

Abstract
Although there are over 1,150 bat species worldwide, the diversity of viruses harbored by bats has only recently come into focus as a result of expanded wildlife surveillance. Such surveys are of importance in determining the potential for novel viruses to emerge in humans, and for optimal management of bats and their habitats. To enhance our knowledge of the viral diversity present in bats, we initially surveyed 415 sera from African and Central American bats. Unbiased high-throughput sequencing revealed the presence of a highly diverse group of bat-derived viruses related to hepaciviruses and pegiviruses within the family Flaviridae. Subsequent PCR screening of 1,258 bat specimens collected worldwide indicated the presence of these viruses also in North America and Asia. A total of 83 bat-derived viruses were identified, representing an infection rate of nearly 5%. Evolutionary analyses revealed that all known hepaciviruses and pegiviruses, including those previously documented in humans and other primates, fall within the phylogenetic diversity of the bat-derived viruses described here. The prevalence, unprecedented viral biodiversity, phylogenetic divergence, and worldwide distribution of the bat-derived viruses suggest that bats are a major and ancient natural reservoir for both hepaciviruses and pegiviruses and provide insights into the evolutionary history of hepatitis C virus and the human GB viruses.
Determining the natural reservoirs for emerging pathogens is critical for disease prediction and prevention. Nearly 60% of emerging infectious diseases in humans are zoonotic, with up to 70% originating from wildlife (1). Bats [order Chiroptera, suborders Yinpterochiroptera and Yangochiroptera (2, 3)] are a natural reservoir for many important zoonotic viruses that cause severe disease in humans, including lyssaviruses, severe acute respiratory syndrome (SARS)-related coronaviruses (SARS-related CoV), filoviruses, henipaviruses, and other paramyxoviruses (4–13). In addition, several viruses establish persistent viral infections in bats, in which apparent clinical signs appear only rarely (5). Bats possess unique characteristics that may contribute to their capacity to function as a major reservoir host for viruses, including long lifespan, high species diversity, unique immune systems, gregarious roosting behaviors, and high spatial mobility and population densities (5).
Although hepatitis C virus (HCV) was discovered more than 20 y ago, its origin is unknown, and, until recently, humans appeared to be its only natural host (14, 15). HCV was initially isolated from the serum of a person with non-A, non-B hepatitis and is now a leading cause of chronic liver disease, cirrhosis, and hepatocellular carcinoma, with ∼3% of the global population infected (16, 17). HCV and its distant relative, GB virus B (GBV-B), belong to the genus Hepacivirus within the family Flaviridae of single-stranded, positive-sense RNA viruses (18, 19). New World monkeys can be experimentally infected with GBV-B, resulting in clinical hepatitis (20). Recently, nonprimate hepaciviruses (NPHV) were discovered in domestic animals (dogs, horses), expanding the natural host range of these agents (21–23). A new flavivirus genus, Pegivirus, has been proposed for GB viruses GBV-A, GBV-C, and GBV-D (19). GBV-A viruses have been identified in nonhuman primates and are not known to infect humans, whereas GBV-C is frequently isolated from humans and chimpanzees (19). GBV-D was recently identified in Old World frugivorous bats (24). No pegiviruses have yet been identified to cause disease in their hosts (19).
Here, we report the discovery of a highly diverse group of bat-derived viruses related to hepaciviruses and pegiviruses. The prevalence, unprecedented viral biodiversity, and broad geographic distribution of these viruses, identified throughout the order Chiroptera, suggest that bats are a key natural reservoir for both hepaciviruses and pegiviruses.
Results
Discovery of Bat-Derived Hepaciviruses and Pegiviruses.
