

Abstract
For the emergence of early life, the formation of biopolymers such as RNA is essential. However, the addition of nucleotide monomers to existing oligonucleotides requires millimolar concentrations. Even in such optimistic settings, no polymerization of RNA longer than about 20 bases could be demonstrated. How then could self-replicating ribozymes appear, for which recent experiments suggest a minimal length of 200 nt? Here, we demonstrate a mechanism to bridge this gap: the escalated polymerization of nucleotides by a spatially confined thermal gradient. The gradient accumulates monomers by thermophoresis and convection while retaining longer polymers exponentially better. Polymerization and accumulation become mutually self-enhancing and result in a hyperexponential escalation of polymer length. We describe this escalation theoretically under the conservative assumption of reversible polymerization. Taking into account the separately measured thermophoretic properties of RNA, we extrapolate the results for primordial RNA polymerization inside a temperature gradient in pores or fissures of rocks. With a dilute, nanomolar concentration of monomers the model predicts that a pore length of 5 cm and a temperature difference of 10 K suffice to polymerize 200-mers of RNA in micromolar concentrations. The probability to generate these long RNAs is raised by a factor of >10600 compared with polymerization in a physical equilibrium. We experimentally validate the theory with the reversible polymerization of DNA blocks in a laser-driven thermal trap. The results confirm that a thermal gradient can significantly enlarge the available sequence space for the emergence of catalytically active polymers.
Keywords: molecular evolution, nonequilibrium, RNA world, (nonenzymatic) emergence of RNA, hydrothermal vents
Polymers are the vital building blocks of all known life forms. According to the central dogma of molecular biology (1), DNA stores the information for how and when to build proteins, which for their part carry out catalytic tasks like the polymerization of DNA. How this self-perpetuating cycle has started is unknown. The RNA-world hypothesis posits that RNA molecules were the central players in prebiotic evolution, because they exhibit both a catalytic function similar to that of proteins and the information storage capabilities of DNA (2). However, how could such an RNA world have emerged from the prebiotic soup?
A key element of the RNA world is a ribozyme that catalyzes RNA replication. Directed in vitro evolution and engineering have shown that such ribozymes exist, but require a length of 200 bases or more, even in favorable high-salt conditions (3). Starting from chemical nonequilibrium conditions with millimolar concentrations of energy-rich nucleotides (4–6) and with the help of catalytic surfaces (7), only the formation of much shorter polynucleotides on the order of 20 bases was demonstrated in the laboratory. Slow kinetics and cleavage due to hydrolysis limit the formation of long polynucleotides and finally lead to a length distribution that decays exponentially in the case of reversible polymerization, i.e., RNA constantly being randomly built up and cleaved. A simple estimate shows that to obtain a traceable concentration of 200-mer RNAs, the concentration of nucleotides has to exceed the effective dissociation constant by at least 100-fold (Fig. S1). Plausible concentrations of nucleotides in primordial settings, e.g., a hydrothermal vent or a warm pond, should be at most in the nanomolar to low micromolar range. However, the binding affinity for nucleotide monomer binding to an oligomer is weak, with estimates for the dissociation constant in the millimolar range (4). Even if dissociation constants in the micromolar range can be reached by specifically activated nucleotides, the nonenzymatic formation of 200-mers seems impossible in a setting that is not supported by an additional physical nonequilibrium.
In this work we demonstrate escalated polymerization in a spatially confined thermal gradient as a possible pathway for RNA to overcome this barrier. The physical nonequilibrium condition in the form of an ordinary temperature gradient will lead to exceedingly long polymers, even under the conservative assumption of reversible polymerization. In an elongated convection cell, thermophoresis accumulates macromolecules like DNA and RNA in a length-selective manner (Fig. 1A). We study how such a thermal molecule trap produces a continuing chemical nonequilibrium condition and thereby affects a weak, reversible polymerization reaction based on bonding of monomers and oligomers and random dissociation (Fig. 1B). Using a combination of experimental and theoretical methods, we identify conditions under which the interplay of these mechanisms dramatically increases the length range and efficiency of the polymerization process (Fig. 1C).
Fig. 1.

Proposed polymerization in a thermal gradient: cross section of a water-filled pore that is exposed to the ocean at the top and subjected to a spatially confined horizontal temperature gradient. (A) Thermal trapping. The temperature gradient drives thermal convection (circular arrows) due to the thermal expansion of water at the hot side. Additionally, solved biomolecules move from hot to cold via thermophoresis with a drift speed 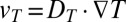 (horizontal arrows). In combination, solved biomolecules are accumulated at the pore bottom, exponentially dependent on their Soret coefficient
(horizontal arrows). In combination, solved biomolecules are accumulated at the pore bottom, exponentially dependent on their Soret coefficient 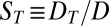 . Because
. Because 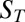 increases with the molecule length, longer molecules are trapped exponentially more efficiently at the optimal trap width. (B) Polymerization. If polymerization and dissociation are in steady state, the concentration of longer polymers decays exponentially with polymer length. The mean length strongly depends on the concentration of monomers and is too short to allow for self-replicating polymers under dilute primordial conditions. (C) Trapping and polymerization. The exponential thermal accumulation can counterbalance the exponentially decaying polymerization. As a result, both the concentration and the mean length of the polymers are enhanced massively.
increases with the molecule length, longer molecules are trapped exponentially more efficiently at the optimal trap width. (B) Polymerization. If polymerization and dissociation are in steady state, the concentration of longer polymers decays exponentially with polymer length. The mean length strongly depends on the concentration of monomers and is too short to allow for self-replicating polymers under dilute primordial conditions. (C) Trapping and polymerization. The exponential thermal accumulation can counterbalance the exponentially decaying polymerization. As a result, both the concentration and the mean length of the polymers are enhanced massively.
A thermal gradient in an elongated compartment arguably was an abundant physical scenario in the prebiotic world. The compartment could be a pore in volcanic rock, a cleft of mud, or a rock fissure with the temperature gradient caused by the vicinity of a warm hydrothermal or volcanic outflow into a colder ocean (8, 9). This could be found in both salty oceanic hydrothermal conditions (10) and the fresh waters near warm water ponds (11). In the laboratory, thermal molecule traps have previously been demonstrated to support accumulation of biomolecules (12, 13), formation of cell-like lipid vesicles (14), and concurrent replication and trapping with a polymerase (15, 16). Additionally, a replication of codon information using tRNA is compatible with the temperature cycling in a thermal molecule trap (17).
