

Abstract
Purpose of review
The Kallikrein-kinin system (KKS) constitutes a complex multi-enzyme cascade that produces several bioactive kinin peptides and their derivatives including bradykinin. In addition to the classical notion of the KKS as a potent vasodilator and a mediator of inflammatory responses, recent studies suggest a link between the KKS and oxidative stress. A number of established mouse model with altered levels of KKS components opened the way to evaluate precise functions of the KKS. Here we review recent findings on the role of the KKS in cardiovascular diseases and chronic kidney diseases, and discuss potential benefits of KKS activation in these diseases.
Recent findings
Deletion of both B1R and B2R in a diabetic mouse model exacerbates its renal phenotypes, suggesting that the KKS exerts protective effects on diabetic nephropathy by suppressing oxidative stress, presumably via nitric oxide (NO) and prostaglandins (PGs).
Summary
Accumulating evidence has highlighted the importance of the KKS as a protective system against oxidative stress and organ damage in the heart and kidney. The activation of the KKS by ACE inhibitors and vasopeptidase inhibitors is likely to be beneficial in senescence-associated cardiovascular diseases and chronic kidney diseases.
Keywords: Kallikrein-kinin system (KKS), angiotensin-I converting enzyme (ACE), oxidative stress, diabetes mellitus complications
Introduction
Kinins are generated by the proteolytic cleavage of kininogens by tissue or plasma kallikreins and other serine proteases. Kinin peptides act via two types of kinin receptors: the bradykinin B1 receptor (B1R/Bdkrb1) and the bradykinin B2 receptor (B2R/Bdkrb2) (Fig. 1). B2R is constitutively expressed in most tissues, whereas B1R is induced under stress conditions including inflammation and diabetes. Both B1R and B2R are seven transmembrane receptors coupled with G-proteins, stimulation of which elevates intracellular Ca++ concentrations and the phospholipase A2 activity and leads to the release of various mediators including nitric oxide (NO), prostaglandins (PGs), arachidonic acids and other inflammatory agents [1–3].
Figure 1. The KKS and suppression of oxidative stress.
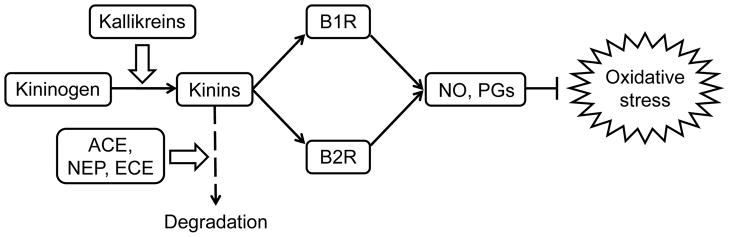
Kinins are generated from kininogens by kallikreins and are inactivated by ACE, NEP and ECE. Binding of kinins to bradykinin receptors (B1R and B2R) leads to the reduction of oxidative stress via NO and PGs. NEP, neprilysin; ECE, endothelin-converting enzyme.
Several mouse models and human epidemiological studies have suggested that KKS activity modifies oxidative stress and disease progression in the heart and kidney. In this review we classify this evidence into three sections according to the modification step the KKS: kinin levels, receptor activity, and mediator production.
Regulation of the kinin levels by angiotensin I-converting enzyme (ACE)
The estimated half-life of kinin peptides is remarkably short (less than 30 seconds in plasma [4]). Active kinins are rapidly cleaved by several kinin peptidases, primarily by angiotensin I-converting enzyme (ACE), which inactivates bradykinin, kallidin (humans), and kallidin-like peptide (rodents) by removing two amino acids from the carboxyl termini of these substrates [2]. ACE also cleaves angiotensin I into an active vasoconstrictor angiotensin II, although it has higher affinity for kinins than that for angiotensin I [5]. Modest changes in ACE levels affect kinin levels more than angiotensin II levels, as supported by computer simulations demonstrating that altered ACE levels have virtually no effect on the levels of products, while they have marked effects on its substrates [6].
