

Summary
Autism spectrum disorders (ASD) are a genetically and phenotypically heterogeneous group of syndromes defined by fundamental impairments in social reciprocity and language development accompanied by highly restrictive interests and/or repetitive behaviors. Recent advances in genetics, genomics, developmental neurobiology, systems biology, monogenic neurodevelopment syndromes, and induced pluripotent stem cells (iPSC) are now offering remarkable insights into their etiologies and converging to provide a clear and immediate path forward from the bench to the bedside.
Leading the charge has been the emergence of reliable genetic findings. Multiple studies have confirmed that rare and de novo point mutations and submicroscopic variations in chromosomal structure contribute to a considerable number of cases and have identified a growing number of specific genomic intervals and genes conferring risks (1–9). These advances provide a critical foundation for the development of a more refined understanding of the biological underpinnings of ASD. The earliest findings in the genetics of idiopathic (nonsyndromic) autism highlighted the role of synaptic adhesion molecules and postsynaptic density proteins (1, 10). A set of newly discovered ASD proteins (see the figure) expands this view, highlighting a role for chromatin modifiers (CHD8), and DNA binding proteins (POGZ), ion channels (SCN2A), microtubule-associated proteins (KATNAL2), neurotransmitter receptors (GRIN2B), and phosphorylation-regulated tyrosine kinases (DYRK1A) (5–9).
Figure 1. Developmental neurobiology of ASD risk genes.
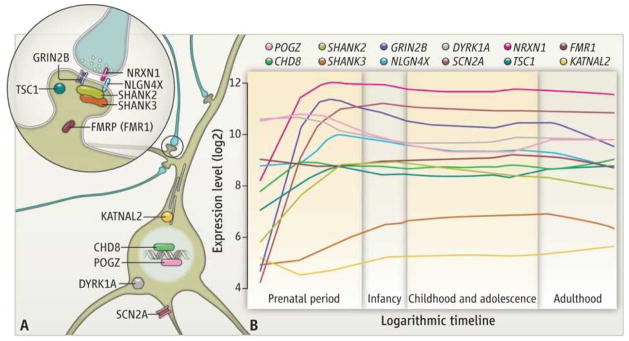
(A) Diverse subcellular distribution and pleiotropic roles for syndromic and idiopathic ASD proteins. (B) Expression profiles of select previously established and newly identified ASD genes during development of the human neocortex. See supplementary materials for methods.
As success in discovery has accelerated, the number of genes predicted to carry risk for ASD has steadily increased, now reaching well into the hundreds (5–9), with no single locus accounting for more than 1% of cases. This “many-to-one” relationship suggests that future studies need to go beyond isolated molecular dissections of single mutations. Another surprising and conceptually challenging observation from recent genetic studies has been the considerable overlap of risks for distinct disorders. Identical highly penetrant variants in different individuals carry large effects but for a wide range of outcomes, including, but likely not limited to, ASD, epilepsy, intellectual disability, and schizophrenia. This “one-to-many” phenomenon, combined with the biological pleiotropy of genes that have so far been implicated in ASD and the presence of core phenotypes that are distinctly human, such as impairments in language development, make the study of ASD in model organisms especially challenging. While modeling mutations in animals or cell culture will reveal perturbations in conserved biological pathways, the key challenge will be to determine which molecular, cellular, and neural circuit–level phenotypes are particularly relevant for ASD.
Ironically, the staggering degree of locus heterogeneity may hold the key to translating genetic findings into a new generation of treatments. The growing set of newly discovered ASD genes should provide a powerful means to use convergence among molecular pathways and cellular processes to identify relevant biological and therapeutic targets. As gene discovery progresses for schizophrenia, epilepsy, and other neurodevelopmental and neuropsychiatric disorders, the ability to study both convergence and divergence of mechanism across conditions will become increasingly feasible. There is already convincing evidence that molecular pathways converge in ASD (9, 10). What is particularly exciting, however, is the new-found opportunity to address not just the question of which molecules and pathways intersect but to specify when and where these events occur within the developing human brain.
The recent emergence of comprehensive maps of spatiotemporal gene expression (11) in the human brain, the ability to construct similar maps of gene-regulatory interactions and chromatin states, and the availability of a growing list of definitive ASD point mutations sets the stage for a powerful developmentally informed approach to studying ASD biology. For example, genes conferring risk can be assessed for spatially and temporally defined gene coexpression and coregulatory networks during human brain development. The identified networks can be evaluated with respect to molecular, anatomical, and developmental convergence, and mechanistic hypotheses can be generated and tested. Confirmation at the bench can leverage model systems, postmortem human tissue, and iPSC-derived neural cells, including isogenic lines carrying engineered mutations and cells generated from patients carrying the mutation(s) of interest. Overall, this type of integration offers immediate opportunities to define relevant molecular and cellular processes and, as more and more genes are identified, will increasingly point to higher-level neural system properties relevant to ASD.