This previous work used the thermal trap as a passive length-selective concentration enhancer without biochemical reactions (12, 13) and replicators with fixed product length (14–16). Here, we show how the length selectivity of thermal traps and a linear polymerization reaction with arbitrary product lengths mutually enhance each other by a coupling between physical and chemical nonequilibrium (Fig. 1C): The thermal trap accumulates monomers and thereby pushes the system out of chemical equilibrium. Longer polymers are created due to the concentration-dependent polymerization. The longer polymers are accumulated more efficiently by the length-selective physical nonequilibrium of the thermal trap, which in turn leads to higher local concentrations. The positive feedback loop allows the formation of long RNA polymers even at low nucleotide concentrations. We validate our theory experimentally in the fast polymerizing regime, using a laser-driven thermal trap filled with a solution of double-stranded DNA blocks, which reversibly polymerize at specifically designed sticky ends. The experiment was designed to fully control all parameters of the system. We record this polymerization process in real time and find a quantitative agreement with our theoretical model.
Theory
Thermal Molecule Trap.
To study the interplay between thermal trapping and polymerization, we first describe the dynamics of each process separately and then consider the coupled processes. In a water-filled compartment, vertical gravitation and a horizontal temperature gradient 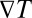 lead to a laminar convective flow due to the thermal expansion of the fluid (Fig. 1A). Additionally, dissolved biomolecules with a thermodiffusion coefficient
lead to a laminar convective flow due to the thermal expansion of the fluid (Fig. 1A). Additionally, dissolved biomolecules with a thermodiffusion coefficient 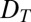 will thermophoretically move along the thermal gradient with a drift speed
will thermophoretically move along the thermal gradient with a drift speed 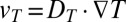 . Charged molecules have a tendency to move toward the cold, a trend that can be understood from local free energy considerations (18–21). In a thermal molecule trap, both effects are combined: The biomolecules are accumulated at the bottom corner of the compartment until a steady state of thermodiffusion and the counteracting diffusion is reached. Such an accumulation effect was first observed in gases by Clusius and Dickel for chlorine isotopes (22).
. Charged molecules have a tendency to move toward the cold, a trend that can be understood from local free energy considerations (18–21). In a thermal molecule trap, both effects are combined: The biomolecules are accumulated at the bottom corner of the compartment until a steady state of thermodiffusion and the counteracting diffusion is reached. Such an accumulation effect was first observed in gases by Clusius and Dickel for chlorine isotopes (22).
We describe the accumulation of molecules inside a thermal trap, using a 2D transport equation of the drift-diffusion type for the concentration 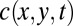 of biomolecules (23),
of biomolecules (23),
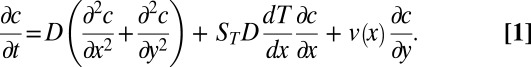 |
Here, x is the horizontal coordinate in Fig. 1A, y is the vertical coordinate, and t denotes time. The three terms on the right account for diffusion, thermodiffusion, and convection. The two latter effects are evoked by the linear temperature gradient 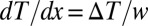 over the horizontal width w of the pore, where
over the horizontal width w of the pore, where  denotes the (fixed) temperature difference between the left and right boundaries. On the one hand, the gradient yields a thermodiffusive flux in the x direction with D the diffusion coefficient and
denotes the (fixed) temperature difference between the left and right boundaries. On the one hand, the gradient yields a thermodiffusive flux in the x direction with D the diffusion coefficient and 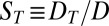 the length-dependent Soret coefficient of the dissolved molecule (18). On the other hand, thermal expansion leads to a vertical convection flow with the velocity profile
the length-dependent Soret coefficient of the dissolved molecule (18). On the other hand, thermal expansion leads to a vertical convection flow with the velocity profile 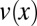 (SI Text A). The characteristic velocity,
(SI Text A). The characteristic velocity, 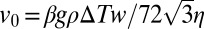 , also depends on the gravitational acceleration g and the density ρ, viscosity η, and thermal expansivity β of the solvent (Fig. S2D).
, also depends on the gravitational acceleration g and the density ρ, viscosity η, and thermal expansivity β of the solvent (Fig. S2D).
Eq. 1 can be solved by separation of variables. In the steady state, the concentration 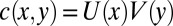 increases exponentially along the vertical direction,
increases exponentially along the vertical direction, 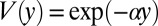 with the accumulation coefficient α fixed by the solution of
with the accumulation coefficient α fixed by the solution of 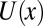 . The detailed calculation (SI Text A) shows that α ∼
. The detailed calculation (SI Text A) shows that α ∼ 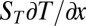 , predicting that the total accumulation in the vertical direction scales exponentially with the Soret coefficient
, predicting that the total accumulation in the vertical direction scales exponentially with the Soret coefficient 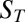 . Because longer DNA or RNA molecules have larger ST, they can be exponentially stronger accumulated in the thermophoretic trap.
. Because longer DNA or RNA molecules have larger ST, they can be exponentially stronger accumulated in the thermophoretic trap.
Polymerization.
We describe the chemical dynamics of a generic polymerization process as a reversible aggregation–fragmentation reaction (24),
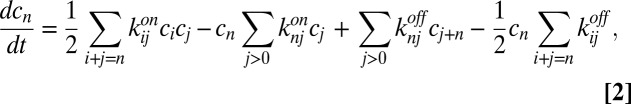 |
for the concentration 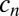 of a polymer with length n. The concentration
of a polymer with length n. The concentration 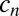 can increase via bonding of two shorter polymers (first term on right-hand side) or via the dissociation of a longer polymer (third term), whereas it can also decrease via bonding (second term) and dissociation (fourth term). Cyclization or formation of nonproductive complexes is not considered here. The dynamics are governed by the on and off rates,
can increase via bonding of two shorter polymers (first term on right-hand side) or via the dissociation of a longer polymer (third term), whereas it can also decrease via bonding (second term) and dissociation (fourth term). Cyclization or formation of nonproductive complexes is not considered here. The dynamics are governed by the on and off rates, 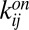 and
and 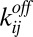 . Under the “worst-case” assumption that these rates satisfy the detailed balance condition (25) (instead of supporting a finite flux driven by a separate chemical activation process), the steady state of Eq. 2 is
. Under the “worst-case” assumption that these rates satisfy the detailed balance condition (25) (instead of supporting a finite flux driven by a separate chemical activation process), the steady state of Eq. 2 is
see SI Text A for details. Via summation over Eq. 3 the concentration of free monomers, 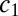 can be related to the total local concentration
can be related to the total local concentration 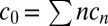 of all monomers inside a finite volume element
of all monomers inside a finite volume element 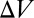 . By inverting this functional dependence numerically, we obtain the polymer length distribution
. By inverting this functional dependence numerically, we obtain the polymer length distribution 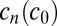 . The
. The 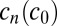 fully characterizes the steady state of the polymerization process. The simplest observable is the mean polymer length
fully characterizes the steady state of the polymerization process. The simplest observable is the mean polymer length 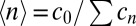 , which corresponds to the ratio of the total monomer concentration to the total concentration of polymer molecules of any size inside
, which corresponds to the ratio of the total monomer concentration to the total concentration of polymer molecules of any size inside 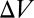 . We will see that the experimental fluorescence resonance energy transfer (FRET) signal considered below is directly related to the mean length via
. We will see that the experimental fluorescence resonance energy transfer (FRET) signal considered below is directly related to the mean length via  .