The systemic effects of quantitatively varied ACE activities were examined by utilizing genetically engineered mice that carry different copy numbers of the ACE gene. The one-copy mice showed 62% of plasma ACE activities of the wild-type (two-copy) mice, and the three copy mice had 144% of normal [7]. A genetic decrease in ACE activity reduced myocardial infarct size after ischemia/reperfusion injury, and the effect was completely suppressed by pretreatment with a selective B2R antagonist [8]. In the streptozotocin (STZ)-induced diabetic model, the onset of the proteinuria was accelerated in three-copy diabetic mice [9]. These results indicate the protective effect of low ACE levels on oxidative stress-associated diseases in the heart and kidney.
A naturally occurring polymorphism in human ACE gene, the I/D polymorphism, is characterized by the insertion (I allele) or the non-insertion (D allele) of a 287-bp Alu retrotransposon in intron 16. The polymorphism is associated with differences in the plasma and tissue levels of ACE (I/I, 76%; I/D, 100%; D/D, 126% of average), and the D allele is associated with higher levels of ACE and kallikrein [10, 11]. Many reports have indicated that the D/D genotype may contribute to increased risks for cardiovascular and renal diseases [12, 13].
Angiotensin-I converting enzyme (ACE) inhibitors are the most important agents that augment KKS activity. While widely recognized as an inhibitor of renin-angiotensin-aldosterone system (RAAS), their primary function is an upregulator of kinins rather than an inhibitor of ACE. Indeed, the minimum effective dose of an ACE inhibitor perindopril for increasing plasma bradykinin levels is much lower than that for reducing angiotensin II levels [14]. This effect, partly explained in the preferential change in kinin levels more than angiotensin levels, could also be mediated by higher affinity of bradykinin binding sites of ACE than that of the angiotensin I binding sites by ACE inhibitor [15]. Moreover, it is proposed that ACE inhibitors are the direct or indirect allosteric enhancers of B1R and B2R [16].
The protective role of ACE inhibitors in cardiac and renal diseases has been well documented [17]. Vasopeptidase inhibitors, which simultaneously inhibit ACE and neprilysin (NEP), a circulating enzyme that degrades vasoactive peptides including natriuretic peptides and kinins, also prevent diabetes and improve diabetic nephropathy in rat models [18, 19]. The beneficial effects of ACE inhibitors and vasopeptidase inhibitors are attenuated by a B2R antagonist in diabetic model animals, indicating that the protective effects of ACE inhibitors are mediated by B2R signaling [19–21].
In addition to the ACE inhibitors, angiotensin receptor blockers (ARBs) are widely used drugs proven to be reno- and cardio-protective. Although ARBs are considered to modulate RAAS, several studies demonstrated that the cardio-protective effect of an ARB losartan is reduced by a selective B2R inhibitor, indicating that some effects of ARBs are mediated through B2R activation [22]. The mechanism remains to be elucidated, however, Bonde et al. recently proposed that losartan directly binds to and activate the B2R [23].
ACE inhibitors might be most effective in diabetic patients with the D allele of ACE gene, however, clinical studies have given conflicting results [13]. Further studies on more homogenous group are needed to assess the therapeutic effects of ACE inhibitors in patients with different ACE genotypes.
Modulation of bradykinin receptor activity
Recent studies suggest the protective role of B2R in oxidative stress-mediated disorders of the heart and kidney [20, 21, 24]. We have previously shown that the lack of B2R worsens diabetic complications and accelerates premature aging in Akita (Ins2Akita/+) diabetic mice [24]. The Akita mice lacking B2R develop senescence-associated phenotypes including alopecia, skin atrophy, kyphosis, osteoporosis, and testicular atrophy. On the other hand, another group reported that the lack of B2R protects against the development of STZ-induced diabetic nephropathy [25]. The opposite results could possibly be caused by differences in strain and age of the mice and in the method of induction of diabetes.
