fuel can be obtained through a wide range of processes involving fermentation or catalytic reactions [9]:
Alcohols; methanol can be obtained from ‘syngas’[10], while ethanol, propanol, or butanol can be obtained through the fermentation of sugars or starches which can be produced from very common crops, such as sugar cane or corn [11][12]. Ways to produce alcohols from cellulose [13][14] or algae [15] have also been proposed. Alcohols are the most usually blended with gasoline. Heavier alcohols such as n-butanol have a higher energy density, a higher boiling point and a lower solubility in water than ethanol (see table 2) which should facilitate their use in spark ignition engines.
Fatty acid methyl and ethyl esters, which are obtained through the trans-esterification of vegetal oils or animal fat with methanol or ethanol, respectively. Vegetal oils mainly include triglycerides (98%), with small amounts of mono and diglycerides. The main fatty acids in rapeseed and soybean oils oleic (C18:1 monounsaturated) and linoleic (C18:2 omega-6 polyunsaturated) acids [16]. Methyl esters can be blended in any proportion with diesel fuel obtained from petroleum [11].
Acyclic ethers (see formulae in Table 1), such as dimethyl ether, which can be obtained from methanol or directly from ‘syngas’, and which has been proposed as an efficient alternative fuel for use in a diesel engine [17], or ethyl ter-butyl ether, which can be obtained by reaction of ethanol with isobutene and which is a common additive to gasoline [18].
Cyclic ethers (see formulae in Table 1) of the family of furans, such as 2,5-dimethylfuran, which can be obtained from fructose which is enzymatically produced from starch [19]. Saturated cyclic ethers such as methyl- or dimethyl-tetrahydrofurans, which can been obtained from carbohydrates or cellulosic biomass by a catalytic process [20], are also considered. These cyclic ethers could be valuable substitutes to fossil fuels for transportation. As shown in Table 2, compared to ethanol, 2,5-dimethylfuran has a higher energy density, a higher boiling point, and is not soluble in water [19].
Figure 1.

Ways to obtain components of fuels derived from biomass.
*See full names and formulae in table 1.
Table 2.
Properties of several oxygenated fuels and conventional fuels.
| Chemical Formula |
MWb (g/mol) |
Density (kg/l) |
Boiling Point (K) |
LHVb (MJ/kg) |
AITb (K) |
RON/MONb | CNb | |
|---|---|---|---|---|---|---|---|---|
| Alcohols | ||||||||
| Methanol | CH3OH | 32.04 | 0.796 [22] | 337.9 [22] | 20.0 [22] | 783 [23] | 126/96 [22] | 0-3 [22] |
| Ethanol | C2H5OH | 46.07 | 0.794 [22] | 351.6 [22] | 26.8 [22] | 763 [23] | 120/99 [22] | 5-8 [24] |
| n-Propanol | C3H7OH | 60.10 | 0.804 [25] | 355.4 [26] | 30.9 [25] | 753 [23] | 112/-- [26] | -- |
| n-Butanol | C4H9OH | 74.12 | 0.810 [25] | 355.9 [26] | 33.1 [27] | 703 [23] | 96/78 [27] | -- |
| n-Pentanol | C5H11OH | 88.15 | 0.811 [25] | 393.0 [28] | 34.8 [25] | 573 [29] | -- | -- |
| Acyclic ethers | ||||||||
| DME d | CH3OCH3 | 46.07 | 0.66 a [11] | 248.3 [11] | 28.6 [11] | 623 [30] | -- | >55 [30] |
| MTBE d | C4H9OCH3 | 88.15 | 0.74 [22] | 328.5 [22] | 35.2 [22] | 733 [31] | 118/101 [22] | <0 [22] |
| ETBE d | C4H9OC2H5 | 102.17 | 0.75 [22] | 346.0 [22] | 35.9 [22] | -- | 117/101 [22] | <0 [22] |
| TAME d | C6H14O | 102.17 | 0.75 [22] | 359.5 [22] | 36.5 [22] | 703 [32] | 114/100 [22] | <0 [22] |
| DMM b | CH3OCH2OCH3 | 76.09 | 0.86 [24] | 315.2 [24] | 22.4 [24] | 510 [24] | -- | 25-30 [24] |
| DEE b | C2H5OC2H5 | 74.12 | 0.71 [30] | 307.6 [30] | 33.9 [30] | 433 [30] | -- | >125 [30] |
| Methyl esters | ||||||||
| DMCb | C3H6O3 | 90.08 | 1.07 [33] | 363.2 [34] | 15.8 [33] | -- | -- | 35.5 [33] |
| Biodiesel c | C12-C22 FAME b | ≈300 | 0.88 [35] | 455-611 [35] | 37.2 [35] | 589 [24] | -- | 48-65 [35] |
| Cyclic ethers | ||||||||
| THF d, e | C4H8O | 72.11 | 0.89 [36] | 339.2 [36] | -- | 594 [36] | -- | -- |
| MTHFd | C5H10O | 86.13 | 0.85 [37] | 353.4 [37] | 32.0 [38] | -- | 86/72 [39] | -- |
| DMTHFd | C6H12O | 100.16 | 0.83 [40] | 364.1 [19] | -- | -- | 82 /-- [19] | -- |
| Furan | C4H4O | 68.07 | 0.94 [31] | 305.2 [31] | -- | 663 [31] | -- | -- |
| DMFd | C6H8O | 96.13 | 0.89 [41] | 366.5 [28] | 33.7 [19] | 559 [42] | 119 /-- [19] | -- |
| MF d | C5H6O | 82.10 | 0.91 [31] | 62.9 [19] | -- | -- | 131 /-- [19] | -- |
| Conventional fuels | ||||||||
| Gasolinec | C4-C14 HC | ≈102 | 0.74 [22] | 303-473 [22] | 42.7 [22] | ≈723 [23] | 95/85 [22] | -- |
| Diesel c | C8-C25 HC | ≈200 | 0.84 [22] | 443-633 [22] | 43.0 [22] | ≈533 [23] | -- | 40-55 [22] |
Notes: Density at 1 atm and 298 K.
LHV-Lower Heating Value (LCV- Lower Calorific Value); RON-Research Octane Number; MON-Motor Octane Number; CN-Cetane Number; AIT-Autoignition Temperature; MW- Molecular Weight; C12-C22 FAME - Methyl Esters of C12 to C22 Fatty Acids; DEE-Diethyl Ether; DMC-Dimethyl Carbonate; DMM-Dimethoxy Methane.
Fuel standard.
See full names and formulae in Table 1.
Not directly considered as a biofuel, but from the same family as potential biofuels.
Table 1.
| Acyclic ethers | Cyclic ethers | ||
|---|---|---|---|
| Species | Formula | Species | Formula |
| Diisopropyl ether (DIPE) |
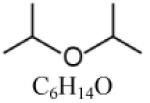
|
2,5- Dimethylfuran (DMF) |
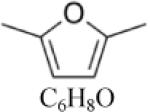
|
| Ethyl tert-butyl ether (ETBE) |
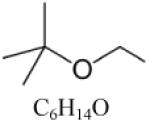
|
2-Methylfuran (MF) |
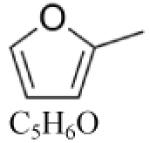
|
| Methyl tert-butyl ether (MTBE) |
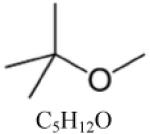
|
2-Methyltetrahydrofuran (MTHF) |
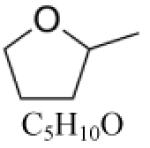
|
| Tertiary amyl methyl ether (TAME) |
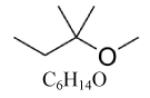
|
2,5-Dimethyltetrahydrofuran (DMTHF) |
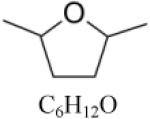
|
| Dimethyl ether (DME) |
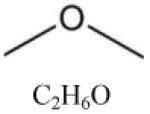
|
Tetrahydrofuran (THF) |
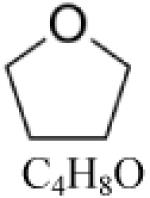
|
| Other species | ||
|---|---|---|
|
|
|
III. DETAILED KINETIC MODELS FOR THE COMBUSTION OF POTENTIAL OXYGENTATED COMPONENTS OF BIOFUELS
Table 3 summarizes the major detailed kinetic models which have been published for the combustion of oxygenated molecules which are considered as potential components of fuels. For each model, the conditions under which they have been validated are detailed. Mixtures have not been considered.
Table 3.
Major models for the combustion of potential oxygenated components of fuel.