.
Polymerization and Trapping.
The precise interplay of polymerization and thermal trapping depends on the relation between their characteristic timescales. A full analysis of the coupled dynamics would require solving Eqs. 1 and 2 simultaneously, which is computationally intensive. For our purposes it suffices to consider the two adiabatic limits of this system: The limit where the polymerization reaction is fast and always remains equilibrated during the accumulation process is adequate to describe the experiments reported below, as well as for the prebiotic scenario of RNA polymerization explored below. The opposite limit where polymerization is slow or quasi-frozen over the timescale of the accumulation is covered in SI Text C.
The characteristic timescale 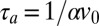 of accumulation is the transport time for a molecule over the accumulation length scale of the trap
of accumulation is the transport time for a molecule over the accumulation length scale of the trap 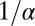 at the characteristic flow velocity
at the characteristic flow velocity 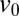 . In the polymerization reaction, a characteristic time
. In the polymerization reaction, a characteristic time 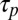 is obtained from the typical on rate and polymer concentration. For fast polymerization (
is obtained from the typical on rate and polymer concentration. For fast polymerization (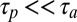 ),
), 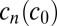 always equilibrates according to the current local concentration
always equilibrates according to the current local concentration 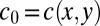 inside the trap. One can then approximate the accumulation process by solving Eq. 1 with an effective diffusion coefficient
inside the trap. One can then approximate the accumulation process by solving Eq. 1 with an effective diffusion coefficient 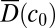 and Soret coefficient
and Soret coefficient 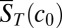 (SI Text A). In the other extreme of slow polymerization (
(SI Text A). In the other extreme of slow polymerization (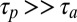 ), the trapped concentrations of the polymer are independently amplified for each length by the accumulation factor of the thermal trap (8).
), the trapped concentrations of the polymer are independently amplified for each length by the accumulation factor of the thermal trap (8).
Trapped RNA Polymerization Under Primordial Conditions.
We extrapolate our theory to the scenario of RNA polymerization inside a thermal trap (8) (Fig. 2). We consider a rectangular elongated compartment and assume a reasonable temperature difference of 10 K. Smaller temperature gradients would require only linearly elongated pores with slower equilibration times (16). We assume that the pore is diffusively coupled to an infinitely large ocean or pond, and therefore we fix the local monomer concentration at the top of the pore at c0. The bottom of the pore is closed and the fluid flow is fully characterized by convection. We assume that the RNA polymerization reaction is in the activation-controlled regime where the diffusive association is faster than the binding reaction and the on rate is independent of length, 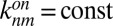 . A constant off rate is a good approximation for short polymers and poses an upper bound for long polymers, because base pairing can significantly enhance the stability of a bond. As a reference, we chose an outside concentration of 1 nM and keep it smaller than the temperature-averaged dissociation constant KD to reflect that only monomers exist in the primordial solution outside the trap.
. A constant off rate is a good approximation for short polymers and poses an upper bound for long polymers, because base pairing can significantly enhance the stability of a bond. As a reference, we chose an outside concentration of 1 nM and keep it smaller than the temperature-averaged dissociation constant KD to reflect that only monomers exist in the primordial solution outside the trap.
Fig. 2.

RNA polymerization: accumulation and polymerization of RNA at the bottom of a primordial geothermal fissure. (A) Length distribution of a fast-reacting polymer (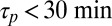 ) inside a 2.5- to 4-cm (black boxes 1–4)-long trap at 100 µm width with a temperature difference of 10 K, an average trap temperature of 55 °C, a dissociation constant of
) inside a 2.5- to 4-cm (black boxes 1–4)-long trap at 100 µm width with a temperature difference of 10 K, an average trap temperature of 55 °C, a dissociation constant of 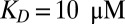 for polymerization, and an outside monomer concentration of 1 nM at physiological salt concentration (150 mM NaCl,
for polymerization, and an outside monomer concentration of 1 nM at physiological salt concentration (150 mM NaCl, 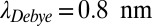 ). Oligomers with the size of active ribozymes are predicted to exist in the trap. (B) Escalation of polymerization. As soon as the total concentration of RNA monomers reaches the KD of polymerization, the polymer length and therefore the effective Soret coefficient of the polymers increase with the trap height
). Oligomers with the size of active ribozymes are predicted to exist in the trap. (B) Escalation of polymerization. As soon as the total concentration of RNA monomers reaches the KD of polymerization, the polymer length and therefore the effective Soret coefficient of the polymers increase with the trap height 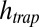 . Therefore,
. Therefore, 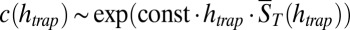 grows hyperexponentially with
grows hyperexponentially with 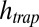 . (C) Mean polymer length vs. pore heights and a broad range of dissociation constants KD. Even for low affinities, a plausible trap height is found at which polymerization escalates and the polymer length diverges. For polymerization slower than the accumulation, a similar behavior is found (Fig. S4, III). We conclude that the escalating growth of RNA length by a thermal trap is a robust phenomenon irrespective of the speed of polymerization.
. (C) Mean polymer length vs. pore heights and a broad range of dissociation constants KD. Even for low affinities, a plausible trap height is found at which polymerization escalates and the polymer length diverges. For polymerization slower than the accumulation, a similar behavior is found (Fig. S4, III). We conclude that the escalating growth of RNA length by a thermal trap is a robust phenomenon irrespective of the speed of polymerization.
Concerning thermophoretic properties of RNA, we use recent measurements of diffusion and Soret coefficients of RNA (20, 26) as documented in SI Text A (Fig. S3 C and D). Temperature and salt concentration play a crucial role because they have a considerable impact on the Soret coefficient of RNA. We investigated three possible primordial scenarios. For a physiological salt concentration (150 mM NaCl, 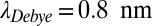 ) we consider an enhanced average trap temperature (55 °C, Fig. 2 and Fig. S4, III) and colder water (25 °C, Fig. S4, I). In addition, a salt-deprived scenario (3 mM NaCl,
) we consider an enhanced average trap temperature (55 °C, Fig. 2 and Fig. S4, III) and colder water (25 °C, Fig. S4, I). In addition, a salt-deprived scenario (3 mM NaCl, 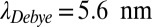 , Fig. S4, II) is analyzed, where the temperature does not change the Soret coefficients sufficiently. For the warm and salty scenario shown in Fig. 2, the experimental scaling laws for diffusion and Soret coefficients were fitted by
, Fig. S4, II) is analyzed, where the temperature does not change the Soret coefficients sufficiently. For the warm and salty scenario shown in Fig. 2, the experimental scaling laws for diffusion and Soret coefficients were fitted by 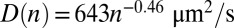 and
and 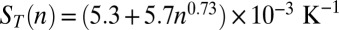 (Fig. S3 C and D). Despite the fact that the Soret coefficients decrease with increasing salt concentration (18, 20), we will see that the exponential trap results in very similar outcomes for our three scenarios, demonstrating the robustness of the escalation effect.