In fact, the available published data on ASD gene expression in the developing human brain already points to some intriguing etiological hypotheses and plausible explanations for the observation that a single genetic risk variant can lead to highly divergent phenotypes, including autism and schizophrenia. Many of the ASD genes noted above exhibit distinctive spatiotemporal expression patterns in the developing brain, including dramatic increases in the cerebral cortex during mid-gestation (see the figure), a developmental period crucial for the formation of early neural circuits (11, 12). The assembly of related cortical neural circuits progresses in an orderly spatiotemporal pattern such that, in general, neurons in the anterior regions are chronologically older, and form synapses earlier, than neurons in the posterior regions. Indeed, signs of early cortical neuronal differentiation and synapse formation are present in the regions of the early and mid-fetal prefrontal and temporal cortices (11, 12) that ultimately give rise to circuits underlying executive control, social affective processing, and language—all functions that are altered in ASD (13). These early regional differences in the timing of synaptogenesis may help explain why the ontogenetically older circuits involved in these processes are particularly vulnerable in ASD, whereas other cortical processes, such as vision, are less affected. The rarity and functional immaturity of nascent synapses in the early and mid-fetal cortex may make them especially susceptible to perturbed function of ASD-related genes, which appear to be dynamically regulated during the same developmental window.
Several lines of evidence indicate that cortical areas and their circuits mature at different rates during postnatal development (14). Higher-order association cortices, including prefrontal and temporal cortices, do not fully mature until late adolescence and early adulthood (14), and some of their maturational trajectories are altered in ASD and schizophrenia (13, 14). Their extended period of maturation may increase the sensitivity of the ontogenetically older frontal and temporal circuits involved in executive control, social affective processing, and language, to both ongoing alterations in the molecular landscape, and interceding environmental insults. In short, the identical genetic risk could lead to variable phenotypes via early developmental and ongoing functional alterations in temporally defined molecular interactions in neural circuits that show both early vulnerability and an extended period of maturational sensitivity.
Although integrated genetic, molecular, and circuit-level analyses are critical for identifying more refined mechanistic hypotheses, important questions will remain unresolved. For instance, what is the nature of the male predominance in ASD? How and why do particular individuals show resilience in the face of highly penetrant genetic risks? What roles do genetic background, somatic mutation, epigenetic modifications, the microbiome, environmental factors, and stochastic events play in determining the emergence and trajectory of symptoms? Fortunately, as the overall genetic landscape of ASD becomes clearer, these difficult questions appear increasingly tractable.
The most pressing issue for patients, families, and physicians at present is what recent findings portend for prevention and treatment. In this regard, demonstrations that aspects of the phenotypes accompanying monogenic neurodevelopmental syndromes are reversible in model organisms (10) provides promise that key features of human neurodevelopmental disorders involve the contribution of dynamic, and therefore potentially treatable, derangements in neural function. Moreover, although the picture of one mutation leading to a diverse array of disorders is conceptually challenging, the model we have proposed suggests that there may be useful analogies to treatment strategies from other areas of medicine. For example, heart disease and stroke prevention both rely in part on the management of hypertension. It may well be that ASD and schizophrenia will increasingly be thought of in a similar light, reflecting divergent manifestations of a shared pathophysiological liability. This would further underscore the importance of clarifying the nature of the molecular and neural circuit–level perturbations underlying these disorders. It would also suggest that future treatment trials will need to be organized around these shared mechanisms, as opposed to traditional psychiatric diagnostic categories. Finally, while the molecular diversity underlying ASD presents a considerable challenge in the search for convergence, it may well also portend, just as it has in cancer, the development of both more personalized and more effective therapies.
Supplementary Material
Acknowledgments
This work was supported by Overlook International Foundation (M.W.S. and N.S.); the Simons Foundation (M.W.S.); and the National Institutes of Health (U01MH081896 and R01NS054273 to N.S.; R01MH081754, P50MH081756, and RC2MH089956 to M.W.S.).
Contributor Information
Matthew W. State, Email: matthew.state@yale.edu.
Nenad Šestan, Email: nenad.sestan@yale.edu.
References and Notes
- Jamain S, et al. Nat Genet. 2003;34:27. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, et al. Science. 2007;316:445. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari P, et al. Nat Genet. 2007;39:319. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- State MW, Levitt P. Nat Neurosci. 2011;14:1499. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, et al. Nat Genet. 2011;43:585. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, et al. Nature. 2012;485:237. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, et al. Nature. 2012;485:242. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, et al. Nature. 2012;485:246. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, et al. Neuron. 2012;74:285. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Bear MF. Cold Spring Harb Perspect Biol. 2012;4:a009886. doi: 10.1101/cshperspect.a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, et al. Nature. 2011;478:483. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, et al. Cell. 2012;149:899. doi: 10.1016/j.cell.2012.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CA, et al. Cell. 2008;135:396. doi: 10.1016/j.cell.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport JL, Gogtay N. Neuropsychopharmacology. 2008;33:181. doi: 10.1038/sj.npp.1301553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.