

Abstract
In contrast with the very well explored concept of structure–activity relationship, similar studies are missing for the dependency between binding kinetics and compound structure of a protein ligand complex, the structure–kinetic relationship. Here, we present a structure–kinetic relationship study of the cyclin-dependent kinase 8 (CDK8)/cyclin C (CycC) complex. The scaffold moiety of the compounds is anchored in the kinase deep pocket and extended with diverse functional groups toward the hinge region and the front pocket. These variations can cause the compounds to change from fast to slow binding kinetics, resulting in an improved residence time. The flip of the DFG motif (“DMG” in CDK8) to the inactive DFG-out conformation appears to have relatively little influence on the velocity of binding. Hydrogen bonding with the kinase hinge region contributes to the residence time but has less impact than hydrophobic complementarities within the kinase front pocket.
Keywords: kinetic profiling, structure-based drug design
The cyclin-dependent kinase 8/cyclin C (CDK8/CycC) complex is a potent oncogene (1–4), involved in transcription and regulation of transcriptional activity (5–10), linked to epigenetic processes (11), and regarded as an attractive drug target. The recent clinical success of the small molecule inhibitors sorafenib (BAY-43006, Bayer Pharma) and imatinib (STI-571, Novartis Pharma AG) has been attributed to their deep pocket binding mode (12). The “deep pocket” (13) is adjacent to the kinase ATP binding site and accessible in protein kinases by the rearrangement of the DFG motif (a short motif composed of the residues Asp-Phe-Gly near the N-terminal region of the activation loop) from the active (DFG-in) to the inactive state (DFG-out) (13, 14). Binding of a type II compound to such a DFG-out conformation often includes slow binding kinetics (15) with a prolonged “residence time,” which is defined as the period for which a target is occupied by a compound (16). Residence time is currently considered to be a key success factor for compound optimization during drug discovery and perhaps as important as the apparent affinity such as half-inhibitory concentration (IC50) or dissociation constant (Kd) (16, 17). Accordingly, residence time could be a key to providing enhanced potency in vivo (18) and a means to improve the correlation of in vitro and in vivo efficacy of drugs (19). Despite the increased acceptance of the need to understand binding kinetics, the interplay between compound–target interactions and binding kinetics is in general too complex and poorly understood to enable prediction of the essential dynamic properties. The impact of the combination of kinetic data of protein–inhibitor interactions with crystallographic studies has been recognized with pioneer studies such as bovine trypsin in complex with the bovine pancreatic trypsin inhibitor (20). The concept of structure–activity relationships describing the interdependency between binding affinity and compound structure has been well explored in the last decades. However, similar studies describing the dependency between binding kinetics and compound structure, the structure–kinetic relationships (SKRs), are, to date, less well explored. The concept of screening for compounds with slow binding kinetics and a beneficial residence time has been developed recently using a reporter displacement assay allowing the high-throughput quantification of the association rate constant (kon) and the dissociation rate constant (koff) of the compound target binding, enabling the determination of residence time (1/koff) (21, 22). Using this concept, we set out to investigate the features of SKRs in the CDK8/CycC complex. We have already presented structural and kinetic data of a deep pocket binder that binds CDK8/CycC (22, 23). Because the morphology of the ATP binding site is very well conserved in the CDK family, ATP-competitive type I compound binding is rather indiscriminative (24). In this respect, as CDK8/CycC is the only known CDK kinase that provides access to its deep pocket, targeting it in a type II binding mode obviously enables its selective inhibition. This structure–kinetic relationship (SKR) study analyzes compounds with short and long residence times, selected from a screen of the Proteros fragment library (18), which share a scaffold moiety with type II binding features. Additionally, compounds were designed according to the “retro-design” concept (18). In this case the inhibitor scaffold is anchored within the CDK8 deep pocket, whereas the groups directed toward the CDK8 hinge region vary in their nature and size. Surprisingly, our results reveal that the flip from the CDK8/CycC DMG-in conformation, as observed in the CDK8/CycC apo form, to DMG-out, as seen in all complex structures presented in this work, does not detectably influence the residence time. Instead, long residence times are triggered by H-bond formation with the hinge region, whereas hydrophobic complementarities within the CDK8 front pocket have the most obvious impact.
Results and Discussion
Discovery of Slowness: Primary Screen and Detection of Binding Kinetics with Slow koff Rates.