Whether B1R is beneficial or detrimental in the heart or kidney remains unclear. Although lack of the B1R does not affect cardiac remodeling after myocardial infarction, blockade of B2R worsened cardiac remodeling and dysfunction in B1R knockout mice but not in the wild type mice [26]. In contrast, B1R knockout mice were protected against cardiac and renal injury [27, 28]. A recent study also shows that activation of the B1R exacerbates glomerulosclerosis [29]. However, as genetic disruptions of either receptor causes enhanced expression of the remaining receptor, these results would not directly reflect the isolated lack of either receptor.
To define the role of each receptor, we generated mice lacking both B1R and B2R [30*, 31**]. Susceptibility to renal ischemia/reperfusion injury was B1RB2R-null > B2R-null > wild type, demonstrating that both B1R and B2R have protective roles in kidney [30*]. Furthermore, the lack of both B1R and B2R in the Akita diabetic mouse exacerbates the pathological changes characteristic of diabetes, such as nephropathy, neuropathy, and osteopathy (Fig. 2). Markedly elevated levels of plasma oxidative stress markers and enhanced renal mitochondrial DNA damage suggest that the increased oxidative stress contributes to the pathological changes in the B1RB2R-null/Akita mice [31**]. These results clearly demonstrate that the KKS plays a protective role in the progression of oxidative stress-related diseases, and suggest a novel therapeutic strategy in that the modulation of the KKS could be beneficial for the treatment or prevention of these diseases.
Figure 2. Pathological changes in the B1RB2R-null/Akita mice.

PAS staining of renal glomeruli in 12-month-old male WT, B2R-null, B1RB2R-null, Akita, B2R-null, and B1RB2R-null/Akita mice. Lack of bradykinin receptors enhances diabetic glomerulosclerosis. Scale bar, 100 μm. Reproduced with permission from: [31**].
In contrast to the deleterious effect of B1R and B2R deficiency in the kidney, no significant changes in cardiac structure or function were observed in B1RB2R-null mice or B1RB2R-null/Akita mice, although some parameters of cardiac dysfunction were decreased in B1RB2R-null/Akita mice [32*, 33]. The different susceptibilities of the heart and kidney imply the tissue-specific distributions and functions of each receptor.
In a human polymorphism in B2R gene (+9/−9 bp insertion/deletion), the insertion (+9) allele is associated with decreased B2R gene transcription. The +9/+9 genotype is correlated with greater cardiovascular risk including increased left ventricular hypertrophic responses and higher systolic blood pressure, implying a harmful effect of the +9 allele [34]. In contrast, the +9 allele is associated with a decreased progression of nephropathy in type 1 and type 2 diabetic patients [35]. The association between +9/−9 polymorphism and risks of the other oxidative stress-related diseases requires further clinical studies.
Intracellular mediator—NO and PGs
Reactive oxygen species (ROS), generated mainly by mitochondrial oxidative phosphorylation, are considered to be the primary cause of aging through their damage to DNA[36]. Dias et al. reported that activation of B1R increases the oxidative stress through the activation of NADPH oxidase in the vasculature in a rat model [37]. In contrast, several studies indicate that the B2R may contribute to reducing ROS. In STZ-treated hyperglycemic rats, bradykinin reduces hydrogen peroxide and a biomarker of oxidative stress. The protective effects of ACE inhibitors against oxidative stress are attenuated by a B2R antagonist [38].
