

Abstract
Current chemotherapy regimens are comprised mostly of single-target drugs which are often plagued by toxic side effects and resistance development. A pharmacological strategy for circumventing these drawbacks could involve designing multivalent ligands that can modulate multiple targets while avoiding the toxicity of a single-targeted agent. Two attractive targets, histone deacetylase (HDAC) and topoisomerase I (Topo I), are cellular modulators that can broadly arrest cancer proliferation through a range of downstream effects. Both are clinically validated targets with multiple inhibitors in therapeutic use. We describe herein the design and synthesis of dual-acting histone deacetylase-topoisomerase I inhibitors. We also show that these dual-acting agents retain activity against HDAC and Topo I, and potently arrest cancer proliferation.
Current chemotherapeutic options for the treatment of cancer are often plagued by debilitating side effects and off-target toxicities. While other pharmacological options, such as gene or immunotherapies, are attaining increasing viability for researchers, most clinical options still center on traditional small molecule chemotherapy. There is considerable interest in designing novel small molecule agents that retain efficacy, while increasing the specificity toward the target of choice, thereby reducing side effects. While single-target drugs remain a popular design endpoint, there has been a recent surge of interest toward multivalent ligand design. It is thought that these drugs could possess a greater therapeutic advantage, by modulating multiple targets and avoiding the side effects of any single agent. Additionally, multivalent ligands are not expected to face the inherent pharmacokinetic and pharmacodynamics disadvantages of administering two or more separate drugs, a common liability that may complicate the outcome of traditional combination therapy.1 The benefit of drugs with multiple targets relative to the conventional combination therapies has only begun to be elucidated, and these therapies are becoming increasingly common across a variety of pharmacological applications.1-5
Cancer offers a unique opportunity for the design of a multifunctional drug due to the multiple pathways contributing to the disease state. One promising pathway for tumor growth inhibition is that of epigenetic and protein acetylation state modulation by histone deacetylases (HDACs). HDACs function within a pathway that was originally discovered to alter the acetylation of histone proteins, leading to a more condensed nucleosome and decreased transcription.6,7 The counterpart enzyme, histone acetyltransferase (HAT), has the opposite effects; acetylating histones and upregulating transcription.8 The proposed cancer-promoting mechanism of HDAC involves transcriptional silencing of tumor suppressors via deacetylation of nucleosomes containing tumor suppressor genes.9,10 However, recent evidence has shown HDAC involvement in the deacetylation of important non-histone regulatory proteins such as p53,11 E2F,12 and tubulin.13 HDACs inhibitors (HDACi) have been shown to cause growth arrest, differentiation, and apoptosis in cancer cells.14-16 Two HDACi, SAHA (Vorinostat) (Figure 1) and FK-228 (Romidepsin), have been approved by the FDA for the treatment of cutaneous T-cell lymphoma,17,18 thus opening the door for HDACi as viable therapeutic agents.19,20 For these reasons, HDACs remain an attractive target for small molecule inhibition.
Figure 1.

Representative HDAC and Topo I inhibitors.
Another proven anticancer target is topoisomerase I (Topo I). The Topo I enzyme relieves the torsional strain on DNA during DNA replication by cutting one strand of the DNA double helix and passing one strand over the other.21,22 Due to the inherent need for rapid replication in cancer, inhibitors of topoisomerases result in DNA strand breaks, cell cycle arrest, and apoptosis.23-27 Many small molecule inhibitors of Topo I have proven clinically effective and are currently FDA-approved for cancer chemotherapy.25 Since both HDAC and Topo I enzymes are localized to the nucleus, the opportunity for dual inhibition from a single agent is a promising possibility. Creating a dual-acting HDAC-Topo I inhibitor could prove beneficial for many reasons. First, HDACi have been shown to act synergistically with Topo I inhibitors, resulting in enhanced apoptosis in cancer.28 Also, since both enzymes are nuclear-localized, dual-acting agents may have better therapeutic indices.