| Components | References of chemical kinetic models |
Conditions of validation | |
|---|---|---|---|
| Alcohols | Ethanol | Natarajan and Bhaskaran 1981 [21] |
- Shock tube (T=1300-1700 K, P=101-203 kPa, Φ=0.5-2.0) [21]. |
| Dunphy et al. 1991 [43] | - Shock tube (T=1080-1660 K, P=182-466 kPa, Φ=0.25-2.0) [44]. | ||
| Norton and Dryer 1992 [45] |
- Flow reactor (T=1100 K, P=101kPa, Φ=0.61-1.24) [45]. | ||
| Marinov et al. 1999 [46] | - Shock tube [21]*, [44]*; Flow reactor [45]*. - Laminar burning velocities in constant volume bomb (P=100-800 kPa, Φ=0.7-1.4) [47] and in counterflow twin-flame (P=101 kPa, Φ=0.55-1.8) [48]. - Jet-stirred reactor (T=1000-1200 K, P=101 kPa, Φ=0.2-2.0) [49]. |
||
| Saxena and Williams 2007 [50] |
- Shock tube [21]*, [44]*; Counterflow twin-flame [48]*. - Counterflow diffusion and partially premixed flames (P=101 kPa) [50]. - Extinction in counterflow diffusion flames (P=101 kPa) [51]. |
||
| Li et al, 2007 [52] | - Shock tube [21]*, [44]*; Laminar burning velocities [47]*, [48]*; Counterflow flames [50]*. - Flow Reactor (T=800-950 K, P=304-1216 kPa, Φ=0.3-1.4) [52]. |
||
| Cancino et al, 2010 [53] | - Shock tube [44]* and (P=1000-5000 kPa, T=750-1220K, Φ=0.3-1.0) [53]. |
||
| Leplat et al. 2011 [54] | - Shock tube [21]*, [44]*; Counterflow flames [50]*. - Jet stirred reactor (P=101 kPa, T=890-1250 K, Φ=0.25-2.0) [54]. - Premixed laminar flat flame (P=5 kPa, Φ=0.75-1.25) [54]. - Laminar burning velocities in constant volume bomb (P=100-1000 kPa, Φ=0.7-1.4) [55][56]. |
||
|
n-and iso- Propanol |
Johnson et al. 2009 [57] | - Shock Tube (T=1350-2000 K, P=101 kPa, Φ=0.5-2.0) [57]. | |
| Frassoldati et al. 2010 [58] |
- Shock tubes [57]*. - Counterflow diffusion flame (P= 101 kPa) [58]. - Flow reactors (P=101 kPa, T=1020-1120 K, Φ=0.61-1.18) [59]. - Premixed laminar flat flame (P=3.3-4.7 kPa, Φ=1.0-1.94) [60]. |
||
| Butanol | Moss et al. 2008 [27] (4 isomers of butanol) |
- Shock tube (T=1200-1800K, P=100-400 kPa, Φ=0.25-1.0) [27]. | |
| Dagaut et al. 2009 [61][62] (n-butanol) |
- Jet stirred reactor (P=101-1010 kPa, T=800-1250 K, Φ=0.25-2.0) [61][62]. - Counterflow diffusion flame (P=101 kPa) [61][62]. - Laminar burning velocities in constant volume bomb (P=90 kPa, Φ=0.8-1.2) [62][63]. |
||
| Black et al. 2010 [64] (n-butanol) |
- Jet stirred reactor [61]*. - Shock Tube (T=1100-1800 K, P=101-811 kPa, Φ=0.5-2) [64]. |
||
| Grana et al.2010 [65] (4 isomers of butanol) |
- Shock tube [27]*, [64]*; Flow reactors [59]*; Jet stirred reactor [61]*,[62]* - Counterflow diffusion flame [62]* and (P=101 kPa) [65]. - Shock tube pyrolysis (P=71-101 kPa, T=1200-1600 K) [66]. |
||
| Harper et al. 2011 [67] (n-butanol) |
- Shock tube [27]*, [64]*; Jet-stirred reactor [61]*, [62]*; Counterflow diffusion flame [62]*. - Shock tube (butanal:T=1200-1600 K, P=142-167 kPa, Φ=1.0-2.0) [68] - Butanol-doped methane diffusion flame (P=101 kPa) [69]. - Pyrolysis experiments in flow reactor (Tinl=673-749 K, Tmax= 923- 1080 K, P=172 kPa) [67]. |
||
| n-Pentanol | Togbé et al. 2011 [70] | - Jet-stirred reactor (P=1013 kPa, T=770-1220 K, Φ=0.35-4.0) [70]. - Laminar burning velocities in constant volume bomb (P=101 kPa, Φ=0.7-1.4) [70]. |
|
| n-Hexanol | Togbé et al. 2010 [71] | - Jet-stirred reactor (P=1013 kPa, T=560-1220 K, Φ=0.5-3.5) [71]. - Laminar burning velocities in constant volume bomb (P=100-1000 kPa, Φ=0.7-1.5) [71]. |
|
| Methyl esters | Dimethyl carbonate |
Glaude et al. 2005 [72] | - Counterflow diffusion flame (P=101 kPa) [73]. |
| Methyl butanoate |
Fisher et al. 2000 [74] | - Static reactor (P=40 kPa, T=541-741 K, Φ=3.25) [75]. | |
| Gaïl et al. 2007 [76], 2008 [77] |
- Jet-stirred reactor (T= 800-1400 K, P= 101 kPa, Φ=0.375-1.13) [76] [77]. - Flow reactor (T=500-900 K, P= 1266 kPa, Φ=0.35-1.5) [76]. - Counterflow diffusion flame (P= 101 kPa ) [76]. |
||
| Dooley et al. 2007 [78], 2008 [79]. |
- Jet-stirred reactor [76]*; Counterflow diffusion flame [76]*; Flow reactor [76]*. - Shock Tube (T= 1100-1760 K, P=101-405 kPa, Φ=0.25-1.5) [79], [78]. - Rapid compression machine (T= 640-949 K, P=1013-4053 kPa, Φ=0.33-1.0) [79]. |
||
| Hakka et al. 2010 [80] | - Shock Tube [78]* and (T=1280-1990K, P=770-922 kPa, Φ=0.25-2.0) [80]. - Rapid compression machine (T= 935–1117 K, P=1000 kPa, Φ=0.4) [81]. - Jet-stirred reactor (T= 800- 850 K, P= 101 kPa, Φ=0.5-1.0) [80]. |
||
| Methyl-2- butenoate |
Gaïl et al. 2008 [77] | - Jet-stirred reactor [77] - Counterflow diffusion flame (P=101 kPa) [77]. |
|
| Bennadji et al. 2011 [82] | - Shock Tube (T=1280-1930K, P=700-965 kPa, Φ=0.25-2.0) [82]. | ||
| Methyl hexanoate |
Dayma et al. 2008 [83] | - Jet-stirred reactor (T=500-1000 K, P=1013 kPa, Φ=0.5-1.5) [83]. | |
| Methyl heptanoate |
Dayma et al.2009 [85] | - Jet-stirred reactor (T=550-1150 K, P=1013 kPa, Φ=0.6-2.0) [85]. | |
| Glaude et al. 2010 [84] | - Jet-stirred reactor [85] * | ||
| Methyl Decanoate |
Herbinet et al. 2008 [86] | No direct validation. | |
| Glaude et al. 2010 [84] | - Jet-stirred reactor (T=500-1100 K, P=106 kPa, Φ=1.0) [84]. | ||
| Methyl Decenoates |
Herbinet at al. 2010 [87] | No direct validation. | |
| Heavier methyl esters |
Naik et al. 2011 [88] (Methyl Stearate, Methyl Oleate) |
No direct validation. | |
| Herbinet et al. 2011 [89] (Saturated C13, C15, C17, C19 ) |
- Jet-stirred reactor (P=106 kPa, T=550–1100 K, Φ=1.0) [90] (Methyl Palmitate/n-decane mixture). |
||
| Westbrook et al. 2010 [91][92]( Saturated C17 and C19 methyl esters, methyl oleate, methyl minoleate, methyl linolenate) |
- Jet-stirred reactor [90]* (Methyl Palmitate/n-decane mixture). - Jet-stirred reactor ((P=106 kPa, T=550–1100 K, Φ=1.0) [92] (Methyl Oleate/n-decane mixture). |
||
| Acyclic ethers | DME** | Dagaut et al. 1996 [93], 1998 [94] |
- Shock tube (T=650-1600 K, P=350-4000 kPa, Φ=0.5-2.0) [94]. - Jet-stirred reactor (P=101-1013 kPa, T=550-1275 K, Φ=0.2-2.5) [93] [94]. |
| Curran et al. 1998 [95], 2000 [96] |
- Jet-stirred reactor [93]*, [94]*. - Shock tube (T=650-1300 K, P=1317-4053 kPa, Φ=1.0) [97]. - Flow reactor (T=550-855 K, P=1216-1824 kPa, Φ=0.7-4.2) [96]. |
||
| Fischer et al. 2000 [98] | - Shock tube [94]*; Jet-stirred reactor [93]*. - Flow reactor pyrolysis (T=1060 K, P=253 kPa), near-pyrolysis (T=1118 K, P=101 kPa) and oxidation (T=1080-1086 K, P=101 kPa, Φ=0.3-3.4) [98] - Flow reactor (T=600-1500 K, P=101 kPa, Φ=0.5) [99]. |
||
| Zhao et al. 2008 [100] | - Shock tube [94]* [97]*; Flow reactor [96]*, [98]*, [99]*; Jet-stirred reactor [93]*, [94]*. - Shock tube (T=1241-1538 K, P=182-197 kPa, Φ=1.0-2.0) [100] - Flow reactor pyrolysis (T=980 K, P=1013 kPa) [100]. - Premixed laminar flat flame (P=4 kPa, Φ=0.98) [101] and (P=101 kPa, Φ=0.67) [102]. - Laminar flame speeds in spherical bomb (P=101-1013 kPa, Φ=0.6- 1.7) [103][104] and stagnation flame burner (P=101 kPa, Φ=0.7-1.4) [105]. |
||
| MTBE** | Brocard et al.1983 [106] | - Static reactor (T=573-773 K, P=13 kPa, Φ=7.5) [106]. | |
| Curran et al.1992 [107] | - Shock tube (T=1100-1900 K, P=355 kPa, Φ=0.15-2.4) [107]. | ||
| Goldaniga et al. 1998 [108] |
- Shock tube [107]*. - Jet-stirred reactor (P=1013 kPa, T=800-1150 K, Φ= 0.5-2.0) [108]. |
||
| Glaude et al. 2000 [109] | - Flow reactor [45]*; Static reactor [106]*. Jet-stirred reactor [108]*. | ||
| Yasunaga et al. 2010 [110] |
- Shock tube pyrolysis (T=900-1500 K, P=101-274 kPa) [110]. - Shock tubes oxidation (T=1400-1800 K, P=142-263 kPa) [110]. |
||
| ETBE** | Goldaniga et al. 1998 [108] |
- Jet-stirred reactor [108]*. | |
| Glaude et al. 2000 [109] | - Jet-stirred reactor [108]*. | ||
| Ogura et al. 2007 [111] | - Jet stirred reactor [108]*. | ||
| Yahyaoui et al. 2008 [112] |
- Shock tube (T=1280-1750 K, P=200-1000 kPa, Φ=0.25-1.5) [112]. - Laminar burning velocity in constant volume bomb (P=101 kPa, Φ=0.5-1.5) [112]. |
||
| Yasunaga et al. 2010 [110] |
- Shock tube pyrolysis [110]*. - Shock tubes oxidation [110]*. |
||
| Diethyl ether |
Yasunaga et al. 2010 [113] |
- Shock tube pyrolysis and oxidation (P=101-405 kPa, T=900-1900 K) [113] |
|
| Dimethoxy methane |
Daly et al. 2001 [114] | - Jet-stirred reactor (P=507 kPa, T=800-1200K, Φ= 0.444-1.778) [114]. |
|
| Dias et al. 2010 [115] | - Premixed laminar flat flame (Φ=0.24-1.72, P=5 kPa) [115]. | ||
| TAME** | Goldaniga et al. 1998 [108] |
- Jet-stirred reactor [108] | |
| DIPE** | Goldaniga et al. 1998 [108] |
- Jet-stirred reactor [108] | |
| Cyclic ethers | THF**, *** |
Dagaut et al. 1998 [116] | - Shock tube (P=203-507 kPa, T=1000-1800 K, Φ=0.5-2.0) [116]. - Jet-stirred reactor (P=101-1013 kPa, T=800-1100 K, Φ=0.5-1.0) [116]. |
| Furan*** | Tian et al. 2011 [117] | - Premixed laminar flat flame (P=4.7 kPa, Φ=1.4-2.2) [117]. - Shock tube pyrolysis (T=1100-1700 K, P=2017 kPa) [[118] (T=1533 K, P=26 kPa) [119]. |
|
Conditions presented above. Φ- equivalence ratio; T- temperature; P- pressure.
See full names and formulae in Table 1.
Not directly considered as a biofuel, but from the same family as potential biofuels.
All the experimental results used for model validation have been obtained for pressures below 10 bar, the major part being measured close to atmospheric pressure, and for equivalence ratios from 0.2 to 4, the major part being measured close to stoichiometry. More experimental studies are certainly needed in order to enable model validations under a wider range of conditions, especially under pressures closer to those usually observed in engines.