To find further compounds with an elongated residence time similar to the CDK8/CycC/sorafenib complex (22, 23), we performed a primary screen (hit rates, Table S1) of a kinase-focused subset of the Proteros library using the Proteros reporter displacement assay with a reporter based on a type I inhibitor for kinases (21, 22). The library screen was performed according to the previously described method for active p38 alpha (21, 22) and in total 4,921 compounds with different molecular weights (MW) (fragment with MW < 350 g/mol and lead-like compounds with MW > 350 g/mol) were tested. The slow binding behavior of nine lead-like compounds was easily detectable in a high-throughput fashion (Fig. S1) including sorafenib and BIRB796 (Doramapimod, 1-[5-tert-butyl-2-(4-methylphenyl)-3-pyrazolyl]-3-[4-[2-(4-morpholinyl)ethoxy]-1-naphthalenyl]urea) as control substances in agreement with previously reported results (22) and seven hits from different scaffolds. The binding kinetics of the hit compounds 1 and 2 already provided clues for an increased residence time (Fig. S1), as they contain a scaffold moiety, which is also found in the fast binding hit compounds 3, 4, and 5 (Fig. 1, Fig. S1). We endeavored to delineate the switch from fast binding and short residence times (“SRTCs;” below the detection limit < 1.4 min) to slow binding with detectable residence times (> 1.4 min) and to long residence times (“LRTCs”). Within a back-to-front approach (18), the different SKR compounds (Fig. 1) were measured with the reporter displacement assay continuously over 8 h (Table 1) as follows: because the large hydrophobic moiety of the hit compounds, 1-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl (“deep pocket binding moiety,” shaded forest green in Fig. 1) is supposed to make van der Waals interactions with residues of the deep pocket (13), this deep pocket binding moiety was combined with an amine group (compound 6) or a urea group (compound 7). Whereas compound 6 obviously did not interact with CDK8 in the concentrations applied in the assay, compound 7 was detected as the “minimal compound” of this SKR compound series with moderate affinity and fast binding kinetics (Table 1, Fig. 1). Our minimal compound is identical to the deep pocket binding moiety of BIRB796 together with its urea linker (25), which forms hydrophilic contacts with the conserved Glu–Lys ion pair of all kinases involving the αC helix, the “roof” of the deep pocket (26), and the backbone of the CDK8 DMG motif. In the further compound building process the minimal compound was combined with a methyl group (compound 8) or a (3-hydroxy-propyl)-group (compound 9), but affinity and binding kinetics were still comparable to the hit compounds 3 and 4, which combine the minimal compound with a (2-hydroxyethyl)-group and a [2-(morpholine-4-yl)ethyl]-group. A detectable residence time was observed only for compounds with longer extensions: 10 and 11, which combine the newly designed (4-hydroxybutyl)-group or (5-hydroxypentyl)-group or the hit compound 5 with a [3-(morpholine-4-yl)propyl]-group. Obviously, residence time increases with the complexity of the hinge-directed moieties, which is in accordance with literature: hinge interaction of a type II inhibitor is present in most, but not all, available kinase structures (27) and regarded as favorable to stabilize the protein–compound interaction (13). An overview of the kinetic parameters of compounds with detectable residence times is visualized on a kinetic map (Fig. 2). There, the kon value is plotted on the x axis against the koff value and thus the residence time (1/koff, Eq. S4) on the y axis, showing both the affinity (Kd=koff/kon, Eq. S4) and the binding kinetics. Compounds with longer residence times are located below compounds with shorter residence times. Compounds with identical Kd values (Kd=koff/kon, Eq. S4) would be located on the same diagonal line. Compounds with high affinity are located in the lower right part of the kinetic map, whereas compounds with low affinity are located in the upper left part of the kinetic map. The kinetic map shows whether compound affinity improves due to increase of the kon rate or due to decrease of the koff rate. Overall, the improvement of selectivity and binding kinetics is mainly driven by the koff rate. For example, compounds 10 and 11 show a similar kon rate (1.85 × 10−3 s−1 ⋅ µM−1; 3.65 × 10−3s−1 ⋅ µM−1), but the koff rate of 11 (2.9 × 10−4 ⋅ s−1) is 1 order of magnitude lower than that of 10 (2.41 × 10−3 ⋅ s−1), increasing its affinity (Kd = 0.08 µM) by a factor ∼16 (Kd = 1.30 µM). Consequently, the residence time of compound 11 is increased eightfold (57 min versus 7 min). Both compounds 2 and 1 show a similarly high affinity (Kd = 0.01 µM and Kd = 0.03 µM) in combination with a favorable koff rate (8.57 × 10−6 ⋅ s−1 and 1.02 × 10−5 ⋅ s−1), which leads to an optimized residence time.
Fig. 1.
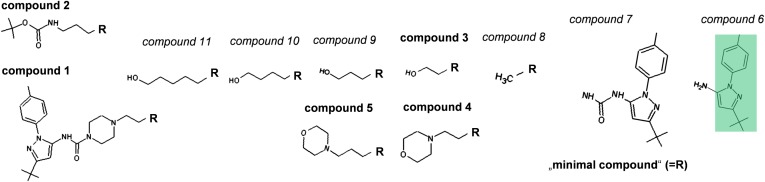
Structures of the relevant compounds used for the SKR study. Within this figure, hit compounds of the library screen are marked with bold letters; newly designed compounds to complete the back-to-front approach are marked with italic letters. The deep pocket binding moiety is shaded forest green. 1-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]amine (compound 6); 1-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]urea (compound 7); 1-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]-3-methylurea (compound 8); 1-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]-3-(2-hydroxyethyl)urea (compound 3); 1-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]-3-[2-(morpholine-4-yl)ethyl]-urea (compound 4); 1-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]-3-(3-hydroxypropyl)urea (compound 9); 1-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]-3-[3-(morpholine-4-yl)propyl]urea (compound 5); 1-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]-3-(4-hydroxybutyl)urea (compound 10); 1-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]-3-(5-hydroxypentyl)urea (compound 11); tert-butyl[3-({[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]carbamoyl}amino)propyl]carbamate (compound 2); N-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]-4-[2-({[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]carbamoyl}amino)ethyl]piperazine-1-carboxamide (compound 1).