Using fused-frameworks design approach,1 we have previously, described dual acting agents derived from an anthracycline, a topoisomerase II (Topo II) inhibitor and SAHA analogs, prototypical HDACi. A subset of these dual acting HDAC-Topo II inhibitors inhibited Topo II and HDAC activities more potently compared to parent anthracycline and SAHA respectively.29 Furthermore, a lead compound from this series was equipotent to daunorubin against selected breast, lung and prostate cancer cell lines. As a follow-up to our work on dual-acting HDAC-Topo II inhibitors, we have designed and synthesized dual-acting HDAC-Topo I inhibitors derived from the camptothecin ring system and the linker region of SAHA-like HDACi. We show here that an alternative designed multiple ligand approach, merged-frameworks strategy,1b proved successful in the design of HDAC-Topo I inhibitors. We present evidence here that these compounds retain inhibitory activities against both target enzymes and inhibit the proliferation of selected cancer cell lines.
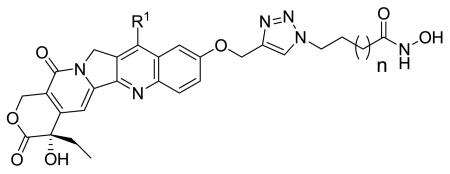
The camptothecin family of Topo I inhibitors are potent anticancer drugs that form a ternary complex at the interface of the cleavage complex, inhibiting dissociation of Topo I from DNA. We chose 10-hydroxycamptothecin and 7-ethyl-10-hydroxycamptothecin (SN-38) (Figure 1) as the Topo I inhibiting templates for the design of the proposed dualacting HDAC-Topo I inhibitors due to their promising activity against a range of tumor types and the presence of a functionalizable phenolic group at their C-10 position. Also, both templates have demonstrated more potency and less toxicity than camptothecin.30-32 From structure-activity relationship (SAR) studies on camptothecins, substitution at the 10-hydroxy group has been found to be tolerable,33 so we used this position as the point of attachment for the HDACi moiety. We have already reported the suitability of 1,2,3-triazole ring as a surface recognition cap group-linking moiety in SAHA-like HDAC inhibitors.34 These studies showed cap group-dependent preference for five to six methylene linkers. In the designed dual-acting compounds, the linker region of SAHA-like HDACi is coupled through a triazole moiety to the camptothecin template, which in turn is anticipated to act as an aromatic surface recognition cap group essential for HDAC inhibition while also retaining its Topo I inhibition activity (Figure 2). We introduced variations into the linker region to test the linker length-dependent potency of the resulting dual-acting agents. Additionally, incorporation of the triazole ring into compound design helped to simplify synthesis and SAR studies.
Figure 2.

Designed dual-acting HDAC-Topo I inhibitors
The reaction route to all the designed compounds is shown in Scheme 1. The phenolic OH-group of 7-ethyl-10-hydroxycamptothecin 1a and 10-hydroxycamptothecin 1b was alkylated with propargyl bromide to yield the corresponding alkyne intermediates 2a and 2b, respectively. Cu-catalyzed Huisgen cycloaddition35 with known azido intermediates 3a-e34,36 afforded trityl-protected compounds 4a-h. Subsequent TFA deprotection of 4a-h yielded the desired compounds 5a-h in good yields with minimal purification required.
Scheme 1.

Synthesis of dual acting HDAC-Topo I inhibitors. Reagents and conditions: (i) propargyl bromide, K2CO3, DMSO, rt, 48 h (ii) 3a-e, CuI, THF:DMSO:Hunig’s base 10:1:0.1 (iii) TFA, thioanisole, CH2Cl2, 0 °C.
Building on our previous observations about the linker length-dependent potency of aryltriazolyl HDACi,34, 36 we first synthesized and evaluated the anti-HDAC activity of 7-ethylcamptothecin-derived compounds 5a-e against HeLa cell nuclear extract HDACs as described previously with a slight modification.34 Briefly, camptothecin has a fluorescence emission (excitation λ = 370 nm, emission λ = 434 nm) close to the wavelength (460 nm) of the fluorescence generated by the HDAC enzyme cleavage of its fluorogenic substrate. To circumvent this potential interference, controls containing the same concentration of the test compound without the enzyme were used, and the background fluorescence of these controls were subtracted from the experimental fluorescence readings. Compound 5a, an analog with a three methylene linker separating the triazole ring and the hydroxamate moiety, has no measurable anti-HDAC activity at concentrations as high as 10 μM (Table 1). The inactivity of 5a may be due to the fact that its linker region is too short to effectively position its hydroxamate moiety within the active site while maintaining the crucial surface residue contacts. Conversely, compounds 5b-e displayed linker length-dependent HDAC inhibitory activities with compound 5d, analog with six methylene linkers, having inhibition activities comparable to SAHA (Table 1). The anti-HDAC activities of these compounds followed linker length-dependence similar to what we observed for other aryltriazolyl HDACi.34,36 This result suggests that the camptothecin ring could function as a cap group, facilitating HDAC inhibition perhaps through interactions with the enzyme surface residues.