The family of molecules for which the largest number of models has been proposed is alcohols. Note that the major part of these studies has been recently performed. The experimental investigations related to alcohols, apart from one for n-hexanol [71], have all been performed at temperature above 770 K. This is mostly due to a lack of reactivity of these compounds at lower temperatures. However in a mixture with more reactive species, such as some of those included in gasoline (e.g. linear alkanes), alcohols could certainly react at lower temperature by reactions which are not considered in the present models. The two alcohols, the behaviors of which have been the more investigated, are ethanol and butanols (mainly n-butanol). Concerning n-butanol, the best validated model is the recent one by Harper et al. [67], but improvements are still needed for modeling some experimental data such as those obtained in a jet-stirred reactor [61].
A fair number of models have recently been published about the combustion of methyl esters. A model has also been proposed for the oxidation of dimethylcarbonate [72]. The most studied species is methyl butanoate, even if its small size prevents it to have a chemistry representative of the large molecules present in biodiesel [80]. However even for this small ester, zones of shadow remain to fully explain its lack of low-temperature reactivity [80]. Most work deals with saturated esters, with very few studies dedicated to unsaturated ones, even if unsaturated esters are the most abundant esters in biodiesel. Note that very few experimental studies can be used to test the predictions of the models proposed for heavy esters representative of those included in biodiesel. This is true to such an extent that several models have been proposed without possibility to be validated at the time of their development. The experimental investigations related to methyl esters have all been performed at temperatures from 550 K. The evolution with temperature of the reactivity of methyl esters from methyl hexanoate exhibits a marked negative temperature coefficient behavior. As illustrated in figure 2 in the case of methyl decanoate, this behavior, well known for alkanes [120], consists in a temperature zone where the reactivity decreases with temperature. As shown in figure 2a, this behavior is well predicted by the current models. These models can also reproduce the formation of products specific to esters, e.g. cyclic ethers with an ester function or unsaturated esters, such as heptenoate as shown in figure 2b. Note that, mainly due to the availability of experimental results, no validation of model of the combustion of esters including more than 5 atoms of carbons has been published for flame or ignition (in shock tube or rapid compression machine) conditions. While many models have been proposed for methyl esters, only a very few studies concern ethyl esters [80][82].
Figure 2.

Simulated (lines) and experimental (points) evolution with temperature of the mole fractions of (a) reactant and (b) methyl-6-heptanoate. Model generated using EXGAS software; experimental mole fraction measured by on-line gas chromatography during the oxidation in a jet-stirred reactor of a stoichiometric mixture of methyl decanoate/O2 diluted in helium at atmospheric pressure [84].
A large number of models have also been published about the combustion of acyclic ethers, but a large part of this work has been made before 2000. This is due to the fact that these compounds have been considered as octane boosters well before the need for using biofuels has been seriously considered [106]. The first ether which has been proposed as anti-knock fuel additive is MTBE (its use has been recently reduced due to environmental problems [121]), followed soon by ETBE. This explains why a significant number of models have been written for the oxidation of these two ethers. While acyclic ethers can also exhibit a low-temperature reactivity [106], most of the proposed models have been validated using experimental results obtained for temperatures above 800 K.
Only two models have been proposed for cyclic ethers, one for tetrahydrofuran by Dagaut et al. [116], and a very recent one by Tian et al. [117] for furan.
Note that some recent models can be very large: the model of Harper et al. [67] for the oxidation of n-butanol consists in 263 species and 3381 reactions and that of Herbinet et al. [89] for the oxidation of methyl palmytate includes 30425 reactions for 4442 species.
IV. SPECIFIC REACTION CLASSES CONSIDERED FOR MODELING THE OXIDATION OF ACYCLIC OXYGENATED COMPONENTS OF BIOFUELS
The kinetic study of the oxidation of acyclic oxygenated compounds is advanced enough for a system of automatic generation of mechanism (EXGAS) [122] to be able to produce validated detailed kinetic models for the oxidation of the three types of acyclic saturated molecules present in biofuels: ethers [109], alcohols [27] and methyl esters (EXGAS-ALKANES-ESTERS1) [84]. The used rules of generation are based on the chemical conclusions which can be derived from the analysis of the mechanisms listed in Table 3.
The aim of this part is to present the specific classes of reactions and kinetic parameters which are considered by this system in the case of these oxygenated molecules. While EXGAS can also generate mechanism for unsaturated reactants (e.g. for unsaturated esters [82]), the reactions of unsaturated molecules will not be presented here.
As shown in figure 3, the presence of the oxygen atoms in an organic molecule involves first a difference in the bond dissociation energies (BDEs) compared to what is observed in hydrocarbon molecules. In the EXGAS software, the thermochemical data are computed using THERGAS [125] based on the group additivity rules developed by Benson [126] and on BDEs mostly proposed by Tsang [127] and Luo [128]. In the case of esters, the BDEs of the C-H, C-O and C-C bonds close to the ester function were updated from the values proposed by El-Nahas et al. [129].
Figure 3.

Examples of bond dissociation energies (in kcal/mol) in ester, ether, and alcohol molecules. R is an alkyl group larger than a methyl one.
Before describing more closely the involved reaction classes, let us recall that EXGAS [122] provides reaction mechanisms made of three parts:
- * A comprehensive primary mechanism including all the reactions of the molecular reactants, the initial organic compounds and oxygen, and of the derived free radicals. The primary mechanism includes:
- – Molecular reactions which will be described further in the text,
-
– Unimolecular and bimolecular initiation steps(e.g. C3H7OH⇒•CH2OH + •C2H5; C3H7OH + O2⇒•CH2CH2CH2OH + •OOH),
- – Reactions leading to alkenes from alkyl radicals
-
Decompositions by beta-scission(e.g. •CH2CH2CH2OH⇒•CH2OH + C2H4),
-
Oxidations to form the conjugated alkene and •OOH(e.g. •CH2CH2CH2OH + O2⇒CH2=CHCH2OH + •OOH),
-
-
– Additions of alkyl and hydroperoxyalkyl radicals on a molecule of oxygen,(e.g. •CH2CH2CH2OH + O2⇒•OOCH2CH2CH2OH;HOOCH2CH2CHOH• + O2⇒HOOCH2CH2CHOHOO•),
-
– Intramolecular isomerizations of alkyl and peroxy radicals involving a cyclic transition state,(e.g. •OOCH2CH2CH2OH⇒HOOCH2CH2CHOH•):
-
– Decompositions of hydroperoxyalkyl and di-hydroperoxyal alkylradicals to form cyclic ethers, alkenes, aldehydes or ketones (oxohydroperoxyalkanes),(e.g. HOOCH2CH2CHOH•⇒cyclic-HOC3H5O + •OH),(e.g. HOOCH2CH2COH(OOH)•⇒HOOCH2CH2C(=O)OH + •OH),
-
– Metathesis reactions to abstract an H atom from the initial reactant(e.g. •OH + C3H7OH⇒H2O + •CH2CH2CH2OH),
- – Termination steps:
-
Combinations of two free radicals(e.g. CH3• + OH•⇒CH3OH),
-
Disproportionations of peroxy radicals with •OOH(e.g. •OOCH2CH2CH2OH + •OOH⇒HOOCH2CH2CH2OH + O2),
-
* A C0-C2 reaction base (core mechanism), including all the reactions involving radicals or molecules containing less than three carbon atoms; but including oxygenated species. The fact that no generic rule can be derived for the generation of the reactions involving very small compounds makes the use of this reaction base necessary. This core mechanism is common to all the generated mechanisms, whatever the reactant.
* A lumped secondary mechanism, containing the reactions consuming the molecular products of the primary mechanism which do not react in the reaction base [130].
For the shortness of this paper, we will not detail the reaction classes involved in the secondary mechanism. Note however that these reactions have to be written for consuming the compounds formed by the primary mechanisms: e.g. unsaturated esters, alcohols or ethers and hydroperoxides, cyclic ethers, ketones and aldehydes bearing an additional ester, alcohol or ether function. They are considered in details by EXGAS.
The only reaction class specific to the primary mechanism of oxygenated molecules is molecular reactions, such dehydrations in the case of alcohols, or alcohols eliminations in the case of ethers. For several others types of reactions (unimolecular initiations, bimolecular initiations and H-abstractions, decompositions of radicals by β-scission, intramolecular isomerizations), only changes in the kinetic parameters have to be taken into account. For the remaining types of reactions, no change was needed.
VI.1. Molecular reactions
Figure 4 presents typical molecular pericyclic reactions in the case of ethers, alcohols and esters. In the case of ethers, a molecular reaction involving the transfer of a H-atom bound to an atom of carbon in β position of the atom of oxygen to give an alcohol molecule and an alkyl radical via a favored four centered cyclic transition state (see first reaction in figure 4) should be taken into account as proposed by Choo et al. [131]. The activation energies of the rate constants used in EXGAS for this reaction class are 52 kcal/mol for ethyl tert-butyl ether and 59 kcal/mol for methyl tert-butyl ether [123].
Figure 4.

Examples of molecular reactions which can be observed in ethers, alcohols and ethyl esters.
Intramolecular dehydration leading to the formation of water and an alkene molecule via a favored four centered cyclic transition state (see figure 4) is an important reaction class for alcohols. Note that if we consider isomers of butanol, there is one possible intramolecular dehydration reaction for 1-butanol (see second reaction in figure 4), iso-butanol and tert-butanol, and two for 2-butanol. Few kinetic parameters have been published concerning these reactions (e.g. [132][133], with activation energy from 62 to 69 kcal/mol) and disagreements can be observed between the proposed rate constants, which have been shown to be sensitive parameters in detailed kinetic modeling [27]. Molecular dehydrogenations of alcohols leading to aldehydes have been recently proposed [65] but are not yet considered in EXGAS.
In the case of esters, El-Nahas et al. [129] have theoretically investigated several types of molecular reactions, but no easy decomposition appeared in the case of methyl esters. The only one having an impact on detailed kinetic modeling is the formation of ethylene and acid from ethyl esters via a favored six-centered cyclic transition state (see third reaction in figure 4). The activation energy of the rate constant used in EXGAS for this reaction class is 47.3 kcal/mol [80].
IV.2. Unimolecular initiations
The rate constants for unimolecular initiations (decompositions) given by EXGAS are obtained from the data on the reverse reactions, i.e. combinations, using thermochemical data calculated with THERGAS software [125]. Activation energies for every combination are taken to be zero. The pre-exponential factor for the combination of two free radicals is estimated from modified collision theory [134].
In the case of ethers and alcohols, the activation energies of unimolecular initiations were also calculated using THERGAS [125]. In the case of esters, the activation energies of unimolecular initiations, which involve the breaking of the C-C bonds located in α- and β-positions from the ester function, have been updated from the BDEs (see figure 3) recently calculated by El-Nahas et al. [129]. For the other unimolecular initiations, the bond energies used in THERGAS [125] were in agreement to about ±1 kcal.mol−1 with those proposed by El-Nahas et al. [129] and were not modified.
