

Abstract
Hereditary deafness is a genetically heterogeneous phenotype for which more than 100 genomic loci have been identified thus far. By analysis of a consanguineous Palestinian family, GPSM2 was recently discovered to be the cause of autosomal recessive nonsyndromic hearing loss DFNB82. Here, we report a second truncating mutation, GPSM2 p.Q562X, identified via autozygosity mapping in a consanguineous Turkish family. This report provides evidence for allelic heterogeneity of GPSM2 and confirms its causative role for nonsyndromic deafness.
Keywords: Autosomal recessive, deafness, GPSM2, hearing loss, homozygosity, genome-wide SNP genotyping
INTRODUCTION
Hearing loss, or deafness in its most severe form, affects more than 28 million persons in the United States and more than 33 million persons in Europe1. The prevalence of permanent bilateral sensorineural hearing loss (SNHL) is approximately 2 per 1000 newborns and 2.7 per 1000 five-year-olds1, 2. Of these cases, 75–80% are autosomal recessive and nonsyndromic3. Mutations in 39 genes have been reported to be associated with autosomal recessive nonsyndromic deafness. (Hereditary Hearing Loss Homepage, http://webh01.ua.ac.be/hhh/).
One of the genes responsible for nonsyndromic SNHL is GPSM2, identified via a truncating mutation, GPSM2 p.R127X, which was homozygous in seven individuals with prelingual SNHL in a large Palestinian kindred4. GPSM2 is essential for the maintenance of cell polarity and spindle orientation4. Here, we report another truncating mutation in GPSM2 in a Turkish family with autosomal recessive nonsyndromic SNHL.
MATERIALS AND METHODS
Subjects
Study subjects were an extended family of Turkish ancestry with three children with congenital SNHL; marriages of the parents of all affected children were consanguineous (Fig. 1A). The study was approved by the human subjects review boards of Ankara University, the University of Miami, and the University of Washington. Audiograms of family members indicated severe to profound SNHL in all affected individuals and normal hearing in their parents (Fig. 1B). The remainder of the physical examinations of all relatives were completely normal, including normal anterior chamber and fundus of the eyes; normal gross motor development; normal balance with no vertigo, dizziness, or spontaneous and positional nystagmus; normal tandem walking and negative Romberg tests. CT scans of the temporal bone in one affected family member were normal. Peripheral leukocytes were sampled from all seven members of the third and fourth generations of the family, and DNA extracted by the phenol chloroform method. Mutations in the GJB2 gene were excluded via Sanger sequencing of both exons.
Figure 1. Homozygosity mapping of sensorineural hearing loss (SNHL) in a consanguineous Turkish family.

A. Haplotypes at chromosome 1p13.1 co-segregate with recessive SNHL in the family. B. Audiograms revealing severe to profound hearing loss in the affected children and normal hearing in their parents.
Molecular Genetics
SNP genotyping was carried out for all seven sampled relatives using Illumina 1M duo beadchips and assays (Illumina, CA). Genome-wide homozygosity mapping was performed as previously described5. Multipoint linkage analysis of the longest autozygous region was conducted using GeneHunter v2.1 included in the easyLINKAGE Plus v5.086 assuming a fully penetrant autosomal recessive phenotype, with population disease allele frequency 0.0001. SNPs spanning the homozygous region were chosen for linkage analysis based on tagging and heterozygosity in the parents. Allele frequencies were obtained from the CEU HapMap.
All coding exons and flanking sequences of GPSM2 (reference sequence NM_013296.4) were sequenced on an ABI 3730xl, as previously described. Primers and reaction conditions are presented in Supplemental Data (Table S1).
RESULTS
Homozygosity mapping revealed that hearing loss in the family segregated with a 13.6 MB autozygous region on chromosome 1p13.1 (Fig. 1A). Boundaries of the autozygous region were chr1: 102,297,496 (rs2039942) and chr1: 115,895,588 bps (rs12081407) (hg19), with a lod score of 3.0 for linkage of SNHL with the region. Other homozygous regions <1.5 MB in length, considerably shorter than the autozygous segment on chromosome 1 (Supplemental TableS2). The chromosome 1p13.1 autozygous segment harbors GPSM2 at chr1:109,417,972–109,473,044 bps. No other known deafness genes were located within this or the other homozygous regions.