Table 1.
Determination of binding kinetics
| ID | Kd, µM | kon, s−1⋅µM−1 | koff, s−1 | Residence time, min | PDB ID | Contact atoms | Quotient* |
| Compounds with a short residence time for CDK8/CycC | |||||||
| 6 | — | n.a. | n.a. | b.d. < 1.4 | — | — | — |
| 7 | 3.24 | a.d. | a.d. | b.d. < 1.4 | 4F6S | 29 | <1.4 |
| 8 | 1.57 | a.d. | a.d. | b.d. < 1.4 | — | — | — |
| 3 | 5.82 | a.d. | a.d. | b.d. < 1.4 | 4F7J | 30 | <1.4 |
| 4 | 1.82 | a.d. | a.d. | b.d. < 1.4 | 4F70 | 32 | <1.4 |
| 9 | 3.53 | a.d. | a.d. | b.d. < 1.4 | — | — | — |
| Compounds with a detectable or long residence time for CDK8/CycC | |||||||
| 10 | 1.30 | 1.85 × 10−3 | 2.41 × 10−3 | 7 | — | — | |
| 5 | 0.70 | 1.68 × 10−3 | 1.18 × 10−3 | 14 | 4F6U | 47 | 0.3 |
| 11 | 0.08 | 3.65 × 10−3 | 2.90 × 10−4 | 57 | 4F7N | 37 | 1.5 |
| 2 | 0.01 | 5.73 × 10−4 | 8.57 × 10−6 | 1,944 | 4F7L | 43 | 45.2 |
| 1 | 0.03 | 2.99 × 10−4 | 1.02 × 10−5 | 1,626 | 4F6W | 58 | 28 |
a.d., above detection limit; b.d. below detection limit; ID, compound-identity; n.a., not applicable.
Quotient was calculated by dividing the residence by the contact atoms.
Fig. 2.

Kinetic map of the compounds with a detectable residence time. Within the kinetic map, the koff rate and thereby the residence time is plotted on the y axis, whereas the kon rate is plotted on the x axis. Compounds with an identical Kd (Kd = koff/kon) lie on the same diagonal line. Arrows indicate that compounds with a low affinity are located in the upper left part of the kinetic map, whereas compounds with a higher affinity are located in the lower right part of the kinetic map. Compounds with a shorter residence time (residence time = 1/koff) are located above compounds with a longer residence time.
Combining Structural with Kinetic Data Reveals That Flip of CDK8 DMG Motif Does Not Influence Kinetic Binding Behavior of Compounds of SKR Series.
The detailed binding modes of the compounds complexed to CDK8/CycC help to explain the observed kinetic data. All compounds were back-soaked into CDK8/CycC crystals cocrystallized with a regular CDK8/CycC DMG-in type I inhibitor (binding kinetics, Table S2) that induces the CDK8 DMG-in conformation. This protocol was chosen to confirm that the DMG-out conformation of CDK8/CycC in the crystals is induced by addition of the SKR compounds. We have also determined the crystal structure of the CDK8/CycC apo form (Tables S3 and S4), which retains the DMG-in conformation after removal of the type I compound by several washing steps (Fig. 3, Fig. S2A). The resulting X-ray structures of CDK8/CycC in complex with compounds with both short and long residence times (Tables S3 and S4) are shown in detailed stereo figures (Fig. S3 A–G), and the most important findings in Fig. 3. For a better overview, the CDK8 residues within the active site were assigned to the specific pockets and kinase regions as follows: (i) the deep pocket and surrounding residues, (ii) the “key” residues of the DMG motif and kinome-wide conserved salt bridge, (iii) the DMG-motif surroundings, (iv) the β1–β2 loop residues, (v) the hinge residues, and (vi) the front pocket and its surroundings.
Fig. 3.