IV.3. Bimolecular initiations and H-abstractions
Bimolecular initiations and H-abstractions are usually the most important for the fuel consumption. In the case of hydrocarbons, the rate constants of these reactions depend only of the type of alkyl H-atoms which can be abstracted: primary -CH3, secondary -CH2 or tertiary -CH. The abstraction of a primary H-atom is the most difficult, that of a tertiary one is the easiest. In oxygenated reactants, more types of H-atoms have to be considered. As shown in figure 3, in all oxygenated compounds, there are C-H bonds close to the oxygen atom which are more easily breakable. This involves easiest bimolecular initiations and H-abstractions.
In the case of ethers, the activation energy used by EXGAS for the bimolecular initiations involving the abstraction of an H-atom from a carbon atom in the α-position of the oxygen atom has been reduced by a factor about 4 kcal/mol compared to the case of alkanes [122] and no change was considered for H-abstractions.
In the case of alcohols, bimolecular initiations and H-abstractions can involve the abstraction of alkylic or alcoholic (-OH) H-atoms. In EXGAS, the rate constants for bimolecular initiations were estimated using the correlation proposed by Ingham et al. [135] for hydrocarbons (the activation energy is taken equal to the reaction enthalpy) and the rate constants for the abstractions were generally those used for the alkanes [122]. However the rate constants for the abstraction of a H-atom from a carbon atom located in the α-position of the alcohol function, the fastest of the H-atom abstraction reactions, have been evaluated using an Evans–Polanyi type correlation from Dean and Bozzelli [136], developed for the abstraction of H-atoms from hydrocarbons:
where nH is the number of abstractable H-atoms; A, n, and E0 are the rate parameters for the case of a metathesis by the considered radical from ethane; ΔH0 is the enthalpy of the metathesis by the considered radical from ethane; ΔH is the enthalpy of the metathesis by the considered radical from the reacting molecule; f is a correlation factor, the values of which are given by Dean and Bozzelli [136] for each considered radical; and R is the gas constant. While, according to Luo [128], the BDE of the O-H bond for the alcohol function is between 102 and 106 kcal.mol−1, a value equal to that of a C-H bond in the case of an alkylic primary H-atom, the kinetic parameters for an H-atom abstraction from an alcohol function used by EXGAS are equal to those for the abstraction of a tertiary alkylic H-atom. That has been shown to be closer to experimental values for H-atom abstractions.
The BDE of the C-H bonds carried by the carbon atom just neighboring an ester function (94.3 kcal/mol [129]) is close to that of a tertiary atom of carbon (e.g., 95.7 kcal/mol in isobutene [128]). Thus, for the abstraction of this type of H-atoms, the correlations were taken by EXGAS the same as those used for the abstraction of tertiary H-atoms from alkanes, except in the case of the abstractions by •O• and •H atoms and by •OH, •HO2 and •CH3 radicals. For these last atoms and radicals, the rate parameters have been evaluated using the Evans–Polanyi type correlation proposed by Dean and Bozzelli [136] previously described.
IV.4. Decompositions of radicals by β-scission
Table 4 shows a summary of the activation energies which are used in EXGAS for the decompositions by β-scission involving the breaking of a C-C or a C-O bond in radicals deriving from saturated oxygenated molecules. At temperature above 800 K, decompositions by β-scission are the major way of consumption of radicals obtained from the fuel molecules by H-abstractions. While the data related to ethers, which have been proposed in 2000 [109], were based on rough estimations, the data related to esters, proposed in 2010 [84], have been obtained from quantum calculations. These quantum calculations were performed for model reactions at the CBS-QB3 level of theory using Gaussian03 [137]. Intrinsic Reaction Coordinate (IRC) calculations have been systematically performed at the B3LYP/6-31G(d) level of theory on transition states, to ensure that they are correctly connected to the desired reactants and products.
Table 4.
Activation energies used for the decompositions by β-scission of oxygenated radicals.
| Reaction class | Ea (kcal/mol) |
|---|---|
|
|
5.1a |
|
|
15.0 b |
|
|
24.0 b |
|
|
29.0 c |
|
|
26.0 d |
|
|
30.7 e |
|
|
31.9a |
|
|
34.9e |
|
|
39.9 f |
|
|
15.6 g |
|
|
49.0 e |
Notes: CBS-QB3 [137] calculation performed for R•= CH3•,
Estimated based on the kinetics of the reverse step of the addition of alkyl radical on a ketone, i.e. 10 kcal/mol according to Benson [126],
from Wilk et al. [138],
Estimated based on the kinetics of the reverse step of the for the addition of an alkoxy radical on an alkene, according to Choo et al. [131], R5 can be an H-atom.
Theoretical calculation performed for R•= H•,
Theoretical calculation performed for R•= C2H5•,
from Mereau et al. [139].
If we considered that the activation energy of the breaking of a C-C bond in saturated hydrocarbons is usually between 26 and 31 kcal/mol [122], table 4 illustrates well that the presence of oxygen atoms in the involved radicals can have a significant impact on the values of the activation energies of the this reaction class.
IV.5. Intramolecular isomerizations
Intramolecular isomerizations involving the transfer of a H-atom through a cyclic transition state are important reactions of alkyl (R•) and peroxyalkyl (RO2•) radicals during the oxidation of hydrocarbons. The rate constants of the isomerizations of peroxyalkyl radicals have been shown to be particularly sensitive parameters during the low-temperature oxidation of alkanes [122]. These reactions need also to be considered in the case of oxygenated reactants. In EXGAS, the activation energy for an isomerization is set equal to the sum of two contributions: the activation energy for H-abstraction from the molecule by analogous radicals and the strain energy involved in the cyclic transition state [122]. For modeling the low-temperature oxidation of ethyl tert-butyl ether and methyl tert-butyl ether [123], the same values for these contributions have been taken as for alkanes. EXGAS has never been used to generate mechanism for the low-temperature oxidation of alcohols.
In the case of esters, some values had to be changed to take into account the influence of the ester group. The activation energy for the internal H-abstraction in α-position from the ester function has been taken equal to that for the abstraction of a tertiary H-atom in the case of alkanes (i.e., 9 kcal.mol−1 for a tertiary H-atom in an alkyl radical and 14 kcal.mol−1 for a tertiary H-atom in peroxy-alkyl radicals [122]). The ring strain energy involved in the transition state of some reactions of isomerization, transferring a H-atom above the ester function, had also to be re-estimated. These isomerizations concern ester alkyl radicals and ester alkyl-peroxy radicals. The ring strain energies of the transition states involved in the reactions of isomerization of ester alkyl radicals were deduced from the enthalpy of formation of the corresponding lactones measured by Wiberg and Waldron [140]. Ring strain energies are (in kcal.mol−1) equal to 9, 11, 11.2, and 12.5 (in kcal.mol−1) for 5, 6, 7, and 8 membered ring lactones, respectively. These values can be compared to the lower ones used for alkyl radicals: 6.3, 1, 6.4, and 9.9 for 5, 6, 7, and 8 membered rings, respectively.
Some isomerizations of peroxyalkyl radicals, shifting a H-atom above the ester function, involve cyclic transition states which contain three oxygen atoms. This kind of isomerizations occurs through seven and eight membered rings as shown in Figure 5. Two configurations are possible according to the position of the peroxy group: either the peroxy group is on the alkyl chain and the shifted H-atom is on the methyl group of the ester function, or the peroxy group in on the methyl part of the ester function and the shifted H-atom in on a carbon atom from the alkyl chain. The strain energies have been considered as equal to the enthalpies of reaction of the isodesmic reactions yielding the cyclic structure from a linear unstrained one (i.e. reactants and products can be decomposed using the same groups according to the group additivity method of Benson [126]) in the case of seven membered ring species [124]. These enthalpies of reaction have been calculated by quantum methods at the CBS-QB3 level of theory using Gaussian03 [137]. Note that the presence of the third oxygen atom in the ring together with the carbonyl group significantly increases the ring strain energy: the calculated strain energies in figure 5 range from 7.7 to 13.6 kcal/mol, while the strain energy for two oxygen cyclic transition states are taken as equal 5 kcal/mol for 7 membered rings and to 4 kcal/mol for 8 membered ring. That involves that the isomerizations of peroxyalkyl radicals is more difficult in the case of esters than for alkanes.
Figure 5.

Reactions of intramolecular isomerization involving transition states including seven and eight membered rings, which contain three oxygen atoms and related to ring strain energies (Erse) in kcal/mol.
V. SPECIFIC REACTIVITY OF OXYGENATED CYCLIC COMPONENTS OF BIOFUELS
Compared to acyclic oxygenated compounds, the kinetic study of the oxidation of cyclic oxygenated compounds is really at its starting point.
V.1. Saturated cyclic ethers
Reaction classes for the development of detailed chemical kinetic models of saturated cyclic ethers, such as tetrahydrofuran derivatives, are similar to those of cyclic alkanes. If reaction classes are identical, rate parameters can be dramatically different because of the presence of oxygen atom in the molecular structure. Recent years have seen the emergence of a large number of detailed chemical kinetic models for the oxidation of cyclic alkanes [141][144]. It was shown that these compounds are characterized by specific reaction classes. Kinetic models for saturated cyclic ethers can be developed using the same reaction rules. These specific reaction classes of cyclic saturated molecular systems can be considered:
Unimolecular initiations of cyclic structures lead to biradical species that further isomerize by internal H-transfer to form an unsaturated compound. In the case of cyclic alkanes, with or without a lateral alkyl chain, the initiation step that opens the ring always leads to the formation of the corresponding 1-alkene. Based on electronic structure calculations, it was shown that activation energies for these processes can be estimated by subtracting the ring strain energy of the cyclic structure to the C-C bond dissociation energy of a corresponding linear alkane [145]. The presence of oxygen in the ring increases the complexity of cyclic ether unimolecular initiations. Two types of biradicals can be obtained as the ring opens: a carbon-carbon biradical (CH2•-(CH2)n-O-(CH2)n-CH2•) or a carbon-oxygen biradical (O•-(CH2)n-CH2•), where n is characteristic of the size of the chain and each H atom could be replaced with an alkyl group in the case of cyclic ethers with a lateral alkyl chain. According to differences in bond dissociation energies for acyclic ethers (figure 3), one can assume that C-O bond fission will be the most favored initiation pathway compared to the CC bond ring opening. In that case, two molecules could be obtained by an internal hydrogen atom transfer from the biradical (O•-(-CHn)-CH2•): an alkene-alcohol and an aldehyde or a ketone. A proposed biradical initiation mechanism for 2-methyltetrahydrofuran is presented in figure 6. Rate coefficients and branching ratios of the products remain unknown in the literature for these processes.