Sequencing of the GPSM2 gene revealed a homozygous mutation c.1684C>T in exon 15, leading to p.Q562X in all three affected individuals (Fig. 2A). All parents were heterozygous for this mutation. GPSM2 p.Q562X is a truncation in the second GoLoco motif of the GPSM2 protein (Fig. 2B). This site was wildtype in 173 hearing controls from the same region of Turkey (Supplemental Figure S1). Analysis of lymphoblast RNA from a heterozygous carrier of p.Q562X indicates that the nonsense allele produces a stable mutant transcript (Supplemental Figure S2).
Figure 2. GPSM2 mutation.
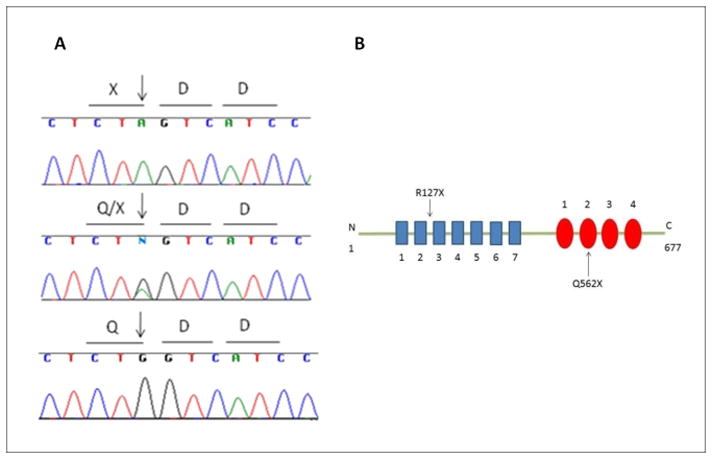
A. Electropherograms of reverse sequence of GPSM2 c.1684C>T (p.Q562X) in an affected child (top), unaffected parent (middle), and unrelated control (bottom). B. GPSM2 has seven tetratricopeptide repeats (TPR) at the amino-terminus (blue boxes) and four Gαi/o-Loco (GoLoco) repeats at the carboxy-terminus (red circles). The previously reported mutation causes a truncation in a TPR whereas p.Q562X causes a truncation in the second GoLoco motif.
DISCUSSION
The second truncating mutation in the GPSM2 gene in a family with SNHL confirms this gene as the cause of DFNB82 hereditary deafness. Both the previously reported family with a mutation in this gene as well as our family had sensorineural, prelingual, bilateral, and severe to profound hearing loss with no additional findings4. In the original report, the causative mutation was GPSM2 p.R127X in an N-terminal tetratricopeptide repeat. The mutation in the family of this study is predicted to cause a more C-terminal truncation in a GoLoco motif.
GPSM2, formerly known as LGN (Leucine-Glycine-Asparagine repeat – enriched protein) and Pins (human homolog of Drosophila, partner of Inscuteable) contains two types of repeats: seven tetratricopeptide repeats (TPR) at the amino-terminus and four Gαi/o-Loco (GoLoco) repeats at the carboxy-terminus4. TPR motifs usually mediate protein-protein interactions7, whereas GoLoco motifs are responsible for association with Gα subunits of heterotrimeric G-proteins8. Interfering with GPSM2 function disrupts cell cycle progression4. GPSM2 plays an important role in spindle pole orientation amd interacts and contributes to the functional activity of Gα proteins9.