(Fig. S3 C and D). Despite the fact that the Soret coefficients decrease with increasing salt concentration (18, 20), we will see that the exponential trap results in very similar outcomes for our three scenarios, demonstrating the robustness of the escalation effect.
Theoretical Results.
The capability of the thermal trap to produce long polymers is shown in Fig. 2. We discuss fast polymerization (Fig. 2 A–C) and slow polymerization (Fig. S4, III) for various dissociation constants of polymerization KD. In both regimes, polymers of considerable size are found inside the trapping region. To stay in the fast reaction regime, the relaxation time of the polymerization process has to obey 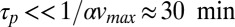 . The concentration distribution inside the compartment is solved using the adiabatic treatment described above, with effective diffusion and Soret coefficients defined according to the polymer length distribution as a function of the total concentration of monomers. Fig. 2A shows the length distribution in the chemical steady state for pore lengths between 2.5 and 4 cm (black boxes 1–4) at fixed width of 100 µm [which is optimal for the trapping of longer polymers (SI Text A)] and with a dissociation constant of
. The concentration distribution inside the compartment is solved using the adiabatic treatment described above, with effective diffusion and Soret coefficients defined according to the polymer length distribution as a function of the total concentration of monomers. Fig. 2A shows the length distribution in the chemical steady state for pore lengths between 2.5 and 4 cm (black boxes 1–4) at fixed width of 100 µm [which is optimal for the trapping of longer polymers (SI Text A)] and with a dissociation constant of 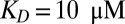 with and without the continuing physical nonequilibrium of a thermal trap. Although dimers are barely existent outside the trap, RNA polymers of more than 100 bases are produced, exceeding even the nanomolar monomer concentration outside the trap. The dependence on trap length is illustrated in Fig. 2B. As soon as the accumulated monomer concentration reaches the threshold KD, long polymers with higher Soret coefficients are formed. Therefore, the effective Soret coefficient
with and without the continuing physical nonequilibrium of a thermal trap. Although dimers are barely existent outside the trap, RNA polymers of more than 100 bases are produced, exceeding even the nanomolar monomer concentration outside the trap. The dependence on trap length is illustrated in Fig. 2B. As soon as the accumulated monomer concentration reaches the threshold KD, long polymers with higher Soret coefficients are formed. Therefore, the effective Soret coefficient 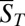 becomes dependent on the trap length
becomes dependent on the trap length 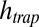 , leading to a hyperexponential escalation of the total RNA concentration with the trap length according to
, leading to a hyperexponential escalation of the total RNA concentration with the trap length according to 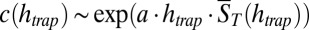 with a being a constant value (Eq. S9a). The time at which hyperexponential trapping is found strongly depends on the trap length and specific shape. A 1,000-fold drop in polymerization affinity is easily balanced by a mere extension of the trap. Also a larger trap aperture presumably leads to faster trap relaxation. Because in literature values of KD for a possible RNA polymerization reaction are only vaguely known, Fig. 2C shows the mean polymer length for a broad range of possible dissociation constants and pore lengths, including a KD of around 10 mM for RNA polymerization measured by the Szostak group (4). In all cases, moderate trap geometries of less than 10 cm lead to polymers longer than the above-mentioned 200 nt needed for a self-replicating ribozyme. Considering the worst-case character of our reversible polymerization, models based on activated monomers are expected to generate longer polymers in even smaller traps.
with a being a constant value (Eq. S9a). The time at which hyperexponential trapping is found strongly depends on the trap length and specific shape. A 1,000-fold drop in polymerization affinity is easily balanced by a mere extension of the trap. Also a larger trap aperture presumably leads to faster trap relaxation. Because in literature values of KD for a possible RNA polymerization reaction are only vaguely known, Fig. 2C shows the mean polymer length for a broad range of possible dissociation constants and pore lengths, including a KD of around 10 mM for RNA polymerization measured by the Szostak group (4). In all cases, moderate trap geometries of less than 10 cm lead to polymers longer than the above-mentioned 200 nt needed for a self-replicating ribozyme. Considering the worst-case character of our reversible polymerization, models based on activated monomers are expected to generate longer polymers in even smaller traps.
For a slow polymerization (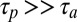 ), we similarly find oligomers of considerable length inside the trap (Fig. S4, III). Therefore, we conclude that the growth of RNA by a thermal trap mechanism is a robust phenomenon, significantly enhancing the range within which RNA can be polymerized, even with inefficient polymerization reactions. The escalation of polymerization by thermal traps persists over a wide range of possible primordial conditions.
), we similarly find oligomers of considerable length inside the trap (Fig. S4, III). Therefore, we conclude that the growth of RNA by a thermal trap mechanism is a robust phenomenon, significantly enhancing the range within which RNA can be polymerized, even with inefficient polymerization reactions. The escalation of polymerization by thermal traps persists over a wide range of possible primordial conditions.
Experiment
Experimental Model System.
To verify the theory of accumulation-enhanced polymerization, we locally accumulated a reversibly polymerizing model system with a laser-driven thermal trap (16). The degree of polymerization as well as the local polymer concentration was measured in real time and comparable with the predicted values from theory and simulation. Care was taken to be able to define and measure all relevant parameters. In existing RNA polymerizing systems the small size of the monomers does not allow for real-time measurements in the picoliter volumes of the trap via, e.g., fluorescence microscopy. We chose larger fluorescently labeled and reversibly polymerizing DNA blocks to implement the polymerization scheme (Fig. 3A). This model system allows for a complete control over all relevant experimental parameters [D, 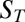 ,
, 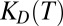 ], which is not possible in an RNA-based system to this date. Despite using DNA, the experimental realization covers all aspects of the used polymerization theory without loss of generality.
], which is not possible in an RNA-based system to this date. Despite using DNA, the experimental realization covers all aspects of the used polymerization theory without loss of generality.
Fig. 3.