The role of a lateral alkyl group becomes critical when the β-scission reaction class of cyclic ether radicals is considered. In the case of cyclic alkyl radicals, it was shown that the presence of a lateral alkyl group allows two types of ring opening reactions: “exo” and “endo” ring openings [146]. If the cyclic structure is free of any lateral alkyl group, then any radical created from the parent compound can only undergo endo ring openings, i.e. the nascent double bond is created inside the cyclic transition state. However, a lateral alkyl group leads to the formation of radicals where the unpaired electron is located on the alkyl group itself, allowing the double bond to be formed outside the cyclic part of the transition state, in an exo ring opening reaction. Figure 6 illustrates an endo and exo ring opening in the case of 2-methyltetrahydrofuran. The impact of the location of the nascent π bond in the transition state (inside or outside the cyclic part of the transition state) on the rate parameters is considerable. In the case of exo ring openings of cyclic alkyl radicals, it was shown that rate parameters can be estimated using an Evans-Polanyi correlation that also describes the C-C bond β-scission of acyclic alkyl radicals. However, endo ring opening reactions exhibited a considerably different behavior characterized by higher activation energies. These activation barriers can be so high that C-H bond β-scissions can be competitive. This was observed, for example, in the case of cyclopentyl radical thermal decomposition. Almost, no information on β-scissions of cyclic ether radicals is available in the literature and although these radicals are expected to behave like cyclic alkyl radicals, the associated rate parameters are largely uncertain.
Isomerization of peroxy radicals formed by the addition of a cyclic radical to O2 is of course not a reaction class specific to cyclic compounds but it leads to specific elementary reactions that involves bicyclic transition states (figure 6). If ab initio calculations are available in the literature for the rate constants of cyclic alkylperoxy radicals [147], rate constants for these processes remain unknown in the case of saturated cyclic ethers.
Figure 6.

Examples of reaction classes (unimolecular initiation, endo and exo β-scissions, and isomerization of a peroxy radical) specific to saturated cyclic ethers.
V.2.Unsaturated cyclic ethers
Reaction classes of unsaturated cyclic ethers such as furan derivatives involve specific processes that are different from saturated cyclic ethers. The presence of a lateral alkyl group on the furan ring has also a considerable impact on the reactivity of these compounds. Simmie and Curran [148] recently pointed out that C-X (with X = H or C) bonds of alkylfurans are among the highest bond dissociation energy known. In alkylfurans, the bond dissociation energy of C-H bonds involving carbon atoms belonging to the ring is as high as 119.6 kcal.mol−1 (to be compared with the values presented in Figure 1 for acyclic species). Such high energies tend to favor unusual pathways for unimolecular initiations involving the formation of carbenes. Similarly, rate parameters for H-abstractions are expected to be affected and decomposition pathways by bond β-scission may be more difficult because of such bond strengths. Indeed, the lowest energy decomposition pathways of furyl derivatives radicals involve hydrogen transfer through strained cyclic transition states and/or successive cyclization-isomerization-ring opening sequences. In more details, the following reaction class specificities can be noted:
Most favored unimolecular initiation mechanisms occur through the formation of carbene intermediates rather than biradical ring opening. Sendt et al. [149] showed that the singlet biradical formed from the ring opening of furan was lying 88.8 kcal.mol−1 above furan (at 0 K) while the energy barrier for hydrogen transfer through a 3 membered ring transition state leading to a carbene was calculated to be 68.9 kcal.mol−1 at several levels of theory (CASPT2, G2MP2). Recent CBS-QB3 calculations from Tian et al. [117] confirmed this 1-2 H-transfer pathway to be favored with a computed barrier of 70.4 kcal.mol−1. As shown in figure 7, the carbene intermediate easily rearranges to formylallene, which in turn decomposes to give CO and propyne through a concerted mechanism involving a hydrogen shift and a C-C bond breaking. It appears that this decomposition pathway is favored because of the ability of the molecule formed from the carbene (formylallene) to easily transfer the aldehydic hydrogen atom. Consequently, this kind of pathway will be largely hindered if a ketone is formed from the carbene intermediate, i.e. a methyl group is bounded to the CO group instead of a H-atom. The presence of a methyl group on the furan ring will therefore have a critical impact on the unimolecular initiation mechanism. Moreover, if a methyl group is bounded to the furan ring one might wonder if a 1-2 CH3 group transfer, leading to a carbene intermediate, can be competitive with the 1-2 H-atom transfer pathway. Calculations performed at the CBS-QB3 level of theory predict a barrier of 81.1 kcal.mol−1 for 1-2 CH3-group transfer in 2,5-dimethylfuran (2,5-DMF). This pathway lies 12 kcal.mol−1 above the energy barrier for the competitive 1-2 H-transfer in 2,5-DMF determined at the same level of theory (see figure 7). It also can be seen that, as already mentioned above, even if 1-2 H-atom transfer is the most favored pathway here, the ketone formed from the carbene intermediate is not able to easily decompose via a concerted reaction because of the methyl group linked to the CO group. Rate parameters for furan unimolecular initiations were proposed by Sendt et al. [149], but no kinetic data is available for methyl- or dimethyl-furan and the presence of methyl groups prevents straightforward analogies with furan kinetic data.
C-O bond β-scissions of furyl radicals were studied by Tian et al. [117] at the CBS-QB3 level of theory. They showed that relatively high energy barriers (above 33 kcal.mol−1) are involved to decompose furyl radicals by β-scissions. Again, the presence of one or two methyl groups on the furan ring is expected to considerably influence the thermal decomposition pathways of methylfuryl radicals. By analogy with saturated cyclic compounds, one could expect that the presence of a lateral methyl group on the furan ring leads to radicals that can undergo exo ring opening with a lower activation energy than that of the endo ring opening of furyl radicals. However a calculation at the CBS-QB3 level of theory predicts the energy barrier to be 8.4 kcal.mol−1 higher than the corresponding endo ring opening (figure 7). Rate constants for these processes remain unknown and the ab initio calculations of Tian et al. [117] on furyl radicals are the only kinetic data available in the literature.
As furan and furan derivatives are unsaturated compounds, the description of addition reactions of small radicals (notably H atoms and OH radicals) to the double bonds is critical to accurately predict the branching ratios of pyrolysis and/or combustion products. It should be emphasized that almost no information is available in the literature for these types of reaction. Tian et al. [117] studied at the CBS-QB3 level of theory the addition of H atom on furan and the decomposition channels of the adduct formed. These data constitute the only kinetic parameters available in the literature. As emphasized above, analogies with results from furan cannot be applied straightforwardly for methyl- and dimethyl-furans and the fate of the adducts formed by H additions remains unknown.
Low temperature oxidation mechanism of furan and its derivatives is yet unknown.
Figure 7.

Specific reactions of unsaturated cyclic ethers: unimolecular initiation mechanisms of furan and dimethyl furan and comparison between endo and exo β-scissions for furan and dimethyl furan. (1) Energies at 0 K from ab initio calculations of Sendt et al. [149]. (2) Enthalpies at 298 K computed at the CBS-QB3 level of theory (present work). (3) Enthalpies at 298 K from ab initio calculations of Tian et al. [117].
VI. CONCLUSION
With the increasing interest to shift from petroleum-based fuels to bio-fuels, many types of oxygenated molecules which could be used as additive to gasoline or diesel fuel have been proposed: alcohols, such as methanol, ethanol, propanols, or butanols, fatty acid methyl esters, acyclic ethers and cyclic ethers of the family of furans. After a first part summarizing the detailed kinetic models existing for the combustion of these molecules, this paper describes the reactions and rates constants which are specific to oxygenated fuels. While a fair number of detailed kinetic models have already been proposed for the combustion and the oxidation of alcohols, methyl esters and acyclic ethers, there are only a very few ones concerning furans and its derivatives. This paper summarizes also the main specificities taken into account in elementary steps and in their kinetic parameters in the case of oxygenated reactants. While the mechanism of the oxidation of saturated cyclic ethers can certainly be close to that of cyclic alkanes, the reactions of unsaturated cyclic ethers have been shown to be very complex and are still very uncertain.
ACKNOWLEDGEMENTS
This work was funded by the European Commission through the “Clean ICE” Advanced Research Grant of the European Research Council.
Footnotes
Software EXGAS-ALKANES-ESTERS automatically generates detailed kinetic mechanisms for the oxidation of linear and branched alkanes, and linear methyl esters and is freely available for academic researchers (valérie.warth@ensic.inpl-nancy.fr).