Table 1.
In vitro HDAC inhibition activity of novel HDAC-Topo I inhibitors.
| Compound | n | R1 | HeLaa IC50 (nM) |
HDAC 1b IC50 (nM) |
HDAC 6b IC50 (nM) |
HDAC 8b IC50 (nM) |
|---|---|---|---|---|---|---|
| 5a | 1 | -CH2CH3 | N.D. | N.D. | 85 ± 34 | 1726 ± 577 |
| 5b | 2 | -CH2CH3 | 155.4 | NT | NT | NT |
| 5c | 3 | -CH2CH3 | 120.7 | 129 ± 33 | 42 ± 6 | N.D. |
| 5d | 4 | -CH2CH3 | 64.65 | 50 ± 7 | 36 ± 5 | N.D. |
| 5e | 5 | -CH2CH3 | 212.3 | 369 ± 111 | 75 ± 34 | 2599 ± 475 |
| 5f | 2 | -H | 144.5 | 116 ± 40 | 260 ± 40 | N.D. |
| 5g | 3 | -H | 112.2 | N.T. | N.T. | N.T. |
| 5h | 4 | -H | 56.2 | 37 ± 7 | 81 ± 26 | 1046 ± 316 |
| SN-38 | - | - | N.D. | N.T. | N.T. | N.T. |
| SAHA | - | - | 65.0 | 38 ± 2 | 27 ± 2 | 1989 ± 156 |
N.D. – Nondeterminable within tested range, 1 nM – 10 μM; N. T – Not tested.
HeLa nuclear extract. Each value is obtained from three independent experiments.
Data obtained through contract arrangement with BPS Bioscience (San Diego, USA; www.bpsbioscience.com). Assays were performed in duplicates at each concentration and data reported with standard error.29, 34c
In order to elucidate the contribution of the ethyl group at the C-7 of 7-ethylcamptothecin on HDAC inhibition activity, we synthesized camptothecin-derived compounds 5f-h, analogs with four, five and six methylene linkers, respectively. Our choice of this linker range is based on the forgoing observation that this range conferred the optimum activity to the 7-ethylcamptothecin derived compounds 5b-d. A comparison of the anti-HDAC activities of 5b-d and 5f-h, against the HeLa cell nuclear extract HDACs, reveals that pairs with the same linker length have nearly identical HDAC inhibition activity (Table 1). These results suggest that the presence or lack thereof of the ethyl group at the C-7 of camptothecin ring system has no significant effect on the inhibition of HeLa cell nuclear extract HDACs. As expected, the standard Topo I inhibitor- SN-38 -had no measurable HDAC inhibition activity.