Experimental model system. (A) Double-stranded DNA with two sticky ends serves as a monomer for the polymerization. The sticky ends have a melting temperature of 55 °C and are labeled with a FRET dye pair (FAM as donor, ROX as acceptor). (B) The absorption of a symmetrically moved IR laser spot in the center of a 50-µm (height) × 100-μm (width) borosilicate capillary creates thermoviscous convection flow and thermal gradients. (C) The fluid flow has four symmetric convection rolls that model four hydrothermal pores connected at their bottom and hot sides. The temperature profile (T = 53–60 °C) is measured using temperature-sensitive fluorescence. The total monomer concentration  is inferred from the acceptor fluorescence and the polymerization is recorded using FRET.
is inferred from the acceptor fluorescence and the polymerization is recorded using FRET.
We chose two single strands of DNA with a 95-bp-long homologous sequence flanked by 25-bp long sticky ends. At experimental trap conditions, this yields a stable block of double-stranded DNA (dsDNA) with two self-complementary binding sites. The dsDNA blocks are able to reversibly bind to each other via hybridization to form polymers. The polymer-binding energies are directly correlated to the length of the sticky ends. With known hybridization kinetics of DNA, polymerization kinetics (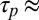 10 s) were faster than the trap kinetics (
10 s) were faster than the trap kinetics (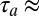 1 h). To measure the degree of polymerization, each sticky end is labeled with a complementary fluorescent dye called donor and acceptor. When a dimer is created, both dyes are brought in close proximity. The donor dye is then quenched by FRET toward the acceptor dye, which will emit the transferred energy as fluorescent light. The normalized efficiency of this transfer is a direct measure for the degree of polymerization, whereas the direct acceptor fluorescence determines the absolute concentration of dsDNA blocks. At a trap temperature of 53 °C, the initial average polymer length equilibrated at a value of 1.7 DNA blocks, which corresponds to a total average length of ∼200 bp.
1 h). To measure the degree of polymerization, each sticky end is labeled with a complementary fluorescent dye called donor and acceptor. When a dimer is created, both dyes are brought in close proximity. The donor dye is then quenched by FRET toward the acceptor dye, which will emit the transferred energy as fluorescent light. The normalized efficiency of this transfer is a direct measure for the degree of polymerization, whereas the direct acceptor fluorescence determines the absolute concentration of dsDNA blocks. At a trap temperature of 53 °C, the initial average polymer length equilibrated at a value of 1.7 DNA blocks, which corresponds to a total average length of ∼200 bp.
We used an improved version of the laser-driven thermal trap described in ref. 16, which allows us to set the convection speed and the temperature gradient independently of the pore width w (Fig. 3B). An infrared laser was focused inside the filled 50-µm (height) × 100-μm (width) borosilicate capillary from below. Light absorption and the rapid movement of the IR laser along the long symmetry axis of the capillary created an elongated (length: 2 × 3,500 µm) and inwardly directed temperature gradient. Additionally, thermoviscous pumping (27) induced four symmetric convection rolls, when an asymmetric laser pattern was applied (Fig. 3C, Flow), which corresponds to four thermal traps that are connected at their hot and bottom sides as symmetric boundaries (Fig. 1A and Fig. S2). Alternating light-emitting diode (LED) excitation provided access to all four excitation and emission channels of the carboxy-X-rhodamine (ROX) and fluorescin amidite (FAM) dye, which was required to measure the fraction of closed bonds and the local concentration of monomers. A typical experimental temperature profile, monomer concentration, and FRET signal at the beginning and end of an experiment are shown in Fig. 3C.
The dsDNA block has three relevant physical parameters: The diffusion coefficient D, the Soret coefficient ST, and the temperature-dependent monomer–monomer dissociation constant, 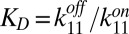 . We determined all three parameters in independent experiments. KD was measured via a melting-curve analysis of a modified DNA that could form only dimers (Fig. S3A). D and ST were both quantified by microscale thermophoresis (18, 26, 28–30), using an unreactive control monomer lacking sticky ends. To obtain the diffusion and Soret coefficients for multimers, we combined the monomer properties with the scaling laws established in ref. 18, yielding
. We determined all three parameters in independent experiments. KD was measured via a melting-curve analysis of a modified DNA that could form only dimers (Fig. S3A). D and ST were both quantified by microscale thermophoresis (18, 26, 28–30), using an unreactive control monomer lacking sticky ends. To obtain the diffusion and Soret coefficients for multimers, we combined the monomer properties with the scaling laws established in ref. 18, yielding 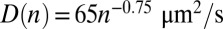 and
and 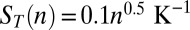 . These experimental constraints turn Eqs. 1 and 2 into a predictive theoretical model for our system. Due to its size, the dsDNA monomer diffuses slowly and reacts quickly via base pairing; we assumed diffusion-controlled association rates between monomers and multimers,
. These experimental constraints turn Eqs. 1 and 2 into a predictive theoretical model for our system. Due to its size, the dsDNA monomer diffuses slowly and reacts quickly via base pairing; we assumed diffusion-controlled association rates between monomers and multimers, 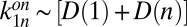 . As all bonds within the polymer are identical, a length-independent dissociation rate can be assumed,
. As all bonds within the polymer are identical, a length-independent dissociation rate can be assumed, 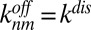 . Taken together, this yields
. Taken together, this yields 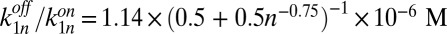 , which allows a theoretical prediction of the steady-state concentrations
, which allows a theoretical prediction of the steady-state concentrations 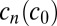 , using Eq. 3.
, using Eq. 3.
Experimental Results.
In Fig. 4A, the time lapse of accumulation in the trap center and at the edge is shown. Laser pumping drove the fluid in circular motion at a speed of 28 µm/s and established a temperature gradient of 7 K. Accumulation sets in immediately, depleting the edges of the trap and filling the center, reaching a constant concentration ratio between the trap center and the edge after 6 h. The edge was subsequently filled via diffusive influx from the outside of the trap, a process that is predicted to take several months (Fig. S3B). Therefore, linear syringe pumping across the capillary was performed during the complete experiment. An average flow speed of 1 µm/s was used to enhance diffusion-driven filling of the trap in a reasonable amount of time. To describe this more complex system, we included the analytical results of Eq. 1 in a 2D finite-element simulation by considering effective diffusion and Soret coefficients 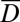 and
and 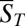 and a temperature-dependent dissociation constant
and a temperature-dependent dissociation constant 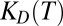 (Fig. S2). Because all relevant parameters [diffusion and Soret coefficients,
(Fig. S2). Because all relevant parameters [diffusion and Soret coefficients, 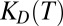 , temperature, and convection flow velocity] were measured separately, no fit parameters were required to describe the experiment by the simulation. The 2D approximation of the 3D experimental system has been verified in previous work (8, 16). Boundary effects can be neglected for large dimensions in the z direction whereas for smaller dimensions, the effect of a parabolic profile is averaged by diffusion.