REFERENCES
- [1].Westbrook CK, Pitz WJ, Curran HJ. Chemical kinetic modeling of the effects of oxygenated hydrocarbons on soot emissions from diesel engines. J Phys Chem A. 2006;110:6912–22. doi: 10.1021/jp056362g. [DOI] [PubMed] [Google Scholar]
- [2].Jacobson MZ. Effect of ethanol (E85) versus gasoline vehicles on cancer mortality in the United States. Environ Sci Technol. 2007;41:4150–57. doi: 10.1021/es062085v. [DOI] [PubMed] [Google Scholar]
- [3].Bünger J, Krahl J, Baum K, Schröder O, Müller M, Westphal G, et al. Cytotoxic and mutagenic effects, particle size and concentration analysis of diesel engine emissions using biodiesel and petro diesel as fuel. Arch Toxicol. 2000;74:490–98. doi: 10.1007/s002040000155. [DOI] [PubMed] [Google Scholar]
- [4].Penner SS, Berlad AL. Fundamental combustion research in support of industrial applications. Energy. 1995;20:311–24. [Google Scholar]
- [5].Schmidt LD, Dauenhauer PJ. Hybrid routes to biofuels. Nature. 2007;447:914–15. doi: 10.1038/447914a. [DOI] [PubMed] [Google Scholar]
- [6].Huber GW, Iborra S, Corma A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem Rev. 2006;106:4044–98. doi: 10.1021/cr068360d. [DOI] [PubMed] [Google Scholar]
- [7].Xu C, Donald J, Byambajav E, Ohtsuka Y. Recent advances in catalysts for hot-gas removal of tar and NH3 from biomass gasification. Fuel. 2010;89:1784–95. [Google Scholar]
- [8].Von Schaub G, Unruh D, Rohde M. Synfuels from biomass via fisher-Tropsch-synthesis - Basic process principles and perspektives. Erdoel Erdgas Kohle. 2004;120:327–31. [Google Scholar]
- [9].Baker E, Keisler JM. Cellulosic biofuels: Expert views on prospects for advancement. Energy. 2011;36:595–605. [Google Scholar]
- [10].Zhu L, Du L, Li X, Li G, Zhang J. Process conditions for preparing methanol from cornstalk gas. J Envir Sci. 2007;19:628–32. doi: 10.1016/s1001-0742(07)60104-1. [DOI] [PubMed] [Google Scholar]
- [11].Agarwal AK. Biofuels (alcohols and biodiesel) applications as fuels for internal combustion engines. Prog Energ Combust Sci. 2007;33:233–71. [Google Scholar]
- [12].Dürre P. Biobutanol: an attractive biofuel. Biotechnol J. 2007;2:1525–34. doi: 10.1002/biot.200700168. [DOI] [PubMed] [Google Scholar]
- [13].He J, Zhang W. Techno-economic evaluation of thermo-chemical biomass-to-ethanol. Applied Energy. 2011;88:1224–32. [Google Scholar]
- [14].Qureshi N, Saha BC, Hector RE, Dien B, Hughes S, Liu S, et al. Production of butanol (a biofuel) from agricultural residues: Part I-Use of barley straw hydrolysates. Biomass and Bioenergy. 2010;34:559–65. [Google Scholar]
- [15].Singh A, Nigam PS, Murphy JD. Renewable fuels from algae: An answer to debatable land based fuels. Bioresource Technology. 2011;102:10–6. doi: 10.1016/j.biortech.2010.06.032. [DOI] [PubMed] [Google Scholar]
- [16].Demirbas A. Biodiesel production from vegetable oils via catalytic and non-catalytic supercritical methanol transesterification methods. Prog Energ Combust Sci. 2005;31:466–87. [Google Scholar]
- [17].Arcoumanis C, Bae C, Crookes R, Kinoshita E. The potential of di-methyl ether (DME) as an alternative fuel for compression-ignition engines: A review. Fuel. 2008;87:1014–30. [Google Scholar]
- [18].Cataluña R, da Silva R, de Menezes E, Ivanov R. Specific consumption of liquid biofuels in gasoline fuelled engines. Fuel. 2008;87:3362–68. [Google Scholar]
- [19].Román-Leshkov Y, Barrett C, Liu Z, Dumesic J. Production of dimethylfuran for liquid fuels from biomass-derived carbohydrates. Nature. 2007;447:982–85. doi: 10.1038/nature05923. [DOI] [PubMed] [Google Scholar]
- [20].Yang W, Sen A. One-step catalytic transformation of carbonhydrates and cellulosic biomass to 2,5-dimethyltetrahydrofuran for liquid fuels. ChemSusChem. 2010;3:597–603. doi: 10.1002/cssc.200900285. [DOI] [PubMed] [Google Scholar]
- [21].Natarajan K, Bhaskaran KA. An Experimental and Analytical Investigation of High Temperature Ignition of Ethanol. Proceedings of the International Symposium on Shock Tubes and Waves (13th); Niagara Falls. July 6-9, 1981. [Google Scholar]
- [22].Ballerini D. Les biocarburants: état des lieux, perspectives et enjeux du développement. Paris: 2006. Edition TECHNIP. [Google Scholar]
- [23].Guibet JC. Carburants et Moteurs. Paris: 1997. Tome 1. Edition TECHNIP. [Google Scholar]
- [24].Lu X, Ji L, Ma J, Huang Z. Improved NOx and smoke emission characteristics of a biodiesel-fueled engine with the port fuel injection of various premixed fuels. Energy Fuels. 2008;22:3798–805. [Google Scholar]
- [25].Yacoub Y, Bata R, Gautam M, Martin D. The performance characteristics of C1-C5 alcohol-gasoline blends with matched oxygen content in a single cylinder SI engine. ACS Division of Fuel Chemistry. 1997;42:723–7. preprints. [Google Scholar]
- [26].Gautam M, Martin DW, II, Carder D. Emissions characteristics of higher alcohol/gasoline blends. Proceedings of the Institution of Mechanical Engineers, Part A: Journal of Power and Energy. 2000;214:165–82. [Google Scholar]
- [27].Moss JT, Berkowitz AM, Oehlschlaeger MA, Biet J, Warth V, Glaude PA, et al. An experimental and kinetic modeling study of the oxidation of the four isomers of butanol. J Phys Chem A. 2008;112:10843–55. doi: 10.1021/jp806464p. [DOI] [PubMed] [Google Scholar]
- [28].Lide DR. Handbook of chemistry and physics. 70th Edition 1989-1990. [Google Scholar]
- [29].≪ 1-pentanol ≫, fiche de sécurité du Programme International sur la Sécurité des Substances Chimiques. http://www.cdc.gov/niosh/ipcsnfrn/nfrn0535.html.
- [30].Bailey B, Eberhardt J, Goguen S, Erwin J. Diethyl ether (DEE) as a renewable diesel fuel. 1997. SAE Technical Paper 972978.
- [31].GESTIS Substance Database of Institute for Occupational Safety and Health of the German Social Accident Insurance (IFA) http://gestis-en.itrust.de/
- [32].National Industrial Chemicals Notification and Assessment Scheme t-Amyl methyl ether (TAME) Full Public Report. 2001 Available electronically at http://www.nicnas.gov.au/publications/car/new/na/nafullr/na0800fr/na878fr.pdf.
- [33].Rounce P, Tsolakis A, Leung P, York APE. A comparison of diesel and biodiesel emissions using dimethyl carbonate as an oxygenated additive. Energy Fuels. 2010;24:4812–9. [Google Scholar]
- [34].Cheung CS, Zhu R, Huang Z. Investigation on the gaseous and particulate emissions of a compression ignition engine fueled with diesel–dimethyl carbonate blends. Science of the Total Environment. 2011;409:523–9. doi: 10.1016/j.scitotenv.2010.10.027. [DOI] [PubMed] [Google Scholar]
- [35].Biodiesel Handling and Use Guide. (Fourth Edition) 2009 Technical Report Number DOE/GO-102008-2658, NREL/TP-540-43672, of National Renewable Energy Laboratory. Available electronically at http://www.osti.gov/bridge.
- [36].Tetrahydrofuran Data Sheet of INVISTA Company http://terathane.invista.com/e-trolley/page_10344/index.html.
- [37].Aycock DF. Solvent Applications of 2-Methyltetrahydrofuran in Organometallic and Biphasic Reactions. Organic Process Research & Development. 2007;11:156–9. [Google Scholar]
- [38].Hayes DJ, Hayes MHB. The role that lignocellulosic feedstocks and various biorefining technologies can play in meeting Ireland’s biofuel targets, Biofuels, Bioprod. Bioref. 2009;3:500–20. [Google Scholar]
- [39].Paul SF. An optimized alternative motor fuel formulation: Natural gas liquids ethanol and a biomass-derived ether. ACS Division of Fuel Chemistry. 1998;43:373–6. Preprints. [Google Scholar]
- [40].DMTHF Data Sheet of Sigma-Aldrich Company http://www.sigmaaldrich.com.
- [41].Daniel R, Tian G, Xu H, Wyszynski ML, Wu X, Huang Z. Effect of spark timing and load on a DISI engine fuelled with 2,5-dimethylfuran. Fuel. 2011;90:449–58. [Google Scholar]
- [42].Zhong S, Daniel R, Xu H, Zhang J, Turner D, Wyszynski ML, Richards P. Combustion and emissions of 2,5-dimethylfuran in a direct-injection spark-ignition engine. Energy and Fuels. 2011;24:2891–9. [Google Scholar]
- [43].Dunphy MP, Patterson PM, Simmie JM. High-temperature oxidation of ethanol. Part 2.-Kinetic modelling. J Chem Soc, Faraday Trans. 1991;87:2549–59. [Google Scholar]
- [44].Dunphy MP, Simmie JM. High-temperature oxidation of ethanol. Part 1.-Ignition delays in shock waves. J Chem Soc Faraday Trans. 1991;87:1691–5. [Google Scholar]
- [45].Norton TS, Dryer FL. An experimental and modeling study of ethanol oxidation kinetics in an atmospheric pressure flow reactor. Int J Chem Kin. 1992;24:319–44. [Google Scholar]
- [46].Marinov NM. A detailed chemical kinetic model for high temperature ethanol oxidation. Int J Chem Kin. 1999;31:183–220. [Google Scholar]
- [47].Gülder OL. Laminar burning velocities of methanol, ethanol and isooctane-air mixtures. Proc Combust Inst. 1982;19:275–81. [Google Scholar]
- [48].Egolfopoulos FN, Du DX, Law CK. A study on ethanol oxidation kinetics in laminar premixed flames, flow reactors, and shock tubes. Symposium (International) on Combustion.1992. pp. 833–41. [Google Scholar]
- [49].Aboussi B. PhD Dissertation. University of Orleans; France: 1991. [Google Scholar]
- [50].Saxena P, Williams FA. Numerical and experimental studies of ethanol flames. Proc Combust Inst. 2007;31:1149–56. [Google Scholar]
- [51].Seiser R, Seshadri K. Western States Section, The Combustion Institute, 2002 Spring Meeting; University of California at San Diego, La Jolla, CA. March 25–26, 2002; 92093-0411. Paper # 02S-27. [Google Scholar]
- [52].Li J, Kazakov A, Chaos M, Dryer FL. Chemical kinetics of ethanol oxidation. 5th US Combustion Meeting; Organized by the Western States Section of the Combustion Institute and Hosted by the University of California at San Diego. March 25-28, 2007. [Google Scholar]
- [53].Cancino LR, Fikri M, Oliveira AAM, Schulz C. Measurement and chemical kinetics modeling of shock-induced ignition of ethanol–air mixtures. Energy Fuels. 2010;24:2830–40. [Google Scholar]