To obtain additional evidence for the specific mode of HDAC inhibition, isoform selectivity was investigated by testing selected compounds against purified HDAC1, HDAC6, and HDAC8. The pattern of the anti-HDAC activities of these compounds against HDAC 1 and HDAC 6 mirrored what was observed for the HeLa cell nuclear extract HDACs with one exception. Specifically, the three methylene-linked compound 5a is inactive against HDAC 1 while it maintains low nanomolar and micromolar IC50’s against HDAC 6 and HDAC 8, respectively (Table 1). In general, these dual-acting agents are more selective for HDAC 6 with modest or no activity against HDAC 8. The preference for HDAC6 over HDAC1 could further explain the inactivity of 5a against HeLa nuclear extract, which is a rich source of HDACs 1 and 2.37
We performed a cell-free DNA plasmid relaxation assay, according to a literature protocol, in order to determine the Topo I inhibition activity of these HDAC-Topo I inhibitors.38, 39 In this assay, a supercoiled plasmid is incubated with Topo I in the presence or absence of Topo I inhibitors. Reactions are terminated by addition of SDS, which denatures Topo I. Reaction mixtures are then electrophoresed in an agarose gel and DNA is visualized using a nucleic acid dye. Stabilized cleavage complexes that are covalently bound to DNA will inhibit migration of DNA in the gel significantly more, relative to unbound, relaxed DNA. SN-38 was used as a positive control for Topo I inhibition and drugs were dosed at 50 μM. The 7-ethylcamptothecin compounds 5a-e inhibited Topo I, evidenced by both the reduction of relaxed plasmid and increase in nicked plasmid compared to uninhibited Topo I, with no apparent drop in activity compared to SN-38 (Figure 3a). Similarly, camptothecin compounds 5f-h inhibited Topo I with similar activities to each other, but are less active relative to SN-38 (Figure 3b). The enhanced Topo I inhibitory activities of 5a-e relative to 5f-h is not unexpected as SN-38, the template for 5a-e, is a more potent Topo I inhibitor compared to 10-hydroxycamptothecin, the template for 5f-h.40,41 These results, taken together with the HDAC inhibition data, showed that these dual-acting HDAC-Topo I inhibitors could function to inhibit either target enzyme and merging of the two inhibiting moieties does not preclude the activities of either parent compound significantly.
Figure 3.

Topoisomerase I-Induced Plasmid Relaxation Assay (a) (Lane 1) PBR322 Plasmid DNA, (Lane 2) DNA and Topo I, (Lanes 3-8) DNA, Topo I, and 50 μM: (3) SN-38, (4) 5a, (5) 5b, (6) 5c, (7) 5d, (8) 5e. (b) (Lane 1) PBR322 Plasmid DNA, (Lane 2) DNA and Topo I, (Lanes 3-6) DNA, Topo I, and 50 μM: (3) SN-38, (4) 5f, (5) 5g, (6) 5h.
To examine the effects of these dual-acting inhibitors on cancer cell proliferation, they were screened against the DU-145 prostate cancer cell line and the inhibition of cell viability was measured. SN-38 and SAHA were used as positive controls with SN-38 potently inhibiting DU-145 viability in the mid-nanomolar range, while SAHA’s IC50 was higher, in the low micromolar range. The bifunctional compounds 5a-h showed linker length dependent anti-proliferative activities with a five methylene linker proving to be optimum for cytotoxicity among the 7-ethylcamptothecin compounds (Table 2, comparing 5a-e). Compound 5a, with the shortest linker of three methylenes, possessed the least potent activity against DU-145. Conversely, the camptothecin compounds 5f-h displayed indistinguishable cytotoxic activity. Comparatively, most of the 7-ethylcamptothecin compounds are generally less active than their camptothecin congeners. One exception is compound 5c which showed cytotoxicity activity that is identical to that of 5g, its direct camptothecin analog. More importantly, the micromolar IC50 values suggest that HDAC inhibition may be the dominating mode of antiproliferative activities of compounds 5a-h.
Table 2.
Whole cell cytotoxicity activity against DU-145 prostate cancer cells, as determined by MTS assay after 72 hours. All values are mean of two experiments performed in triplicate as measured by the MTS assay (Promega).