The KKS may contribute to the reduction of oxidative stress via NO and prostaglandin (PGs) synthesis (Fig. 1). NO negatively regulates mitochondrial oxidative phosphorylation by inhibiting cytochrome c oxidase, which is a key enzyme in the electron transport chain [39]. Both the stimulation of B1R and B2R activates endothelial nitric oxide synthase (eNOS/NOS3) leading to low and transient production of NO, and activates inducible NOS (iNOS/NOS2), facilitating higher and prolonged release of NO [2, 3, 40]. Indeed, urinary NO metabolites in B2R-null mice are lower than in WT [41], and the plasma NO metabolites are lower in B1RB2R-null mice than in WT [30]. A polymorphism in the intron 4 of human eNOS gene is associated with an increased risk for diabetic nephropathy, and a modest decrease in eNOS enhances the development of nephropathy in diabetic mice [42]. Bradykinin reduces mitochondrial superoxide generation in human vascular endothelial cells, which is partly reversed by a NOS inhibitor [30]. Bradykinin also protects bovine aorta endothelial cells from ROS-induced DNA damage and senescence through B2R-mediated NO production [43]. Therefore, bradykinin-mediated NO production has the potential of preventing ROS-induced pathogenesis.
Recent studies also suggest the protective effect of PGs against oxidative stress. PGI2 overexpression in renal tubular cells decreases aminoglycoside-induced production of ROS and prevents apoptosis of these cells [44]. A PGI2 agonist cicaprost attenuates the progression of diabetic renal injury in STZ-treated rats [45]. Therefore, the protective effect of KKS may partially be mediated by NO and PGs.
Conclusions
ACE inhibitors have proven effective in long-term usage for the treatment of a constellation of cardiac and renal diseases. Although the renin-angiotensin-aldosterone system has been implicated in the pathogenesis, recent studies indicate that the benefits of the ACE inhibitors are independent of the antihypertensive properties [46]. Meanwhile, accumulating evidence suggests that potentiating the activity of KKS can protect against the progression of these diseases, by suppressing oxidative stress presumably via production of NO and PGs. Thus, the cardio- and reno-protective effect of ACE inhibitors could largely exerted by the activation of the KKS.
Studies using B1RB2R-null mice indicate that non-selective stimulation of both receptors is likely to have the higher therapeutic efficacies on diabetic nephropathy, while other studies propose that the selective activation of the B2R and inhibition of B1R could be beneficial in cardiovascular diseases. Further experimental and clinical investigations are warranted to evaluate the long-term efficacy of the selective and non-selective KKS modulation.
Key points.
Recent evidences suggest that the KKS has a protective role against oxidative stress and organ damage in the heart and kidney.
Phenotypes of B1R2R-null Akita mice, which lack both of the bradykinin receptors in the diabetic background, indicate that both receptors protect against the progression of diabetic nephropathy via the production of NO and PGs.
Activation of the KKS by ACE inhibitors and vasopeptidase inhibitors is highly beneficial in patients with diabetic complications.
Acknowledgments
Funding: This work was funded by National Institutes of Health Grants DK76131 and HL49277 to O.S., and by Career Development Award 2006-2-106 from Juvenile Diabetes Research Foundation to M.K.
References and recommended reading
- 1.Morand-Contant M, Anand-Srivastava MB, Couture R. Kinin B1 receptor upregulation by angiotensin II and endothelin-1 in rat vascular smooth muscle cells: Receptors and mechanisms. Am J Physiol Heart Circ Physiol. 2010;299:H1625–1632. doi: 10.1152/ajpheart.00735.2009. [DOI] [PubMed] [Google Scholar]