- [54].Leplat N, Dagaut P, Togbé C, Vandooren J. Numerical and experimental study of ethanol combustion and oxidation in laminar premixed flames and in jet-stirred reactor. Combust Flame. 2011 in press. [Google Scholar]
- [55].Liao SY, Jiang DM, Huang ZH, Zeng K, Cheng Q. Determination of the laminar burning velocities for mixtures of ethanol and air at elevated temperatures. Applied Thermal Engineering. 2007;27:374–80. [Google Scholar]
- [56].Bradley D, Lawes M, Mansour MS. Explosion bomb measurements of ethanol–air laminar gaseous flame characteristics at pressures up to 1.4 MPa. Combust Flame. 2009;156:1462–70. [Google Scholar]
- [57].Johnson MV, Goldsborough SS, Serinyel Z, O’Toole P, Larkin E, O’Malley G, et al. A Shock Tube Study of n- and iso-Propanol Ignition. Energy Fuels. 2009;23:5886–98. [Google Scholar]
- [58].Frassoldati A, Cuoci A, Faravelli T, Niemann U, Ranzi E, Seiser R, et al. An experimental and kinetic modeling study of n-propanol and iso-propanol combustion. Combust Flame. 2010;175:2–16. [Google Scholar]
- [59].Norton TS, Dryer FL. The flow reactor oxidation of C1–C4 alcohols and MTBE. Proc Combust Inst. 1990;23:179–85. [Google Scholar]
- [60].Kasper T, Oßwald P, Struckmeier U, Kohse-Höinghaus K, Taatjes CA, Wang J, et al. Combustion chemistry of the propanol isomers-investigated by electron ionization and VUV-photoionization molecular-beam mass spectrometry. Combust Flame. 2009;156:1181–201. [Google Scholar]
- [61].Dagaut P, Sarathy SM, Thomson MJ. A chemical kinetic study of n-butanol oxidation at elevated pressure in a jet stirred reactor. Proc Combust Inst. 2009;32:229–37. [Google Scholar]
- [62].Sarathy SM, Thomson MJ, Togbé C, Dagaut P, Halter F, Mounaim-Rousselle C. An experimental and kinetic modeling study of n-butanol combustion. Combust Flame. 2009;156:852–64. [Google Scholar]
- [63].Roberts AF. Burning velocities of the alcohols. J Imp Coll Chem Eng. Soc. 1959;12:58–73. [Google Scholar]
- [64].Black G, Curran HJ, Pichon S, Simmie JM, Zhukov V. Bio-butanol: Combustion properties and detailed chemical kinetic model. Combust Flame. 2010;157:363–73. [Google Scholar]
- [65].Grana R, Frassoldati A, Faravelli T, Niemann U, Ranzi E, Seiser R, et al. An experimental and kinetic modeling study of combustion of isomers of butanol. Combust Flame. 2010;157:2137–54. [Google Scholar]
- [66].Choudhury TK, Lin MC, Sanders WA. Thermal decomposition of t-butyl alcohol in shock waves. Combust Sci and Tech. 1990;71:219–32. [Google Scholar]
- [67].Harper MR, Van Geem KM, Pyl SP, Marin GB, Green WH. Comprehensive reaction mechanism for n-butanol pyrolysis and combustion. Combust Flame. 2011;158:16–41. [Google Scholar]
- [68].Davidson DF, Ranganath SC, Lam KY, Liaw M, Hong Z, Hanson RK. Ignition Delay Time Measurements of Normal Alkanes and Simple Oxygenates. Journal of Propulsion and Power. 2010;26:280–7. [Google Scholar]
- [69].McEnally CS, Pfefferle LD. Fuel decomposition and hydrocarbon growth processes for oxygenated hydrocarbons: butyl alcohols. Proc Combust Inst. 2005;30:1363–70. [Google Scholar]
- [70].Togbé C, Halter F, Foucher F, Mounaim-Rousselle C, Dagaut P. Experimental and detailed kinetic modeling study of 1-pentanol oxidation in a JSR and combustion in a bomb. Proc Combust Inst. 2011;33:367–74. [Google Scholar]
- [71].Togbé C, Dagaut P, Mzé-Ahmed A, Diévart P. Experimental and Detailed Kinetic Modeling Study of 1-Hexanol Oxidation in a Pressurized Jet-Stirred Reactor and a Combustion Bomb. Energy Fuels. 2010;24:5859–75. [Google Scholar]
- [72].Glaude PA, Pitz WJ, Thomson MJ. Chemical kinetic modeling of dimethyl carbonate in an opposed-flow diffusion flame. Proc Combust Inst. 2005;30:1111–8. [Google Scholar]
- [73].Sinha A, Thomson MJ. The chemical structures of opposed flow diffusion flames of C3 oxygenated hydrocarbons (isopropanol, dimethoxy methane, and dimethyl carbonate) and their mixtures. Combust Flame. 2004;136:548–56. [Google Scholar]
- [74].Fisher EM, Pitz WJ, Curran HJ, Westbrook CK. Detailed chemical kinetic mechanisms for combustion of oxygenated fuels. Proc Combust Inst. 2000;28:1579–86. [Google Scholar]
- [75].Parsons BI, Hinshelwood C. The Oxidation of Hydrocarbons and Their Derivatives. Part II. Structural Effects in the Ester Series. J Chem Soc. 1956:1799–803. [Google Scholar]
- [76].Gaïl S, Thomson MJ, Sarathy SM, Syed SA, Dagaut P, Diévart P, et al. A wide-ranging kinetic modeling study of methyl butanoate combustion. Proc Combust Inst. 2007;31:305–11. [Google Scholar]
- [77].Gaïl S, Sarathy SM, Thomson MJ, Diévart P, Dagaut P. Experimental and chemical kinetic modeling study of small methyl esters oxidation : Methyl (E)-2-butenoate and methyl butanoate. Combust Flame. 2008;155:635–50. [Google Scholar]
- [78].Metcalfe WK, Dooley S, Curran HJ, Simmie JM, El-Nahas AM, Navarro MV. Experimental and Modeling Study of C5H10O2 Ethyl and Methyl Esters. J Phys Chem A. 2007;111:4001–4. doi: 10.1021/jp067582c. [DOI] [PubMed] [Google Scholar]
- [79].Dooley S, Curran HJ, Simmie JM. Autoignition measurements and a validated kinetic model for the biodiesel surrogate, methyl butanoate. Combust Flame. 2008;153:2–32. [Google Scholar]
- [80].Hakka MH, Bennadji H, Biet J, Yahyaoui M, Sirjean B, Warth V, et al. Oxidation of methyl and ethyl butanoates. Int J Chem. 2010;42:226–52. [Google Scholar]
- [81].Walton SM, Wooldridge MS, Westbrook CK. An experimental investigation of structural effects on the auto-ignition properties of two C5 esters. Proc Combust Inst. 2009;32:255–62. [Google Scholar]
- [82].Bennadji H, Coniglio L, Billaud F, Bounaceur R, Warth V, Glaude PA, et al. Oxidation of small unsaturated methyl and ethyl esters. Int J Chem. 2011;43:204–18. [Google Scholar]
- [83].Dayma G, Gaïl S, Dagaut P. Experimental and Kinetic Modeling Study of the Oxidation of Methyl Hexanoate. Energy Fuels. 2008;22:1469–79. [Google Scholar]
- [84].Glaude PA, Herbinet O, Bax S, Biet J, Warth V, Battin-Leclerc F. Modeling of the oxidation of methyl esters-Validation for methyl hexanoate, methyl heptanoate, and methyl decanoate in a jet-stirred reactor. Combust Flame. 2010;157:2035–50. doi: 10.1016/j.combustflame.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Dayma G, Togbé C, Dagaut P. Detailed kinetic mechanism for the oxidation of vegetable oil methyl esters: New evidence from methyl heptanoate. Energy Fuels. 2009;23:4254–68. [Google Scholar]
- [86].Herbinet O, Pitz JW, Westbrook KC. Detailed chemical kinetic oxidation mechanism for a biodiesel surrogate. Combust Flame. 2008;154:507–28. [Google Scholar]
- [87].Herbinet O, Pitz WJ, Westbrook CK. Detailed chemical kinetic mechanism for the oxidation of biodiesel fuels blend surrogate. Combust Flame. 2010;157:893–908. [Google Scholar]
- [88].Naik CV, Westbrook CK, Herbinet O, Pitz WJ, Mehl M. Detailed chemical kinetic reaction mechanism for biodiesel components methyl stearate and methyl oleate. Proc Combust Inst. 2011;33:383–9. [Google Scholar]
- [89].Herbinet O, Biet J, Hakka MH, Warth V, Glaude PA, Nicolle A, et al. Modeling study of the low-temperature oxidation of large methyl esters from C11 to C19. Proc Combust Inst. 2011;33:391–8. doi: 10.1016/j.proci.2010.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Hakka MH, Glaude PA, Herbinet O, Battin-Leclerc F. Experimental study of the oxidation of large surrogates for diesel and biodiesel fuels. Combust Flame. 2009;156:2129–44. [Google Scholar]
- [91].Westbrook CK, Naik CV, Herbinet O, Pitz WJ, Mehl M, Sarathy SM, et al. Detailed chemical kinetic reaction mechanisms for soy and rapeseed biodiesel fuels. Combust Flame. 2010 in press. [Google Scholar]
- [92].Bax S, Hakka MH, Glaude PA, Herbinet O, Battin-Leclerc F. Experimental study of the oxidation of methyl oleate in a jet-stirred reactor. Combust Flame. 2010;157:1220–9. doi: 10.1016/j.combustflame.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Dagaut P, Boettner JC, Cathonnet M. Chemical kinetic study of dimethylether oxidation in a jet stirred reactor from 1 to 10 ATM: Experiments and kinetic modeling. Twenty-Sixth Symposium (International) on Combustion; Pittsburgh: The Combustion Institute; 1996. pp. 627–32. [Google Scholar]
- [94].Dagaut P, Daly C, Simmie JM, Cathonnet M. Oxidation and ignition of dimethylether from low to high temperature (500-1600 K): Experiments and kinetic modeling. Twenty-Seventh Symposium (International) on Combustion; Pittsburgh, PA: The Combustion Institute; 1998. pp. 361–69. [Google Scholar]
- [95].Curran HJ, Pitz WJ, Westbrook CK, Dagaut P, Boettner JC, Cathonnet M. A Wide Range Modeling Study of Dimethyl Ether Oxidation. Int J Chem Kin. 1998;30:229–41. [Google Scholar]
- [96].Curran HJ, Fischer SL, Dryer FL. Reaction kinetics of dimethyl ether. II: low-temperature oxidation in flow reactors. Int J Chem Kin. 2000;32:741–59. [Google Scholar]
- [97].Pfahl U, Fieweger K, Adomeit G. Self-ignition of diesel-relevant hydrocarbon-air mixtures under engine conditions. Twenty-Sixth Symposium (International) on Combustion; Pittsburgh, PA: The Combustion Institute; 1996. pp. 781–9. [Google Scholar]
- [98].Fischer SL, Dryer FL, Curran HJ. Reaction kinetics of dimethyl ether. I: high-temperature pyrolysis and oxidation in flow reactors. Int J Chem Kin. 2000;32:713–40. [Google Scholar]
- [99].Aluzeta MU, Muro J, Bilbao R, Glarborg P. Oxidation of dimethyl ether and its interaction with nitrogen oxides. Israel J Chem. 1999;39:73–86. [Google Scholar]
- [100].Zhao Z, Chaos M, Kazakov A, Dryer FL. Thermal Decomposition Reaction and a Comprehensive Kinetic Model of Dimethyl Ether. Int J Chem Kin. 2008;40:1–18. [Google Scholar]
- [101].McIlroy A, Hain TD, Michelsen HA, Cool TA. A laser and molecular beam mass spectrometer study of low-pressure dimethyl ether flames. Proc Combust Inst. 2000;28:1647–53. [Google Scholar]