Structure–kinetic relationship. (Top and Middle) Active site of the crystal structure of human CDK8/CycC in complex with the SKR compounds; residues of the CDK8 specific pockets and kinase regions are indicated with different colors, compounds are shown as sticks with their carbons colored yellow. H bonds are shown for compound/CDK8 interaction (blue dotted lines), water molecules that bridge interactions as blue nonbound spheres. Secondary structural elements are labeled black. Positioning of the figures was chosen to allow best view depending on the compound’s binding mode. Residence time and affinity are indicated. CDK8/CycC apo form in the DMG-in conformation: within the CDK8/CycC DMG-in conformation, M174CDK8 is in close contact with F176CDK8 in the deep pocket beyond the CDK8 αC helix. Y32CDK8 side chain and activation segment (185–196) are disordered. Detection of residence time is reconciled in the compounds’ binding mode by H bonding with the hinge region (5 and 11); compounds with a long residence time form contacts within the front pocket (2, 1). Compound 7: M174CDK8 is rotated outside the deep pocket to enable compound binding. Activation segment (178–194) is disordered. Compound 5: activation segment (177–193) is disordered; compound 11: activation segment (177–193) is disordered; compound 2: nearest distance to hinge region 5.1 Å; Y32CDK8 side chain and activation segment (176–195) are disordered; compound 1: activation segment residues 176–195 are disordered. (Bottom) Interactions of compounds in relation to their residence time; the sum of the hydrophobic contacts per atom is plotted on the y axis; compound structures at the respective CDK8 kinase site, Kd values [µM], and residence time [min] are indicated. H bonds are marked in red numbers in the respective regions where they appear. As compound 11 forms no hydrophobic contact with the hinge region but two H bonds, the red label is shaded turquoise. The hydrophobic contacts were calculated as described in the text and SI Text. Parts of the kinase are indicated by color. Residues belonging to the same color are listed next to the plot.
Even though the minimal compound 7 as the basic scaffold moiety has a short residence time (below the detection limit <1.4 min), it induces a flip of the CDK8 DMG motif to the out conformation (Fig. 3, Fig. S3A). As postulated previously, its deep pocket binding moiety is anchored within the CDK8 deep pocket and forms hydrophobic contacts with neighboring key residues of the DMG motif and its surroundings, and Tyr32CDK8 (β1–β2 loop). The urea linker contacts the DMG motif via hydrogen bonding and the essential Glu66CDK8 (αC helix), but does not disrupt the salt bridge of the kinome-wide conserved Glu66CDK8–Lys52CDK8 ion pair. The structure of compound 7 allows no interaction with the CDK8 hinge region. The other SRTCs 3 (Fig. S3B) and 4 (Fig. S3C) are found in a similar binding mode. Thus, the displacement of the T loop does not influence the residence time in an obvious way (below the detection limit <1.4 min). More strikingly, a hinge-directed anchor improves the residence time but does not improve the affinity significantly. Compared with compound 4, the additional methyl group in the hinge-directed moiety of compound 5 enables hinge interaction by the terminal [3-(morpholine-4-yl)propyl]-group (Fig. 3, Fig. S3D, refined 2Fo-Fc omit map, Fig. S2B) via H bonding and results in a detectable residence time (14 min) but comparable binding affinity (Kd: 0.7 µM versus Kd: 1.72 µM). The improvement of the residence time for compound 11 (Fig. 3, Fig. S3E, 57 min) by a factor of 4 relative to compound 5 (14 min) is obviously driven by a second H bond with the hinge region. However, the optimization of residence time for the LRTC 2 (Fig. 3, Fig. S3F, 1,944 min) is not easily explained by an even tighter hinge interaction, as its hinge-directed tert-butyl-propylcarbamate-moiety does not contact the hinge region at all. The tertiary butyl of compound 2 is stabilized in a van der Waals distance of Arg356CDK8 that becomes involved in this front pocket interaction. Similarly, the hinge-directed moiety of the LRTC 1 (Fig. 3, Fig. S3G, refined 2Fo-Fc omit map Fig. S2B, 1,626 min) does not interact with the hinge region. Based on the hydrophobic interaction of the piperazine ring with Tyr32CDK8, the hinge-directed N-[3-tert-butyl-1-(4-methylphenyl)-1H-pyrazol-5-yl]amide extends further into the front pocket and forms extensive contacts with Arg356CDK8 and surrounding residues. However, the different binding mode of compound 1 does not further improve its residence time, which is still in the range of compound 2.
In summary, the binding kinetics of the compounds are neither influenced in an obvious way by the flip to the CDK8/CycC DMG-out kinase conformation nor by the degree of disorder and displacement of the CDK8 T loop or the position of Tyr32CDK8, which normally caps the site of phosphate transfer (28, 29).
Crystal Structures of CDK8/CycC with Compounds with Short, Detectable, and Long Residence Times Indicate That Main Contributions to Residence Time for This Compound Series Are Conserved Hydrogen-Bonding and Hydrophobic Contacts Within CDK8 Front Pocket.
Based on the observation that both SRTCs and LRTCs induce the DMG-out flip even within the CDK8/CycC crystals, we set up a working hypothesis to identify parameters that influence the velocity of compound binding independent from a conformational change. To measure the impact of the SKR compound/CDK8/CycC interaction surface on the optimization of the residence time, the number of contacts within the binding surface was analyzed for each interacting atom of CDK8/CycC (Table 1, Fig. 3). The sum of the binding interactions correlates with the extension of residence time as SRTCs form fewer hydrogen bonds and hydrophobic contacts with CDK8/CycC (compounds 7: 51, 3: 49, and 4: 53; interactions were calculated at a van der Waals distance of 3.5–4 Å ± 0.2 Å, Table S5) than LRTCs (compounds 5: 70, 11: 63, 2: 78, and 1: 99, Table S5). This applies equally to the switch from binding kinetics with a short to a detectable residence time as exemplified for compounds 4 (<1.4 min) and 5 (14 min): compound 5 forms an additional H bond (in sum 4) but also increased hydrophobic contacts (66 compared with 50). Consequently, the two H bonds that are formed additionally (making a total of five) by compound 11 appear to trigger its fourfold optimized residence time. However, the significantly improved residence time of compounds 1 and 2 appears to be independent of additional H bonds. Further H-bond formation involving water molecules was not considered due to experimental uncertainties at the resolution of our analysis (30). It may have a profound influence though.