- 2.Kuhr F, Lowry J, Zhang Y, et al. Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides. 2010;44:145–154. doi: 10.1016/j.npep.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kakoki M, Smithies O. The kallikrein-kinin system in health and in diseases of the kidney. Kidney Int. 2009;75:1019–1030. doi: 10.1038/ki.2008.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saameli K, Eskes TK. Bradykinin and cardiovascular system: estimation of half-life. Am J Physiol. 1962;203:261–265. doi: 10.1152/ajplegacy.1962.203.2.261. [DOI] [PubMed] [Google Scholar]
- 5.Jaspard E, Wei L, Alhenc-Gelas F. Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J Biol Chem. 1993;268:9496–9503. [PubMed] [Google Scholar]
- 6.Smithies O, Kim HS, Takahashi N, Edgell MH. Importance of quantitative genetic variations in the etiology of hypertension. Kidney Int. 2000;58:2265–2280. doi: 10.1046/j.1523-1755.2000.00411.x. [DOI] [PubMed] [Google Scholar]
- 7.Krege JH, Kim HS, Moyer JS, et al. Angiotensin-converting enzyme gene mutations, blood pressures, and cardiovascular homeostasis. Hypertension. 1997;29:150–157. doi: 10.1161/01.hyp.29.1.150. [DOI] [PubMed] [Google Scholar]
- 8.Messadi E, Vincent MP, Griol-Charhbili V, et al. Genetically determined angiotensin converting enzyme level and myocardial tolerance to ischemia. FASEB J. 2010;24:4691–4700. doi: 10.1096/fj.10-165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang W, Gallois Y, Bouby N, et al. Genetically increased angiotensin I-converting enzyme level and renal complications in the diabetic mouse. Proc Natl Acad Sci U S A. 2001;98:13330–13334. doi: 10.1073/pnas.231476798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rigat B, Hubert C, Alhenc-Gelas F, et al. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343–1346. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Almeida SS, Barros CC, Moraes MR, et al. Plasma kallikrein and angiotensin I-converting enzyme N- and C-terminal domain activities are modulated by the insertion/deletion polymorphism. Neuropeptides. 2010;44:139–143. doi: 10.1016/j.npep.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Ruggenenti P, Bettinaglio P, Pinares F, Remuzzi G. Angiotensin converting enzyme insertion/deletion polymorphism and renoprotection in diabetic and nondiabetic nephropathies. Clin J Am Soc Nephrol. 2008;3:1511–1525. doi: 10.2215/CJN.04140907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rudnicki M, Mayer G. Significance of genetic polymorphisms of the renin-angiotensin-aldosterone system in cardiovascular and renal disease. Pharmacogenomics. 2009;10:463–476. doi: 10.2217/14622416.10.3.463. [DOI] [PubMed] [Google Scholar]
- 14.Jaspard E, Wei L, Alhenc-Gelas F. Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J Biol Chem. 1993;268:9496–9503. [PubMed] [Google Scholar]
- 15.Ceconi C, Francolini G, Olivares A, et al. Angiotensin-converting enzyme (ACE) inhibitors have different selectivity for bradykinin binding sites of human somatic ACE. Eur J Pharmacol. 2007 Dec;22(577):1–6. doi: 10.1016/j.ejphar.2007.07.061. [DOI] [PubMed] [Google Scholar]
- 16.Erdös EG, Tan F, Skidgel RA. Angiotensin I-converting enzyme inhibitors are allosteric enhancers of kinin B1 and B2 receptor function. Hypertension. 2010;55:214–220. doi: 10.1161/HYPERTENSIONAHA.109.144600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruggenenti P, Cravedi P, Remuzzi G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat Rev Nephrol. 2010;6:319–330. doi: 10.1038/nrneph.2010.58. [DOI] [PubMed] [Google Scholar]
- 18.Wang CH, Leung N, Lapointe N, et al. Vasopeptidase inhibitor omapatrilat induces profound insulin sensitization and increases myocardial glucose uptake in Zucker fatty rats: Studies comparing a vasopeptidase inhibitor, angiotensin-converting enzyme inhibitor, and angiotensin II type I receptor blocker. Circulation. 2003;107:1923–1929. doi: 10.1161/01.CIR.0000062646.09566.CC. [DOI] [PubMed] [Google Scholar]