| Compound | n | R1 | IC50 (μM) |
|---|---|---|---|
| 5a | 1 | -CH2CH3 | 6.27 |
| 5b | 2 | -CH2CH3 | 4.25 |
| 5c | 3 | -CH2CH3 | 2.05 |
| 5d | 4 | -CH2CH3 | 3.11 |
| 5e | 5 | -CH2CH3 | 3.51 |
| 5f | 2 | -H | 2.50 |
| 5g | 3 | -H | 1.95 |
| 5h | 4 | -H | 2.03 |
| SN-38 | - | - | 0.11 |
| SAHA | - | - | 2.12 |
Since the anticancer activities displayed by these dual-acting HDAC-Topo I inhibitors against DU-145 appeared to be largely driven by HDACi-based mechanisms, we profiled the contribution of intracellular HDAC inhibition through the level of p21waf1 expression.34b Compounds 5c and 5g were used, premised on the fact that they are representative examples from each of the two Topo I inhibiting templates with identical linker lengths and anticancer activities. Inhibitors were dosed at concentrations near the determined IC50’s in DU-145 (Table 2) and p21waf1 expression was probed via immunoblot (Figure 4). Equivalent protein loading was demonstrated using an anti-actin antibody (Figure 4, top). Both SAHA and SN-38 resulted in marked upregulation of p21waf1 expression levels with 24 hour treatment (Figure 4, bottom, lanes 2-5). Gratifyingly, we observed that the dual-acting compounds 5c and 5g resulted in substantial upregulation of p21waf1 expression in a concentration dependent manner with 5g causing upregulation at levels comparable to SN-38 (Figure 4, bottom panel, lanes 6-9). These results suggest that compounds 5c and 5g derived their cytotoxic activity, in part, through HDAC inhibition. It is unclear at present how much of the p21waf1-dependent anticancer activity is contributed by each inhibiting moiety as both SAHA and SN-38 significantly increased p21waf1 expression. Subsequent investigation into the expression levels of other cellular markers could clarify the driving force behind the cellular effects observed.
Figure 4.

Western blot probing for actin and p21 in the DU-145 cell line. Lanes: (1) Control, (2) SAHA, 2.5 μM, (3) SAHA 5.0 μM, (4) SN-38, 0.1 μM, (5) SN-38 0.5 μM, (6) 5c, 2.5 μM, (7) 5c, 5.0 μM, (8) 5g, 2.5 μM, (9) 5g, 5.0 μM.
A new class of dual-acting HDAC-Topo I inhibitors has been created from camptothecin and SAHA-like templates. Two types of camptothecin templates were used and both were connected through their 10-hydroxy moieties to alklyltriazolyl hydroxamates that we have shown possess enhanced HDAC inhibition activity.34 Results from cell-free and whole cell studies showed that these compounds possess inhibition activities against both target enzymes and inhibit the growth of DU-145 prostate carcinoma cells. Relative to the camptothecin standard SN-38, the functionalization of the 10-hydroxy moiety presented no observable deleterious effect on the Topo I inhibition by 7-ethyl-10-hydroxycamptothecin-derived conjugates 5a-e and only minor attenuation in the inhibitory activities of 10-hydroxycamptothecin-derived conjugates 5f-h at the concentration tested (50 μM). Despite their potent Topo I inhibition activities in cell-free DNA plasmid relaxation assays, these compounds displayed anticancer activities against DU-145 cells at levels more comparable to the HDACi standard SAHA. One plausible explanation for this observation is that the functionalization of the 10-hydroxy moiety may negatively impact the binding of these conjugates to Topo I as crystallographic evidence suggests that the 10-hydroxy group is involved in a hydrogen bonding interaction with a water molecule oxygen at the Topo I active site.42 Alternatively, the ability of these conjugates to interact with other tumor growth-inhibiting secondary targets of camptothecins43,44 may be compromised.
Overall, these compounds show promise as potent anticancer agents with the potential to broadly arrest tumor growth by inhibiting two essential enzymes. Ongoing efforts in our laboratory are on the design of a second generation conjugates which retain the 10-hydroxy moiety of camptothecinin order to better understand the mechanism of antiproliferative activity of this class of compounds.
Supplementary Material
Acknowledgements
This work was financially supported by NIH Grant R01CA131217. W.G. and J.C.C. are recipients of the GAANN predoctoral fellowship from the Georgia Tech Center for Drug Design, Development, and Delivery. R.H. is a Beckman Undergraduate Research Fellow.