The impact of the sum of compound/CDK8/CycC contacts on residence time is quantified by the quotient of the residence time divided by the sum of each CDK8 atom interacting with the compound (“residence time per CDK8 atom,” Table 1, Fig. 3). A quotient of 0.3 calculated for the binding kinetics of compound 5 with a detectable residence time (47 CDK8 atoms, 14 min) is increased to 1.5 for compound 11 (37 CDK8 atoms, 57 min) up to a value of 28 and 45.2 for the LRTCs 1 (58 CDK8 atoms, 1,626 min) and 2 (43 CDK8 atoms, 1,944 min), respectively. Apparently, not only the sum of the interacting atoms in CDK8 (as observed for compound 1) but also their properties and location within the kinase favor efficient binding of a compound. The analysis of site-specific CDK8/CycC interaction with the compounds (Table 1, Fig. 3) reveals an optimization of residence time by including further interaction surfaces. Besides the conserved hydrogen bonding, the main van der Waals contacts of the SRTCs (Table 1, Fig. 3: compounds 7, 3, and 4) are formed with the key residues of the DMG motif and the Glu–Lys ion bridge (44%, 43%, 52% of total) and explain the conformational flip of the DMG motif. The different conformations of Tyr32CDK8 (β1–β2 loop) cause a variable fraction of a β1–β2 loop-based van der Waals interaction but do not influence the compound binding kinetics. In contrast, binding kinetics with a detectable residence time requires at least H bonding with the hinge region, whereas the increased ratio of hydrophobic interactions within the front pocket by the LRTCs 2 (14% of total; 1,944 min; Fig. 3) and 1 (30% of total; 1,626 min; Fig. 3) has the most obvious impact on residence time. Nevertheless, H bonding appears to exert a profound influence as the second H bond with the hinge region of compound 11 compensates for the fewer hydrophobic contacts and results in an increased residence time compared with compound 5. As the minimal compound’s binding mode is conserved for all complex structures (Fig. S4), the detectable residence time of compound 10 (7 min), which has not been crystallized, may be explained by the shortened hinge-directed 4-hydroxybutyl group compared with compound 11 and therefore is expected to form one H bond with the hinge region, similar to compound 5 (14 min), and one less than compound 11 (Fig. 3).
In summary, our SKR series suggests that hydrogen bonding with the hinge region is indispensable to evoke a slow koff rate but only large hydrophobic complementarities within the front pocket significantly optimize the residence time of a compound.
Discussion
This SKR study reveals that factors other than conformational change of the CDK8 DMG motif increase the residence time of the CDK8/CycC specific compounds. We observe the standard H bonding between the urea linker of the SKR compounds with CDK8/CycC as reported for DFG-out binders such as BIRB796 (12, 13, 25, 31), but only further H bonding with the hinge region triggers a detectable residence time. This is consistent with literature reporting that solvent-shielded H bonding slows down dissociation (32). Previously, in the case of BIRB796, it was thought that a DFG-out binding inhibitor would cause a slow dissociation rate (25), but we observe short residence times (<1.4 min) for some of our DMG-out binders. Indeed, BIRB796 is also in contact with the hinge region of p38 alpha, which helps to explain its long residence time. As the crystal structure of the CDK8/CycC apo form shows the DMG-in conformation, the observed DMG-out conformation in all inhibitor complexes described in this study is a consequence of compound binding irrespective of their binding kinetics. Conformational flipping appears to require the combination of the deep pocket binding moiety with a urea linker (compound 7). Compound 6, with only an amine linked to the deep pocket binding moiety, did not bind to CDK8/CycC. The “key H bonding” between the urea linker and the αC helix/DMG backbone seems to be essential for this process.