- 19.Schäfer S, Schmidts HL, Bleich M, et al. Nephroprotection in Zucker diabetic fatty rats by vasopeptidase inhibition is partly bradykinin B2 receptor dependent. Br J Pharmacol. 2004;143:27–32. doi: 10.1038/sj.bjp.0705884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buléon M, Allard J, Jaafar A, et al. Pharmacological blockade of B2-kinin receptor reduces renal protective effect of angiotensin-converting enzyme inhibition in db/db mice model. Am J Physiol Renal Physiol. 2008;294:F1249–56. doi: 10.1152/ajprenal.00501.2007. [DOI] [PubMed] [Google Scholar]
- 21.Manolis AJ, Marketou ME, Gavras I, Gavras H. Cardioprotective properties of bradykinin: role of the B(2) receptor. Hypertens Res. 2010;33:772–777. doi: 10.1038/hr.2010.82. [DOI] [PubMed] [Google Scholar]
- 22.Sato M, Engelman RM, Otani H, et al. Myocardial protection by preconditioning of heart with Losartan, an angiotensin II type 1-receptor blocker: implication of bradykinin-dependent and bradykinin-independent mechanisms. Circulation. 2000;102:346–351. doi: 10.1161/01.cir.102.suppl_3.iii-346. [DOI] [PubMed] [Google Scholar]
- 23.Bonde MM, Olsen KB, Erikstrup N, Speerschneider T, Lyngsø C, Haunsø S, Nielsen MS, Sheikh SP, Hansen JL. The angiotensin II type 1 receptor antagonist Losartan binds and activates bradykinin B2 receptor signaling. Regul Pept. 2011;167:21–25. doi: 10.1016/j.regpep.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 24.Kakoki M, Kizer CM, Yi X, et al. Senescence-associated phenotypes in Akita diabetic mice are enhanced by absence of bradykinin B2 receptors. J Clin Invest. 2006;116:1302–1309. doi: 10.1172/JCI26958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan Y, Keum JS, Wang B, et al. Targeted deletion of B2-kinin receptors protects against the development of diabetic nephropathy. Am J Physiol Renal Physiol. 2007;293:F1026–1035. doi: 10.1152/ajprenal.00203.2007. [DOI] [PubMed] [Google Scholar]
- 26.Xu J, Carretero OA, Sun Y, et al. Role of the B1 kinin receptor in the regulation of cardiac function and remodeling after myocardial infarction. Hypertension. 2005;45:747–753. doi: 10.1161/01.HYP.0000153322.04859.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westermann D, Walther T, Savvatis K, et al. Gene deletion of the kinin receptor B1 attenuates cardiac inflammation and fibrosis during the development of experimental diabetic cardiomyopathy. Diabetes. 2009;58:1373–1381. doi: 10.2337/db08-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein J, Gonzalez J, Duchene J, et al. Delayed blockade of the kinin B1 receptor reduces renal inflammation and fibrosis in obstructive nephropathy. FASEB J. 2009;23:134–142. doi: 10.1096/fj.08-115600. [DOI] [PubMed] [Google Scholar]
- 29.Pereira RL, Buscariollo BN, Correa-Costa M, et al. Bradykinin receptor 1 activation exacerbates experimental focal and segmental glomerulosclerosis. Kidney Int. 2011;79:1217–1227. doi: 10.1038/ki.2011.14. [DOI] [PubMed] [Google Scholar]
- 30*.Kakoki M, McGarrah RW, Kim HS, Smithies O. Bradykinin B1 and B2 receptors both have protective roles in renal ischemia/reperfusion injury. Proc Natl Acad Sci USA. 2007;104:7576–7581. doi: 10.1073/pnas.0701617104. First report of B1R B2R-null mice generated by deleting the genomic region encoding the two receptors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31**.Kakoki M, Sullivan KA, Backus C, et al. Lack of both bradykinin B1 and B2 receptors enhances nephropathy, neuropathy, and bone mineral loss in Akita diabetic mice. Proc Natl Acad Sci U S A. 2010;107:10190–10195. doi: 10.1073/pnas.1005144107. Deletion of both B1R and B2R increases oxidative stress and aggravates nephropathy in the type I diabetes mouse model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Cayla C, Todiras M, Iliescu R, et al. Mice deficient for both kinin receptors are normotensive and protected from endotoxin-induced hypotension. FASEB J. 2007;21:1689–1698. doi: 10.1096/fj.06-7175com. Another B1RB2R-null mouse model generated by disrupting B1R using embryonic stem cells obtained from B2R-null mice. [DOI] [PubMed] [Google Scholar]