- [102].Kaiser EW, Wallington TJ, Hurley MD, Platz J, Curran HJ, Pitz WJ, et al. Experimental and modeling study of premixed atmospheric-pressure dimethyl ether - air flames. J Phys Chem A. 2000;104:8194–06. [Google Scholar]
- [103].Qin X, Ju Y. Measurements of burning velocities of dimethyl ether and air premixed flames at elevated pressures. Proc Combust Inst. 2005;30:233–40. [Google Scholar]
- [104].Daly CA, Simmie JM, Würmel J, Djebaiili N, Paillard C. Burning velocities of dimethyl ether and air. Combust Flame. 2001;125:1329–40. [Google Scholar]
- [105].Zhao Z, Kazakov A, Dryer FL. Measurements of dimethyl ether/air mixture burning velocities by using particle image velocimetry. Combust Flame. 2004;139:52–60. [Google Scholar]
- [106].Brocard JC, Baronnet F, O’Neal HE. Chemical kinetics of the oxidation of methyl tert-butyl ether (MTBE) Combust Flame. 1983;52:25–35. [Google Scholar]
- [107].Curran HJ, Dunphy MP, Simmie JM, Westbrook CK, Pitz WJ. Shock tube ignition of ethanol, isobutene and MTBE: Experiments and modeling. Twenty-Fourth Symposium (International) on Combustion/The Combustion Institute.1992. pp. 769–76. [Google Scholar]
- [108].Goldaniga A, Faravelli T, Ranzi E, Dagaut P, Cathonnet M. Oxidation of oxygenated octane improvers: MTBE, ETBE, DIPE, and TAME. Twenty-Seventh Symposium (International) on Combustion; Pittsburgh: The Combustion Institute; 1998. pp. 353–60. [Google Scholar]
- [109].Glaude PA, Battin-Leclerc F, Judenherc B, Warth V, Fournet R, Côme GM, et al. Experimental and Modeling Study of the Gas-Phase Oxidation of Methyl and Ethyl Tertiary Butyl Ethers. Combust Flame. 2000;121:345–55. [Google Scholar]
- [110].Yasunaga K, Simmie JM, Curran HJ, Koike T, Takahashi O, Kuraguchi Y, et al. Detailed chemical kinetic mechanisms of ethyl methyl, methyl tert-butyl and ethyl tert-butyl ethers: The importance of uni-molecular elimination reactions. Combust Flame. 2010 in press. [Google Scholar]
- [111].Ogura T, Sakai Y, Miyoshi A, Koshi M, Dagaut P. Modeling of the oxidation of primary reference fuel in the presence of oxygenated octane improvers: Ethyl tert-butyl ether and ethanol. Energy Fuels. 2007;21:3233–9. [Google Scholar]
- [112].Yahyaoui M, Djebaili-Chaumeix N, Dagaut P, Paillard CE. Ethyl tertiary butyl ether ignition and combustion using a shock tube and spherical bomb. Energy Fuels. 2008;22:3701–8. [Google Scholar]
- [113].Yasunaga K, Gillespie F, Simmie JM, Curran HJ, Kuraguchi Y, Hoshikawa H, Yamane M, Hidaka Y. A multiple shock tube and chemical kinetic modeling study of diethyl ether pyrolysis and oxidation. J Phys Chem A. 2010;114:9098–109. doi: 10.1021/jp104070a. [DOI] [PubMed] [Google Scholar]
- [114].Daly CA, Simmie JM, Dagaut P, Cathonnet M. Oxidation of dimethoxymethane in a jet-stirred reactor. Combust Flame. 2001;125:1106–17. [Google Scholar]
- [115].Dias V, Lories X, Vandooren J. Lean and rich premixed dimethoxymethane/oxygen/argon flames: Experimental and modeling. Combust Sci and Tech. 2010;182:350–64. [Google Scholar]
- [116].Dagaut P, McGuinness M, Simmie JM, Cathonnet M. The ignition and oxidation of tetrahydrofuran: Experiments and kinetic modeling. Combust Sci and Tech. 1998;135:3–29. [Google Scholar]
- [117].Tian Z, Yuan T, Fournet R, Glaude PA, Sirjean B, Battin-Leclerc F, et al. An experimental and kinetic investigation of premixed furan/oxygen/argon flames. Combust Flame. 2011;158:756–73. doi: 10.1016/j.combustflame.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Organ PP, Mackie JC. Kinetics of pyrolysis of furan. J Chem Soc, Faraday Trans. 1991;87:815–23. [Google Scholar]
- [119].Fulle D, Dib A, Kiefer JH, Zhang Q, Yao J, Kern RD. Pyrolysis of furan at low pressures: Vibrational relaxation, unimolecular dissociation, and incubation times. J Phys Chem A. 1998;102:7480–6. [Google Scholar]
- [120].Pollard RT. Hydrocarbons. In: Bamford CH, Tipper CFH, editors. Comprehensive chemical kinetics: gas-phase combustion. Elsevier; Amsterdam: 1977. p. 17. [Google Scholar]
- [121].Kolb A, Püttmann W. Methyl tert-butyl ether (MTBE) in finished drinking water in Germany. Environ Pollution. 2006;140:294–303. doi: 10.1016/j.envpol.2005.07.008. [DOI] [PubMed] [Google Scholar]
- [122].Buda F, Bounaceur R, Warth V, Glaude PA, Fournet R, Battin-Leclerc F. Progress towards an unified detailed kinetic model for the autoignition of alkanes from C4 to C10 between 600 and 1200 K. Combust Flame. 2005;142:170–86. [Google Scholar]
- [123].Glaude PA, Battin-Leclerc F, Judenherc B, Warth V, Fournet R, Côme GM, et al. Experimental and modeling study of the gas-phase oxidation of methyl and ethyl tertiary butyl ethers. Combust Flame. 2000;121:345–55. [Google Scholar]
- [124].Glaude PA, Herbinet O, Bax S, Biet J, Warth V, Battin-Leclerc F. Modeling of the oxidation of methyl esters–validation for methyl hexanoate, methyl heptanoate, and methyl decanoate in a jet-stirred reactor. Combust Flame. 2010;157:2035–50. doi: 10.1016/j.combustflame.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Muller C, Michel V, Scacchi G, Côme GM. A computer program for the evaluation of thermochemical data of molecules and free radicals in the gas phase. J Chim Phys. 1995;92:1154–77. [Google Scholar]
- [126].Benson SW. Thermochemical Kinetics. 2nd ed John Wiley; New York: 1976. [Google Scholar]
- [127].Tsang W. Heat of Formation of Organic Radicals by Kinetic Methods. In: Simões JAM, Greenberg A, Liebman JF, editors. Energetics of organic free radicals. 4 Ed Kluwer Academic Publishers; Dordrecht: 1996. [Google Scholar]
- [128].Luo YR. Handbook of bond dissociation energies in organic compounds. CRC Press; Boca Raton: 2003. [Google Scholar]
- [129].El-Nahas AM, Navarro MV, Simmie JM, Bozzelli JW, Curran HJ, Dooley S, Metcalfe W. Enthalpies of formation, bond dissociation energies and reaction paths for the decomposition of model biofuels: ethyl propanoate and methyl butanoate. J Phys Chem A. 2007;111:3727–39. doi: 10.1021/jp067413s. [DOI] [PubMed] [Google Scholar]
- [130].Biet J, Hakka MH, Warth V, Glaude P, Battin-Leclerc F. Experimental and modeling study of the low-temperature oxidation of large alkanes. Energ Fuels. 2008;22:2258–69. [Google Scholar]
- [131].Choo KY, Golden DM, Benson SW. Very low-pressure pyrolysis (VLPP) of t-butylmethyl ether. Int J Chem Kin. 1974;6:631–41. [Google Scholar]
- [132].Tsang W. Energy transfer effects during the multichannel decomposition of ethanol. Int J Chem Kin. 2004;36:456–65. [Google Scholar]
- [133].Bui B, Zhu R, Lin M. Thermal decomposition of iso-propanol: first-principles prediction of total and product-branching rate constants. J Chem Phys. 2002;117:11188–95. [Google Scholar]
- [134].Warth V, Stef N, Glaude PA, Battin-Leclerc F, Scacchi G, Côme GM. Computed aided design of gas-phase oxidation mechanisms: Application to the modelling of normal-butane oxidation. Combust Flame. 1998;114:81–102. [Google Scholar]
- [135].Ingham T, Walker RW, Woolford RE. Kinetic parameters for the initiation reaction RH + O2 → R + HO2. Proc Combust Inst. 1994;25:767–74. [Google Scholar]
- [136].Dean AM, Bozzelli JW. Combustion Chemistry of Nitrogen. In: Gardiner WC, editor. Gas-phase Combustion Chemistry. Springer-Verlag; New York: 2000. [Google Scholar]
- [137].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian03. revision B05 Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- [138].Wilk RD, Cernansky NP, Pitz WJ, Westbrook CK. Combust Flame. 1989;77:145–70. [Google Scholar]
- [139].Mereau R, Rayez M-T, Rayez J-C, Caralp F, Lesclaux R. Theoretical study on the atmospheric fate of carbonyl radicals: kinetics of decomposition reactions. Phys Chem Chem Phys. 2001;3:4712–17. [Google Scholar]
- [140].Wiberg K, Waldron R. Lactones. 2. Enthalpies of hydrolysis, reduction, and formation of the C4-C13 monocyclic lactones. Strain energies and conformations. J Am Chem Soc. 1991;113:7697–705. [Google Scholar]
- [141].Cavallotti C, Rota R, Faravelli T, Ranzi E. Ab initio evaluation of primary cyclo-hexane oxidation reaction rates. Proc Combust Inst. 2007;31:201–9. [Google Scholar]
- [142].Silke EJ, Pitz WJ, Westbrook CK, Ribaucour M. Detailed Chemical Kinetic Modeling of Cyclohexane Oxidation. J Phys Chem A. 2007;111:3761–75. doi: 10.1021/jp067592d. [DOI] [PubMed] [Google Scholar]
- [143].Sirjean B, Buda F, Hakka H, Glaude PA, Fournet R, Warth V, et al. The autoignition of cyclopentane and cyclohexane in a shock tube. Proc Combust Inst. 2007;31:277–84. [Google Scholar]
- [144].Buda F, Heyberger B, Fournet R, Glaude PA, Warth V, Battin-Leclerc F. Modeling of the Gas-Phase Oxidation of Cyclohexane. Energ Fuels. 2006;20:1450–59. [Google Scholar]
- [145].Sirjean B, Glaude PA, Ruiz-Lopez MF, Fournet R. Detailed kinetic study of the ring opening of cycloalkanes by CBS-QB3 calculations. J Phys Chem A. 2006;110:12693–704. doi: 10.1021/jp0651081. [DOI] [PubMed] [Google Scholar]
- [146].Sirjean B, Glaude PA, Ruiz-Lopez MF, Fournet R. Theoretical kinetic study of thermal unimolecular decomposition of cyclic alkyl radicals. J Phys Chem A. 2008;112:11598–610. doi: 10.1021/jp805640s. [DOI] [PubMed] [Google Scholar]
- [147].Sirjean B, Glaude PA, Ruiz-Lopez MF, Fournet R. Theoretical kinetic study of the reactions of cycloalkylperoxy radicals. J Phys Chem A. 2009;113:6924–35. doi: 10.1021/jp901492e. [DOI] [PubMed] [Google Scholar]
- [148].Simmie JM, Curran HJ. Formation enthalpies and bond dissociation energies of alkylfurans. The strongest C-X bonds known? J Phys Chem A. 2009;113:5128–37. doi: 10.1021/jp810315n. [DOI] [PubMed] [Google Scholar]
- [149].Sendt K, Bacskay GB, Mackie JC. Pyrolysis of furan: Ab initio quantum chemical and kinetic modeling studies. J Phys Chem A. 2000;104:1861–75. [Google Scholar]