CDK8/CycC specific features may contribute to our results. The effect of the change of Phe to Met within the DFG motif in the activation loop can be evaluated by superimposing p38α (DFG) and CDK8/CycC (DMG) both complexed with sorafenib. No significant difference in the conformation of the DFG/DMG motifs is seen (23). However, the CDK8-specific Phe176CDK8, two residues beyond DMG (Leu in other CDKs), might allow a higher flexibility to the activation loop compared with other members of the CDK family. Within the CDK8/CycC DMG-in conformation, Phe176CDK8 is in close contact with Met174CDK8 in the deep pocket (Fig. 3, Fig. S2). More importantly, the CDK8 kinase is activated via a phosphorylation-independent mechanism (23), which might be one important difference that explains why CDK8 is still the only known member of the CDK family that provides access to its deep pocket, explaining the observation that a type II compound binds to the DMG-out conformation without an increased residence time. The interaction of CDK8 with CycC is likely to be key to this unique feature, as the association of the cyclin with the CDK8 αC helix is expected to stabilize a certain CDK8 kinase conformation in the general CDK activation by cyclins (33, 34). In the complex, the equilibrium between active and inactive kinase conformations may be shifted from which type II binders select the DMG-out conformation. For other kinases the slow binding kinetic signature is believed to derive from conformational selection of the inactive T-loop kinase conformation from a preexisting equilibrium (13).
Independent of CDK8/CycC complex-specific features, we presented evidence that H bonding with the hinge region triggers detectable residence times. Moreover, the quotient of residence time divided by the sum of interacting atoms of CDK8 was shown to be a suitable measure to quantify the optimization of the CDK8/CycC compounds. Previously, “kinetic efficiency” (KE) metrics were described for binding kinetics of compounds without structural information to rank the physiochemical properties of a compound series with similar MW and residence time. The kinetic parameters for the binding interaction per atom of a compound have been described as KE = τ/NHA = t1/2/(0.693 × NHA); where τ is the residence time and NHA is the number of nonhydrogen atoms of a compound (35). Subsequently, the sum of interactions needs to be fine-tuned within the compound/target interaction surface in terms of distinctive kinase sites. In our study, targeting the CDK8 front pocket, where its C-terminal segment gets involved with Arg356CDK8, optimizes the residence time. A kinase-site-dependent increase in affinity was as well reported for type I compounds that bind the CDK front pocket, the so-called “Lys89-pocket” (reviewed by Lolli, ref. 36). Importantly, the C terminus of CDK9 within the CDK9/CycT complex was reported to interact with the CDK9 hinge region and contributes to an increased compound affinity (37). The extension of the residence time by hydrophobic interactions within the hinge region/front pocket appears also to be rationalized in part by kinase dynamics descriptions such as the “hydrophobic spine” model (18, 38). The core of the hydrophobic spine consists of four amino acids that are considered to build a hydrophobic network/spine allowing the kinase “breathing motion” between the N lobe and the C lobe to enable nucleotide binding and release (39). In the case of the CDK/cyclin family, this mechanism is modified as exemplified for the CDK2/CycA complex (38), which positions the CDK loop accurately for the active kinase conformation even in the absence of an activatory T-loop phosphorylation (Fig. S5B) (38, 39). Only in compounds that additionally interact with the hinge region of CDK8 or even more efficiently with the CDK8 front pocket as “anchor point,” the CDK8 T-loop/CycC interaction surface (Fig. S5A) may be destabilized. This possibly results in a fast dissociation rate of SRTCs, whereas a more efficient reduction of the kinase motions could explain the slower dissociation of LRTCs. Analogously, binding of inhibitors changed the internal motion of isolated CDK2 probably by a blocked hinge movement between its N lobe and C lobe (40). Although X-ray crystallography provides only a static model to detect reduced kinase motions, the combination of X-ray structural data and kinetic data of compound binding within this SKR study suggests that residence time may be influenced by hindered kinase motion. This SKR study reveals a behavior of the CDK8/CycC kinase inhibitors that deviates in some aspects from the general paradigm of drug design within the kinase family and adds a promising tool and approach of drug design such as the back-to-front approach. Generalization of our observations to the other kinase families is possible but requires SKR studies, as the parameters that influence the binding kinetics of compounds in those may differ in detail.
Methods
Protein Expression and Purification.
Protein expression and purification of the corresponding CDK8(Hs1-403)/CycC(Hs1-283) constructs was performed according to the previously published protocols (23). Protein crystals were obtained within the hanging drop setup at 20 °C in 20% (wt/vol) polyethylene glycol 3350 and 0.2 M sodium formate. Tested hit compounds and newly designed compounds were synthesized by Vichem Chemie Ltd. with a purity ≥95%.
Protein Crystallography.
For complex formation with CDK8/CycC, compounds were back-soaked into crystals within a concentration range of 1–10 mM from 4 h to 7 d. These crystals had been prepared by co-crystallization with a type I compound (N-(2-phenylethyl)quinazolin-4-amine) that induces the DMG-in conformation. The crystal structure of this complex has also been solved and is deposited (PDB-ID 4F7S). For the CDK8/CycC apo form, the ligand was removed by several washing steps. Crystals were shock-frozen using 25% (vol/vol) ethylene glycol as cryoprotectant. Data sets were taken at Swiss Light Source, Paul Scherer Institute at beamline X06SA. Data were processed using XDS and XSCALE (41) (4F6S, 4F6U, 4F6W, 4F7S, and 4F7L) or XDS and SCALA/Collaborative Computational Project, number 4 (42) (4F70, 4F7J, 4F7N, and 4G6L). Molecular replacement was achieved by MOLREP (43) and refinement cycles were calculated using REFMAC5 (44). Building was performed with COOT (45) and graphical figures were prepared with PYMOL (46).