- 33.Wende AR, Soto J, Olsen CD, et al. Loss of bradykinin signaling does not accelerate the development of cardiac dysfunction in type 1 diabetic Akita mice. Endocrinology. 2010;151:3536–3542. doi: 10.1210/en.2010-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dhamrait SS, Payne JR, Li P, et al. Variation in bradykinin receptor genes increases the cardiovascular risk associated with hypertension. Eur Heart J. 2003;24:1672–1680. doi: 10.1016/s0195-668x(03)00441-x. [DOI] [PubMed] [Google Scholar]
- 35.Maltais I, Bachvarova M, Maheux P, et al. Bradykinin B2 receptor gene polymorphism is associated with altered urinary albumin/creatinine values in diabetic patients. Can J Physiol Pharmacol. 2002;80:323–327. doi: 10.1139/y02-036. [DOI] [PubMed] [Google Scholar]
- 36.Lu T, Finkel T. Free radicals and senescence. Exp Cell Res. 2008;314:1918–1922. doi: 10.1016/j.yexcr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dias JP, Talbot S, Sénécal J, et al. Kinin B1 receptor enhances the oxidative stress in a rat model of insulin resistance: outcome in hypertension, allodynia and metabolic complications. PLoS One. 2010;5:e12622. doi: 10.1371/journal.pone.0012622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mikrut K, Paluszak J, Kozlik J, et al. The effect of bradykinin on the oxidative state of rats with acute hyperglycemia. Diabetes Res Clin Pract. 2001;51:79–85. doi: 10.1016/s0168-8227(00)00222-9. [DOI] [PubMed] [Google Scholar]
- 39.Brunori M, Forte E, Arese M, et al. Nitric oxide and the respiratory enzyme. Biochim Biophys Acta. 2006;1757:1144–1154. doi: 10.1016/j.bbabio.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 40.Brovkovych V, Zhang Y, Brovkovych S, et al. A novel pathway for receptor-mediated post-translational activation of inducible nitric oxide synthase. J Cell Mol Med. 2011;15:258–269. doi: 10.1111/j.1582-4934.2009.00992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schanstra JP, Duchene J, Praddaude F, et al. Decreased renal NO excretion and reduced glomerular tuft area in mice lacking the bradykinin B2 receptor. Am J Physiol Heart Circ Physiol. 2003;284:H1904–1908. doi: 10.1152/ajpheart.01150.2002. [DOI] [PubMed] [Google Scholar]
- 42.Wang CH, Li F, Hiller S, et al. A modest decrease in endothelial NOS in mice comparable to that associated with human NOS3 variants exacerbates diabetic nephropathy. Proc Natl Acad Sci U S A. 2011;108:2070–2075. doi: 10.1073/pnas.1018766108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oeseburg H, Iusuf D, van der Harst P, et al. Bradykinin protects against oxidative stress-induced endothelial cell senescence. Hypertension. 2009;53:417–422. doi: 10.1161/HYPERTENSIONAHA.108.123729. [DOI] [PubMed] [Google Scholar]
- 44.Hsu YH, Chen CH, Hou CC, et al. Prostacyclin protects renal tubular cells from gentamicin-induced apoptosis via a PPARalpha-dependent pathway. Kidney Int. 2008;73:578–587. doi: 10.1038/sj.ki.5002704. [DOI] [PubMed] [Google Scholar]
- 45.Villa E, Rabano A, Ruilope LM, et al. Effects of cicaprost and fosinopril on the progression of rat diabetic nephropathy. Am J Hypertens. 1997;10:202–208. doi: 10.1016/s0895-7061(96)00319-6. [DOI] [PubMed] [Google Scholar]
- 46.Nobakht N, Kamgar M, Rastogi A, Schrier RW. Limitations of angiotensin inhibition. Nat Rev Nephrol. 2011;7:356–359. doi: 10.1038/nrneph.2011.29. [DOI] [PubMed] [Google Scholar]