Abbreviations
- HDAC
Histone deacetylase
- HAT
Histone acetyltransferase
- HDACi
Histone deacetylase inhibitors
- SAHA
Suberoylanilide hydroxamic acid
- TSA
Trichostatin A
- Topo I
Topoisomerase I class of enzymes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Morphy R, Kay C, Rankovic Z. Drug Discov. Today. 2004;9:641. doi: 10.1016/S1359-6446(04)03163-0. [DOI] [PubMed] [Google Scholar]; (b) Morphy R, Rankovic Z. J. Med. Chem [Google Scholar]; (c) Gangjee A, Lin X, Kisliuk RL, McGuire JJ. J. Med. Chem. 2005;48:7215. doi: 10.1021/jm058234m. [DOI] [PubMed] [Google Scholar]
- 2.Kerr DJ, La Thangue NB. Ann. Oncol. 2004;15:1727. doi: 10.1093/annonc/mdh480. [DOI] [PubMed] [Google Scholar]
- 3.Zhou J, Goh BC, Albert DH, Chen CS. J. Hematol. Oncol. 2009;2:33. doi: 10.1186/1756-8722-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KWH, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ, Heinrich MC, Cherrington JM. Blood. 2003;101:3597. doi: 10.1182/blood-2002-07-2307. [DOI] [PubMed] [Google Scholar]
- 5.Kogen H, Toda N, Tago K, Marumoto S, Takami K, Ori M, Yamada N, Koyama K, Naruto S, Abe K, Yamazaki R, Hara T, Aoyagi A, Abe Y, Kaneko T. Org. Lett. 2002;4:3359. doi: 10.1021/ol026418e. [DOI] [PubMed] [Google Scholar]
- 6.Davie JR, Hendzel MJ. J. Cell. Biochem. 1994;55:98. doi: 10.1002/jcb.240550112. [DOI] [PubMed] [Google Scholar]
- 7.Wolffe AP. Science. 1996;272(5260):371. doi: 10.1126/science.272.5260.371. [DOI] [PubMed] [Google Scholar]
- 8.Braunstein M, Rose AB, Holmes SG, Allis CD, Broach JR. Genes Dev. 1993:592. doi: 10.1101/gad.7.4.592. [DOI] [PubMed] [Google Scholar]
- 9.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, Kim CW, Kim KW. Nat. Med. 2001;7:437. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 10.Cress WD, Seto E. J. Cell. Physiol. 2000;184:1. doi: 10.1002/(SICI)1097-4652(200007)184:1<1::AID-JCP1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 11.Gu W, Roeder RG. Cell. 1997;90:595. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 12.Marzio G, Wagener C, Gutierrez MI, Cartwright P, Helin K, Giacca M. J. Biol. Chem. 2000;275:10887. doi: 10.1074/jbc.275.15.10887. [DOI] [PubMed] [Google Scholar]
- 13.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. Nature. 2002;417:455. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 14.Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. Proc. Natl. Acad. Sci. USA. 1999;96:4592. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glick RD, Swindemen SL, Coffey DC, Rifkind RA, Marks PA, Richon VM, La Quaglia MP. Cancer Res. 1999;59:4392. [PubMed] [Google Scholar]
- 16.Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA, Richon VM. Cancer Res. 2000;60:5165. [PubMed] [Google Scholar]
- 17. http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm094952.htm.
- 18. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2009/ucm189629.htm.
- 19.Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, Lin TS, Liu S, Sklenar AR, Davis ME, Lucas DM, Fischer B, Shank R, Tejaswi SL, Binkley P, Wright J, Chan KK, Grever MR. Blood. 2005;105:959. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- 20.Gryder BE, Sodji QH, Oyelere AK. Future Med. Chem. 2012;4:505. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang JC. Annu. Rev. Biochem. 1985;54:665. doi: 10.1146/annurev.bi.54.070185.003313. [DOI] [PubMed] [Google Scholar]
- 22.Champoux JJ. Annu. Rev. Biochem. 2001;70:369. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 23.Chen AY, Liu LF. Annu. Rev. Pharmacol. Toxicol. 1994;34:191. doi: 10.1146/annurev.pa.34.040194.001203. [DOI] [PubMed] [Google Scholar]