, temperature, and convection flow velocity] were measured separately, no fit parameters were required to describe the experiment by the simulation. The 2D approximation of the 3D experimental system has been verified in previous work (8, 16). Boundary effects can be neglected for large dimensions in the z direction whereas for smaller dimensions, the effect of a parabolic profile is averaged by diffusion.
Fig. 4.

Experimental data compared with simulation. (A) Simulated and measured monomer concentration in the center and at the edge of the trap with a flow-through drift of 1 µm/s. The initial monomer concentration was 2 μM (time-point A). The separation ratio 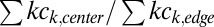 is increased (separation phase) until the steady state of concentrations within the trap is reached after 6 h (time-point C). The monomer concentration in the trap edges is depleted by two orders of magnitude. By drift and diffusion from the surrounding bulk reservoir, the trap is filled until the trap edge reaches the concentration of the outside bulk reservoir (filling phase). Due to the restricted one-dimensional geometry outside the trap, filling is inefficient and requires several weeks (time-point D). (B) The FRET signal is a direct measure for the mean length of the polymer, plotted against the total monomer concentration in the trap center. During the complete experiment, the relationship between concentration and mean length matches the polymer theory without fitting parameters. The same relationship is obtained by titration in bulk solution (open squares). (C and D) The polymerization theory describes the time evolution of length distribution and polymer concentration. The mean polymer length increases from 1.7 (equivalent to ∼200 bp, time-point A) to 24 monomers (equivalent to ∼2,880 bp, time-point D).
is increased (separation phase) until the steady state of concentrations within the trap is reached after 6 h (time-point C). The monomer concentration in the trap edges is depleted by two orders of magnitude. By drift and diffusion from the surrounding bulk reservoir, the trap is filled until the trap edge reaches the concentration of the outside bulk reservoir (filling phase). Due to the restricted one-dimensional geometry outside the trap, filling is inefficient and requires several weeks (time-point D). (B) The FRET signal is a direct measure for the mean length of the polymer, plotted against the total monomer concentration in the trap center. During the complete experiment, the relationship between concentration and mean length matches the polymer theory without fitting parameters. The same relationship is obtained by titration in bulk solution (open squares). (C and D) The polymerization theory describes the time evolution of length distribution and polymer concentration. The mean polymer length increases from 1.7 (equivalent to ∼200 bp, time-point A) to 24 monomers (equivalent to ∼2,880 bp, time-point D).
Four different time points are indicated by letters A–D in Fig. 4 and Fig. S3B. After 6 h (C in Fig. 4 and Fig. S3B), the concentration ratio between the trap center and the edge reached a fixed value by reducing the monomer concentration at the trap edge. Only at a later time point (D in Fig. 4 and Fig. S3B), also the absolute monomer concentration inside the trap reached its steady state. At this time, monomers are accumulated more than 1,000-fold inside the trap. The physical nonequilibrium of the thermal trap is still required after time-point D to maintain the length-enhanced chemical equilibrium of polymerization. In Fig. 4 C and D, the polymer concentration and length distribution at the respective time points A–D of the experiment are shown. The trapped polymerization results in the concentration of 11-mers being larger than the concentration of monomers. In the final state (D in Fig. 4 and Fig. S3B), polymers consisting of 100 monomers or a corresponding length of 12,000 bp are expected. Fig. 4B shows the FRET signal recorded during the accumulation experiment (solid line). It precisely matches the theoretical prediction of our polymerization theory (dashed line), which we had calibrated using the FRET signals obtained by titration in a heated, nonconvecting, nonaccumulating trap (open squares).
Assumptions, such as the omission of cyclization reactions and the diffusion-controlled limit for the experimental system, are supported by the agreement of Eq. 2 to the titration values. Furthermore, the agreement between the dynamic and static experiments (Fig. 4B) shows that at every time point polymerization is faster than the thermal trap as assumed in theory. This corresponds to a small perturbation of the chemical equilibrium of polymerization by the continuing physical nonequilibrium of the thermal trap until the final length distribution is reached. This gives us the confidence to extrapolate the findings toward a prebiotic scenario, implemented solely by using the experimentally verified theory of accumulation-enhanced polymerization, but with RNA parameters and a gravitationally driven trap instead of a laser-driven one.
Discussion
Our detailed theory to describe accumulation and polymerization in a thermal gradient is based on realistic assumptions and uses experimentally measured parameters. It connects physical and chemical nonequilibrium systems that are plausible in a primordial setting. The polymerization of RNA in thermal traps can be predicted because reliable measurements of the thermophoretic parameters of RNA for different lengths at various salt concentrations are available. In various scenarios, a critical length of the trap is found for which the accumulation reaches the dissociation constant of polymerization. Beyond this critical trap length, the trapped concentration diverges hyperexponentially and the RNA length distribution reaches lengths beyond 100 bases (Fig. 2C). Because the thermophoretic coefficients depend only moderately on concentration, significant reductions of the ionic contributions to thermophoresis are expected only well beyond millimolar concentrations (18) and are unlikely to vanish due to hydrophobic contributions.
The kinetics of the trap can require weeks and years in the simple linear geometry discussed here. As the outside molecules have to access the trap by diffusion, the equilibration timescale of the trap increases linearly with the concentration ratios. Reaching the point (2) in Fig. 2 A–C by pure linear diffusion demands 100 y. However, alternative geometries with, e.g., a larger trap aperture for increased diffusive influx or with a continuous overflow of ocean water perpendicular to the hydrothermal convection allow reaching the steady state much faster.
The proposed mechanism of trapped polymerization is well suited for low salt concentrations used, for example, in RNA polymerization from cyclic nucleotides (5). The polymerization requires cGMP in millimolar concentrations, which could be enriched from catalytically active rock surfaces (31). In general, surface catalysis on rock particles of the micrometer scale such as clay (7) is highly compatible, because larger particles show only minor accumulation due to their slow diffusional response time. For example, 1-µm beads cycle under conditions at which DNA strongly accumulates (Movie S1), forcing molecules to reversibly attach and detach at the particle surface.