Kinetic Measurements.
Binding kinetics of the compounds was measured with the Proteros reporter displacement assay (21, 22). A brief description, equations, error estimation, and correlation coefficients are available in SI Text.
Supplementary Material
Acknowledgments
We thank Sabine Höppner, who identified the target, and Gerhard Müller and Timothy Woodcock for discussions before the idea of starting this SKR study was developed. We also thank Allegra Ritscher, Birgit Flicke, Theresa Wolfram, Sonja Lernet, Ursula Heunisch, and others for their introduction to various techniques. We are grateful to Alexander Duschek and Günther Jerg for the analysis of compound masses, Michael Blaesse and Mario Mörtl for their introduction to X-ray programs and support thereof, Konstanze von König for indispensable scientific discussions in the screening field, Russell Thomas for critically reading the manuscript, as well as Torsten Neuefeind for continuous support. This work was financially supported by the Robert Huber Nobelpreisträgerstipendium.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4F6S, 4F7J, 4F70, 4F6U, 4F7N, 4F7L, 4F6W, 4G6L, and 4F7S).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305378110/-/DCSupplemental.
References
- 1.Firestein R, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455(7212):547–551. doi: 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Firestein R, Hahn WC. Revving the Throttle on an oncogene: CDK8 takes the driver seat. Cancer Res. 2009;69(20):7899–7901. doi: 10.1158/0008-5472.CAN-09-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris EJ, et al. E2F1 represses beta-catenin transcription and is antagonized by both pRB and CDK8. Nature. 2008;455(7212):552–556. doi: 10.1038/nature07310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He S-B, et al. Effects of cyclin-dependent kinase 8 specific siRNA on the proliferation and apoptosis of colon cancer cells. J Exp Clin Cancer Res. 2011;30:109. doi: 10.1186/1756-9966-30-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinhero R, Liaw P, Bertens K, Yankulov K. Three cyclin-dependent kinases preferentially phosphorylate different parts of the C-terminal domain of the large subunit of RNA polymerase II. Eur J Biochem. 2004;271(5):1004–1014. doi: 10.1111/j.1432-1033.2004.04002.x. [DOI] [PubMed] [Google Scholar]
- 6.Hengartner CJ, et al. Temporal regulation of RNA polymerase II by Srb10 and Kin28 cyclin-dependent kinases. Mol Cell. 1998;2(1):43–53. doi: 10.1016/s1097-2765(00)80112-4. [DOI] [PubMed] [Google Scholar]
- 7.Akoulitchev S, Chuikov S, Reinberg D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature. 2000;407(6800):102–106. doi: 10.1038/35024111. [DOI] [PubMed] [Google Scholar]
- 8.Donner AJ, Szostek S, Hoover JM, Espinosa JM. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol Cell. 2007;27(1):121–133. doi: 10.1016/j.molcel.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donner AJ, Ebmeier CC, Taatjes DJ, Espinosa JM. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol. 2010;17(2):194–201. doi: 10.1038/nsmb.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galbraith MD, Donner AJ, Espinosa JM. CDK8: A positive regulator of transcription. Transcription. 2010;1(1):4–12. doi: 10.4161/trns.1.1.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kapoor A, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468(7327):1105–1109. doi: 10.1038/nature09590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dietrich J, Hulme C, Hurley LH. The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: A structural analysis of the binding interactions of Gleevec, Nexavar, and BIRB-796. Bioorg Med Chem. 2010;18(15):5738–5748. doi: 10.1016/j.bmc.2010.05.063. [DOI] [PubMed] [Google Scholar]
- 13.Backes AC, Zech B, Felber B, Klebl B, Müller G. Small-molecule inhibitors binding to protein kinase. Part II: The novel pharmacophore approach of type II and type III inhibition. Expert Opin Drug Discov. 2008;3(12):1427–1449. doi: 10.1517/17460440802580106. [DOI] [PubMed] [Google Scholar]
- 14.Zuccotto F, Ardini E, Casale E, Angiolini M. Through the “gatekeeper door”: Exploiting the active kinase conformation. J Med Chem. 2010;53(7):2681–2694. doi: 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
- 15.Kroe RR, et al. Thermal denaturation: A method to rank slow binding, high-affinity P38alpha MAP kinase inhibitors. J Med Chem. 2003;46(22):4669–4675. doi: 10.1021/jm030120s. [DOI] [PubMed] [Google Scholar]
- 16.Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5(9):730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 17.Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008;47(20):5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- 18.Müller G, Sennhenn PC, Woodcock T, Neumann L. The ‘retro-design’ concept for novel kinase inhibitors. IDrugs. 2010;13(7):457–466. [PubMed] [Google Scholar]
- 19.Lu H, Tonge PJ. Drug-target residence time: Critical information for lead optimization. Curr Opin Chem Biol. 2010;14(4):467–474. doi: 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huber R, et al. Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor. II. Crystallographic refinement at 1.9 A resolution. J Mol Biol. 1974;89(1):73–101. doi: 10.1016/0022-2836(74)90163-6. [DOI] [PubMed] [Google Scholar]