GPSM2 also stabilizes the apical complex during neuroblast divisions9. Neuroblasts divide to generate progeny with two different fates. One daughter cell self-renews to maintain the neuroblast pool, whereas the other differentiates to populate the central nervous system. The difference in fate arises from the asymmetric distribution of proteins that specify either self-renewal or differentiation, which is brought about by their polarization into separate apical and basal cortical domains during mitosis. Neuroblast polarization is a dynamic process that results in the formation of two mutually exclusive cortical domains, a basal domain that contains fate determinants that specify differentiation and an apical domain that contains polarity regulatory factors10. Therefore, stabilization of apical complex during neuroblast divisions is crucial. Neuroepithelial progenitors undergo GPSM2-dependent planar divisions to maintain self-renewability during mammalian neurogenesis11. The GPSM2–Gα complex is involved in vesicle trafficking and functions in targeting proteins to dendritic spines12. In the mouse inner ear, the expression of GPSM2 is detected at the apical surfaces of hair and supporting cells of the cochlea, utricle, saccule and cristae in the early stages of development4. Its expression in these tissues decreases in later stages of development suggesting that GPSM2 is crucial for the early development of hearing.
The truncation caused by p.Q562X disrupts the GoLoco motif repeat in the carboxy terminus of GPSM2 and may interfere with the association of GPSM2 with Gα subunits of heterotrimeric G-proteins. In a mouse model of GPSM2 with deletion of the entire C-terminal domain containing all GoLoco motifs, mitotic orientations were randomized within the apical divisions of the dorsolateral brain11. These results indicate that mitotic spindle orientation is controlled by GPSM2 with involvement of GoLoco motifs. The association of deafness with two truncating mutations that truncate the GoLoco motifs of GPSM2 suggests that one mechanism of hearing loss is inadequate cell polarity and hence impaired hair cell transduction in the inner ear.
Supplementary Material
Acknowledgments
This work was supported in part by grants R01DC009645 and R01DC005641 from the National Institute of Deafness and other Communication Disorders of the NIH with a supplement from the American Recovery and Reinvestment Act.
Footnotes
Conflict of interest
The authors have no conflicts of interest to declare.
References
- 1.Dror AA, Avraham KB. Hearing Loss: Mechanisms Revealed by Genetics and Cell Biology. Annu Rev Genet. 2009;43:411–437. doi: 10.1146/annurev-genet-102108-134135. [DOI] [PubMed] [Google Scholar]
- 2.Morton CC, Nance WE. Newborn hearing screening - a silent revolution. N Engl J Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 3.Hilgert N, Smith RJH, Van Camp G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat Res. 2009;681(2–3):189–196. doi: 10.1016/j.mrrev.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh T, Shahin H, Elkan-Miller T, et al. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet. 2010;87(1):90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sirmaci A, Erbek S, Price J, et al. A truncating mutation in SERPINB6 is associated with autosomal-recessive nonsyndromic sensorineural hearing loss. Am J Hum Genet. 2010;86(5):797–804. doi: 10.1016/j.ajhg.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindner TH, Hoffmann K. easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21(3):405–407. doi: 10.1093/bioinformatics/bti009. [DOI] [PubMed] [Google Scholar]
- 7.Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 8.Siderovski DP, Diverse-Pierluissi M, De Vries L. TheGoLoco motif: a Galphai/o binding motif and potential guanine nucleotide exchange factor. Trends Biochem Sci. 1999;24:340–341. doi: 10.1016/s0968-0004(99)01441-3. [DOI] [PubMed] [Google Scholar]
- 9.Yasumi M, Sakisaka T, Hoshino T, et al. Direct binding of Lgl2 to LGN during mitosis and its requirement for normal cell division. J Biol Chem. 2005;280(8):6761–6765. doi: 10.1074/jbc.C400440200. [DOI] [PubMed] [Google Scholar]
- 10.Prehoda KE. Polarization of Drosophila Neuroblasts During Asymmetric Division. Cold Spring Harb Perspect Biol. 2009 Aug;1(2):1–12. doi: 10.1101/cshperspect.a001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konno D, Shioi G, Shitamukai A, et al. Neuroepithelial progenitors undergo LGN-dependent planar divisions to maintain self-renewability during mammalian neurogenesis. Nat Cell Biol. 2008;10(1):93–101. doi: 10.1038/ncb1673. [DOI] [PubMed] [Google Scholar]
- 12.Knoblich JA. Pins for spines. Nat Cell Biol. 2005;7(12):1157–1158. doi: 10.1038/ncb1205-1057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.