The thermal gradient and convection not only support accumulation and polymerization but also are capable of triggering and preserving the propagation of information, a vital requirement of the RNA world. When following a convection path, oligonucleotides are exposed to temperature oscillations that can trigger exponential replication reactions (15, 16). Compared with the isothermal case, replication is not faster for shorter oligomers and thus does not lead to the degeneration of information by shortening of the replicates as demonstrated by Spiegelmann’s pioneering experiments (32). Thermally triggered replication (PCR) is compatible with thermal trapping: In previous work, DNA was replicated with a polymerase every 30 s in a laser-driven thermal molecule trap (16). Also the protein-free replication of codon information can be approached by using truncated tRNA molecules in a thermal oscillation (17). Other physical nonequilibrium mechanisms to drive replication have also been explored, such as a mechanical disequilibrium-induced pattern of replication in larger DNA complexes (33). Isothermal RNA ligation could be used for a mutual ligation chain reaction (34). Techniques from DNA machines could be used to form oligomers (35) and trigger sequence-determined polymer synthesis (36). Interestingly, important steps in peptide synthesis can be catalyzed by simple RNA molecules under physiological conditions (37). In addition, it was recently shown theoretically by modeling RNA ligation in a thermophoretic trap that sequence information transmission can be initiated by differential degradation in a thermal oscillation (38). More generally, the framework of nonequilibrium statistical thermodynamics can be used as a unifying theoretical framework to describe the generation of sequence information, using chemical and physical nonequilibrium processes (39). An exciting experimental prospect is the implementation of autonomous molecular evolution in a thermal trap—a molecular Darwinian process with replication driven by thermal cycling and selection driven by the length-sensitive trapping.
Materials and Methods
The trapping geometry and laser heating were described previously (16). Trap length was increased to 3.5 mm on each side (aspect ratio of the trap 70 from each side) at a lower temperature difference by plan correcting the scanning optics (specifications provided in SI Text B). A capillary with a rectangular cross section of 100 × 50 μm was thermally coupled to a Peltier element with sapphire and silicon. Double-stranded DNA with a length of 95 bp and with sticky ends 25 bases in length was allowed to reversibly polymerize by hybridization. Physiological salt concentrations (1× PBS: 137 mM NaCl, 2.7 mM KCl, 10 mM 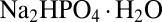 , 2 mM
, 2 mM 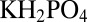 , pH 7.4) were used throughout the experiment. Heating and thermoviscous flow were provided by a 1,940-nm IR laser (20 W; IPG Photonics) and calibrated with the temperature-dependent fluorescence of the FRET signal and silica beads, respectively. DNA binding was recorded with fluorescence energy transfer (Table S1 and S2) (40) in an alternative dual excitation and permanent dual emission (Optosplit II; Cairn Research) microscopy setup (Axiotech Vario; Zeiss) with a 40×, 0.9 NA objective (Zeiss). Multiple images at modulated LED currents were used to enhance the dynamic range of the 12-bit CCD camera (PCO imaging). The thermophoretic properties (diffusion and Soret coefficient) of the 95-bp DNA were measured as described previously (20). To extrapolate the experiments into an RNA-world scenario, short single-stranded RNAs with lengths of 5–50 bases were measured over various salt concentrations and temperatures (SI Text C). Soret coefficients vs. RNA over length were fitted with power laws for use in theory and finite-element simulations.
, pH 7.4) were used throughout the experiment. Heating and thermoviscous flow were provided by a 1,940-nm IR laser (20 W; IPG Photonics) and calibrated with the temperature-dependent fluorescence of the FRET signal and silica beads, respectively. DNA binding was recorded with fluorescence energy transfer (Table S1 and S2) (40) in an alternative dual excitation and permanent dual emission (Optosplit II; Cairn Research) microscopy setup (Axiotech Vario; Zeiss) with a 40×, 0.9 NA objective (Zeiss). Multiple images at modulated LED currents were used to enhance the dynamic range of the 12-bit CCD camera (PCO imaging). The thermophoretic properties (diffusion and Soret coefficient) of the 95-bp DNA were measured as described previously (20). To extrapolate the experiments into an RNA-world scenario, short single-stranded RNAs with lengths of 5–50 bases were measured over various salt concentrations and temperatures (SI Text C). Soret coefficients vs. RNA over length were fitted with power laws for use in theory and finite-element simulations.
Supplementary Material
Acknowledgments
We thank Mario Herzog for measuring the thermophoretic properties of short nucleotides. Michael Nash, David Smith, Moritz Kreysing, and Ernesto di Mauro gave valuable comments on the manuscript at various stages. We thank Ingo Stein and Philip Tinnefeld in the analysis of FRET efficiencies. Financial support from the NanoSystems Initiative Munich, the Ludwig-Maximilians-Universität Munich Initiative Functional Nanosystems, the International Doctorate Program NanoBioTechnology, and the European Research Council Starting Grant is acknowledged.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303222110/-/DCSupplemental.
References
- 1.Crick F. Central dogma of molecular biology. Nature. 1970;227(5258):561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert W. Origin of life: The RNA world. Nature. 1986;319:618. [Google Scholar]
- 3.Wochner A, Attwater J, Coulson A, Holliger P. Ribozyme-catalyzed transcription of an active ribozyme. Science. 2011;332(6026):209–212. doi: 10.1126/science.1200752. [DOI] [PubMed] [Google Scholar]
- 4.Zhang N, Zhang S, Szostak JW. Activated ribonucleotides undergo a sugar pucker switch upon binding to a single-stranded RNA template. J Am Chem Soc. 2012;134(8):3691–3694. doi: 10.1021/ja212027q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costanzo G, Pino S, Ciciriello F, Di Mauro E. Generation of long RNA chains in water. J Biol Chem. 2009;284(48):33206–33216. doi: 10.1074/jbc.M109.041905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costanzo G, et al. Generation of RNA molecules by a base-catalysed click-like reaction. ChemBioChem. 2012;13(7):999–1008. doi: 10.1002/cbic.201200068. [DOI] [PubMed] [Google Scholar]