- 21.Neumann L, Ritscher A, Müller G, Hafenbradl D. Fragment-based lead generation: Identification of seed fragments by a highly efficient fragment screening technology. J Comput Aided Mol Des. 2009;23(8):501–511. doi: 10.1007/s10822-009-9288-x. [DOI] [PubMed] [Google Scholar]
- 22.Neumann L, von König K, Ullmann D. HTS reporter displacement assay for fragment screening and fragment evolution toward leads with optimized binding kinetics, binding selectivity, and thermodynamic signature. Methods Enzymol. 2011;493:299–320. doi: 10.1016/B978-0-12-381274-2.00012-1. [DOI] [PubMed] [Google Scholar]
- 23.Schneider EV, et al. The structure of CDK8/CycC implicates specificity in the CDK/cyclin family and reveals interaction with a deep pocket binder. J Mol Biol. 2011;412(2):251–266. doi: 10.1016/j.jmb.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 24.Echalier A, Endicott JA, Noble ME. Recent developments in cyclin-dependent kinase biochemical and structural studies. Biochim Biophys Acta. 2010;1804(3):511–519. doi: 10.1016/j.bbapap.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Pargellis C, et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9(4):268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 26.Simard JR, et al. Development of a fluorescent-tagged kinase assay system for the detection and characterization of allosteric kinase inhibitors. J Am Chem Soc. 2009;131(37):13286–13296. doi: 10.1021/ja902010p. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2(7):358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 28.Bossemeyer D, Engh RA, Kinzel V, Ponstingl H, Huber R. Phosphotransferase and substrate binding mechanism of the cAMP-dependent protein kinase catalytic subunit from porcine heart as deduced from the 2.0 A structure of the complex with Mn2+ adenylyl imidodiphosphate and inhibitor peptide PKI(5-24) EMBO J. 1993;12(3):849–859. doi: 10.1002/j.1460-2075.1993.tb05725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109(3):275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 30.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285(5):2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 31.Regan J, et al. Structure-activity relationships of the p38alpha MAP kinase inhibitor 1-(5-tert-butyl-2-p-tolyl-2H-pyrazol-3-yl)-3-[4-(2-morpholin-4-yl-ethoxy)naph- thalen-1-yl]urea (BIRB 796) J Med Chem. 2003;46(22):4676–4686. doi: 10.1021/jm030121k. [DOI] [PubMed] [Google Scholar]
- 32.Schmidtke P, Luque FJ, Murray JB, Barril X. Shielded hydrogen bonds as structural determinants of binding kinetics: Application in drug design. J Am Chem Soc. 2011;133(46):18903–18910. doi: 10.1021/ja207494u. [DOI] [PubMed] [Google Scholar]
- 33.Jeffrey PD, et al. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376(6538):313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 34.Pavletich NP. Mechanisms of cyclin-dependent kinase regulation: Structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol. 1999;287(5):821–828. doi: 10.1006/jmbi.1999.2640. [DOI] [PubMed] [Google Scholar]
- 35.Holdgate GA, Gill AL. Kinetic efficiency: The missing metric for enhancing compound quality? Drug Discov Today. 2011;16(21-22):910–913. doi: 10.1016/j.drudis.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 36.Lolli G. Structural dissection of cyclin dependent kinases regulation and protein recognition properties. Cell Cycle. 2010;9(8):1551–1561. doi: 10.4161/cc.9.8.11195. [DOI] [PubMed] [Google Scholar]
- 37.Baumli S, Endicott JA, Johnson LN. Halogen bonds form the basis for selective P-TEFb inhibition by DRB. Chem Biol. 2010;17(9):931–936. doi: 10.1016/j.chembiol.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 38.Kornev AP, Haste NM, Taylor SS, Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci USA. 2006;103(47):17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noble M, et al. Exploiting structural principles to design cyclin-dependent kinase inhibitors. Biochim Biophys Acta. 2005;1754(1-2):58–64. doi: 10.1016/j.bbapap.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 40.Otyepka M, Kríz Z, Koca J. Dynamics and binding modes of free cdk2 and its two complexes with inhibitors studied by computer simulations. J Biomol Struct Dyn. 2002;20(2):141–154. doi: 10.1080/07391102.2002.10506830. [DOI] [PubMed] [Google Scholar]
- 41.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dodson EJ, Winn M, Ralph A. Collaborative Computational Project, number 4: Providing programs for protein crystallography. Methods Enzymol. 1997;277:620–633. doi: 10.1016/s0076-6879(97)77034-4. [DOI] [PubMed] [Google Scholar]
- 43.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 44.Vagin AA, et al. 2004. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr 60(Pt 12 Pt 1):2184–2195.
- 45.Emsley P, Cowtan K. 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60(Pt 12 Pt 1):2126–2132.
- 46. The PyMOL Molecular Graphics System (Schrödinger, New York), Version 1.2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.