- 24.Froelich-Ammon SJ, Osheroff N. J. Biol. Chem. 1995;270:21429. doi: 10.1074/jbc.270.37.21429. [DOI] [PubMed] [Google Scholar]
- 25.Pommier Y. Chem. Rev. 2009;109:2894. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsiang YH, Lihou MG, Liu LF. Cancer Res. 1989;49:5077. [PubMed] [Google Scholar]
- 27.Matsukawa Y, Marui N, Sakai T, Satomi Y, Yoshida M, Matsumoto K, Nishino H, Aoike A. Cancer Res. 1993;53:1328. [PubMed] [Google Scholar]
- 28.Bevins RL, Zimmer SG. Cancer Res. 2005;65(15):6957. doi: 10.1158/0008-5472.CAN-05-0836. [DOI] [PubMed] [Google Scholar]
- 29.Guerrant W, Patil V, Canzoneri JC, Oyelere AK. J. Med. Chem. 2012;55:1465. doi: 10.1021/jm200799p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang R, Li Y, Cai Q, Liu T, Sun H, Chambless B. Cancer Chemother. Pharmacol. 1998;41:257. doi: 10.1007/s002800050738. [DOI] [PubMed] [Google Scholar]
- 31.Chen ZS, Furukawa T, Sumizawa T, Ono K, Ueda K, Seto K, Akiyama SI. Mol. Pharmacol. 1999;55:921. [PubMed] [Google Scholar]
- 32.Sugimori M, Ejima A, Ohsuki S, Uoto K, Mitsui I, Matsumoto K, Kawato Y, Yasuoka M, Sato K, Tagawa H, Terasawa H. J. Med. Chem. 1994;37:3033. doi: 10.1021/jm00045a007. [DOI] [PubMed] [Google Scholar]
- 33.(a) Leu YL, Chen CS, Wu YJ, Chern JW. J. Med. Chem. 2008;51:1740. doi: 10.1021/jm701151c. [DOI] [PubMed] [Google Scholar]; (b) Ulukan H, Swaan PW. Drugs. 2002;62:2039. doi: 10.2165/00003495-200262140-00004. [DOI] [PubMed] [Google Scholar]
- 34.(a) Chen PC, Patil V, Guerrant W, Green P, Oyelere AK. Bioorg. Med. Chem. 2008;16:4839. doi: 10.1016/j.bmc.2008.03.050. [DOI] [PubMed] [Google Scholar]; (b) Oyelere AK, Chen PC, Guerrant W, Mwakwari SC, Hood R, Zhang Y, Fan Y. J. Med. Chem. 2009;52:456. doi: 10.1021/jm801128g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mwakwari SC, Guerrant W, Patil V, Khan SI, Tekwani BL, Gurard-Levin ZA, Mrksich M, Oyelere AK. J. Med. Chem. 2010;53:6100. doi: 10.1021/jm100507q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (b) Tornoe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 36.Patil V, Guerrant W, Chen PC, Gryder B, Benicewicz D, Khan S, Tekwani B, Oyelere AK. Bioorg. Med. Chem. 2010;18:415. doi: 10.1016/j.bmc.2009.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.HDAC Fluorimetric Assay/Drug Discovery Kit. AK-500 Manual. Fluorescent Assay System; Enzo Life Sciences; Plymouth Meeting; PA. 2005; L.P.: BIOMOL International; [Google Scholar]
- 38.Madhavaiah C, Verma S. Bioorg. Med. Chem. Lett. 2003;13:923. doi: 10.1016/s0960-894x(02)01068-5. [DOI] [PubMed] [Google Scholar]
- 39.Dexheimer TS, Pommier Y. Nat. Protoc. 2008;3:1736. doi: 10.1038/nprot.2008.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawato Y, Aonuma M, Hirota Y, Kuga H;, Sato K. Cancer Res. 1991;51:4187. [PubMed] [Google Scholar]
- 41.Larsen AK, Gilbert C, Chyzak G, Plisov SY, Naguibneva I, Lavergne O, Lesueur-Ginot L, Bigg DCH. Cancer Res. 2001;61:2161. [PubMed] [Google Scholar]
- 42.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. Proc. Natl. Acad. Sci. USA. 2002;99:15387. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bredholt T, Dimba EAO, Hagland HR, Wergeland L, Skavland J, Fossan KO, Tronstad KJ, Johannessen AC, Vintermyr OK, Gjertsen BT. Mol. Cancer. 2009;8:101. doi: 10.1186/1476-4598-8-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chatterjee D, Schmitz I, Krueger A, Yeung K, Kirchoff S, Krammer PH, Peter ME, Wyche JH, Pantazis P. Cancer Res. 2001;61:7148. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.