- 7.Ferris JP, Hill AR, Jr, Liu R, Orgel LE. Synthesis of long prebiotic oligomers on mineral surfaces. Nature. 1996;381(6577):59–61. doi: 10.1038/381059a0. [DOI] [PubMed] [Google Scholar]
- 8.Baaske P, et al. Extreme accumulation of nucleotides in simulated hydrothermal pore systems. Proc Natl Acad Sci USA. 2007;104(22):9346–9351. doi: 10.1073/pnas.0609592104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mielke RE, et al. Iron-sulfide-bearing chimneys as potential catalytic energy traps at life’s emergence. Astrobiology. 2011;11(10):933–950. doi: 10.1089/ast.2011.0667. [DOI] [PubMed] [Google Scholar]
- 10.Martin W, Baross J, Kelley D, Russell MJ. Hydrothermal vents and the origin of life. Nat Rev Microbiol. 2008;6(11):805–814. doi: 10.1038/nrmicro1991. [DOI] [PubMed] [Google Scholar]
- 11.Morgan L, et al. Exploration and discovery in Yellowstone Lake: Results from high-resolution sonar imaging, seismic reflection profiling, and submersible studies. J Volcanol Geotherm Res. 2003;122:221–242. [Google Scholar]
- 12.Weinert FM, Braun D. An optical conveyor for molecules. Nano Lett. 2009;9(12):4264–4267. doi: 10.1021/nl902503c. [DOI] [PubMed] [Google Scholar]
- 13.Braun D, Libchaber A. Trapping of DNA by thermophoretic depletion and convection. Phys Rev Lett. 2002;89(18):188103. doi: 10.1103/PhysRevLett.89.188103. [DOI] [PubMed] [Google Scholar]
- 14.Budin I, Bruckner RJ, Szostak JW. Formation of protocell-like vesicles in a thermal diffusion column. J Am Chem Soc. 2009;131(28):9628–9629. doi: 10.1021/ja9029818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braun D, Goddard NL, Libchaber A. Exponential DNA replication by laminar convection. Phys Rev Lett. 2003;91(15):158103. doi: 10.1103/PhysRevLett.91.158103. [DOI] [PubMed] [Google Scholar]
- 16.Mast CB, Braun D. Thermal trap for DNA replication. Phys Rev Lett. 2010;104(18):188102. doi: 10.1103/PhysRevLett.104.188102. [DOI] [PubMed] [Google Scholar]
- 17.Krammer H, Möller FM, Braun D. Thermal, autonomous replicator made from transfer RNA. Phys Rev Lett. 2012;108(23):238104. doi: 10.1103/PhysRevLett.108.238104. [DOI] [PubMed] [Google Scholar]
- 18.Duhr S, Braun D. Why molecules move along a temperature gradient. Proc Natl Acad Sci USA. 2006;103(52):19678–19682. doi: 10.1073/pnas.0603873103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhont JKG, Wiegand S, Duhr S, Braun D. Thermodiffusion of charged colloids: Single-particle diffusion. Langmuir. 2007;23(4):1674–1683. doi: 10.1021/la062184m. [DOI] [PubMed] [Google Scholar]
- 20.Reineck P, Wienken CJ, Braun D. Thermophoresis of single stranded DNA. Electrophoresis. 2010;31(2):279–286. doi: 10.1002/elps.200900505. [DOI] [PubMed] [Google Scholar]
- 21.Ludwig C. 1856. Diffusion zwischen ungleich erwärmten Orten gleich zusammengesetzter Lösungen [Diffusion of homogeneous fluids between regions of different temperature]. Sitzungsbericht Kaiser Akad Wiss, ed Braunmüller W (Mathem-Naturwiss Cl, Vienna), Vol 65, p 539. German.
- 22.Clusius K, Dickel G. Neues verfahren zur gasentmischung und isotopentrennung [A new method for the separation of different gases and isotopes] Naturwissenschaften. 1938;26:546. German. [Google Scholar]
- 23.Debye P. Zur theorie des clusiusschen trennungsverfahrens [Theory about the Clusius separation process] Ann Phys. 1939;428:284–294. German. [Google Scholar]
- 24.Krapivsky PL, Redner S, Ben-Naim E. A Kinetic View of Statistical Physics. Cambridge, UK: Cambridge Univ Press; 2010. [Google Scholar]
- 25.van Kampen NG. Stochastic Processes in Physics and Chemistry. Amsterdam: North-Holland Personal Library; 1992. [Google Scholar]
- 26.Wang Z, Kriegs H, Wiegand S. Thermal diffusion of nucleotides. J Phys Chem B. 2012;116(25):7463–7469. doi: 10.1021/jp3032644. [DOI] [PubMed] [Google Scholar]
- 27.Weinert FM, Braun D. Optically driven fluid flow along arbitrary microscale patterns using thermoviscous expansion. J Appl Phys. 2008;104:104701-1–104701-10. [Google Scholar]
- 28.Duhr S, Braun D. Optothermal molecule trapping by opposing fluid flow with thermophoretic drift. Phys Rev Lett. 2006;97(3):038103. doi: 10.1103/PhysRevLett.97.038103. [DOI] [PubMed] [Google Scholar]
- 29.Seidel SAI, et al. Label-free microscale thermophoresis discriminates sites and affinity of protein-ligand binding. Angew Chem Int Ed Engl. 2012;51(42):10656–10659. doi: 10.1002/anie.201204268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X, et al. Peptide surfactants for cell-free production of functional G protein-coupled receptors. Proc Natl Acad Sci USA. 2011;108(22):9049–9054. doi: 10.1073/pnas.1018185108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saladino R, Botta G, Pino S, Costanzo G, Di Mauro E. Genetics first or metabolism first? The formamide clue. Chem Soc Rev. 2012;41(16):5526–5565. doi: 10.1039/c2cs35066a. [DOI] [PubMed] [Google Scholar]
- 32.Spiegelman S, Haruna I, Holland IB, Beaudreau G, Mills D. The synthesis of a self-propagating and infectious nucleic acid with a purified enzyme. Proc Natl Acad Sci USA. 1965;54(3):919–927. doi: 10.1073/pnas.54.3.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulman R, Yurke B, Winfree E. Robust self-replication of combinatorial information via crystal growth and scission. Proc Natl Acad Sci USA. 2012;109(17):6405–6410. doi: 10.1073/pnas.1117813109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lincoln TA, Joyce GF. Self-sustained replication of an RNA enzyme. Science. 2009;323(5918):1229–1232. doi: 10.1126/science.1167856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lubrich D, Green SJ, Turberfield AJ. Kinetically controlled self-assembly of DNA oligomers. J Am Chem Soc. 2009;131(7):2422–2423. doi: 10.1021/ja807765v. [DOI] [PubMed] [Google Scholar]
- 36.McKee ML, et al. Multistep DNA-templated reactions for the synthesis of functional sequence controlled oligomers. Angew Chem Int Ed Engl. 2010;49(43):7948–7951. doi: 10.1002/anie.201002721. [DOI] [PubMed] [Google Scholar]
- 37.Turk RM, Chumachenko NV, Yarus M. Multiple translational products from a five-nucleotide ribozyme. Proc Natl Acad Sci USA. 2010;107(10):4585–4589. doi: 10.1073/pnas.0912895107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Obermayer B, Krammer H, Braun D, Gerland U. Emergence of information transmission in a prebiotic RNA reactor. Phys Rev Lett. 2011;107(1):018101. doi: 10.1103/PhysRevLett.107.018101. [DOI] [PubMed] [Google Scholar]
- 39.Andrieux D, Gaspard P. Nonequilibrium generation of information in copolymerization processes. Proc Natl Acad Sci USA. 2008;105(28):9516–9521. doi: 10.1073/pnas.0802049105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Förster T. Zwischenmolekulare Energiewanderung und Fluoreszenz [Intermolecular energy transfer and fluorescence] Ann Phys. 1948;437:55–75. German. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.