We surveyed serum specimens from 415 apparently healthy bats captured from five different countries (Guatemala, Cameroon, Nigeria, Democratic Republic of the Congo, and Kenya) that represent 33 species, 26 genera, and seven families (SI Appendix, Tables S1 and S2). Aliquots of sera were combined into 43 pools for nucleic acid extraction; total RNA was randomly amplified and subjected to unbiased high-throughput sequencing (UHTS) (25). After removal of host sequences, each pool yielded between 87 and 3,098 unique reads and 1–128 assembled contiguous sequences. The analysis of these sequence data at the nucleotide (nt) and amino acid (aa) levels revealed the presence of sequences with distant similarity to flaviviruses in 29 sera pools. Between 1 and 716 flavivirus sequences, ranging from 50 to 10,803 nt in length, were identified in the positive pools. The sequences showed aa sequence similarities ranging from 24% to 100% to members of the genus Hepacivirus or Pegivirus. Specific and degenerate primers targeting a conserved motif in the RNA-dependent RNA polymerase gene (RdRp or NS5B) were designed from the UHTS data and used to confirm the initial findings, and further for screening all individual serum specimens in the positive pools. Using this approach, genetic material from a total of 58 bat-derived viruses was detected (SI Appendix, Tables S3 and S4).
To further explore the distribution of these viruses in other countries and bat species, an additional 1,166 sera/plasma specimens and 83 tissue specimens from 34 bats (kidney, liver, and lung), representing 40 species, 31 genera, and seven families of bats from Nigeria, Bangladesh, and Mexico were screened (SI Appendix, Table S2). Twenty-three sera/plasma and one lung specimen were positive. Additional specimens from 9 of the bats from Mexico with positive sera specimens, consisting of seven oral and two rectal swabs were also tested. One positive rectal swab was detected (SI Appendix, Tables S3 and S4).
In total, 1,673 specimens, collected between 2007 and 2011 from 1,615 bats representing 58 species, 44 genera, and eight families in seven countries worldwide were screened (SI Appendix, Table S2). The genetic material of bat-derived viruses was detected in six of the eight families of bats tested, representing all major phylogenetic lineages of the chiroptera. A total of 78 positive sera/plasma, one lung specimen, and one rectal swab were tested positive, representing 21 species, 20 genera, and six families of bats (SI Appendix, Tables S3, S4, and S5). Quantitative PCR assays indicated the presence of 103 to 108 RNA copies/mL of these viruses in the sera or plasma of infected bats (SI Appendix, Table S4). A total of 83 bat-derived viruses, which potentially can represent 22 novel viral species (3 within the Hepacivirus genus and 19 within the proposed Pegivirus genus) were identified (SI Appendix, Table S6).
Genetic Diversity of Bat-Derived Hepaciviruses and Pegiviruses.
Preliminary phylogenetic analysis of a conserved 300-nt region of the RdRp gene revealed the presence of a highly diverse group of viruses that clustered within the Hepacivirus and Pegivirus genera (Fig. 1). Phylogenetic trees based on the complete helicase and RdRp coding sequences of a representative subset of these viruses, along with previously known hepaci- and pegivirus sequences, supported the clustering of these two genera into 11 strongly supported clades [Bayesian posterior probability (BPP) values in all cases ≥0.9] (SI Appendix, Fig. S1). Importantly, these analyses show that, in both hepaciviruses and pegiviruses, all previously described viruses fall within the phylogenetic diversity of the bat-derived viruses described here, indicating that bats are a major and ancient reservoir for genetic diversity in both genera. Within the Hepacivirus genus, the viruses fell into three highly divergent clades composed solely of viruses from two species of African bats (Hipposideros vittatus, Otomops martiensseni) (BPP = 1.0). Clade A viruses were distinct from, but most closely related to GBV-B whereas clade C and D viruses occupied a basal position relative to the clades containing NPHV (clade E) and HCV (clade F). The bat-derived pegiviruses identified in this study formed three distinct lineages (BPP = 1.0). Viruses from the clades G and K formed phylogenetically distinct clusters within the genus whereas clade H viruses clustered with the previously identified GBV-D viruses (24). Notably, the African, North American, and Asian bat pegivirus lineages were all paraphyletic, suggesting a long history of diversification and cross-species transmission. Although we were unable to detect a history of recombination in our dataset, coinfections were identified in individuals from three species of African bats (Mops condylurus, O. martiensseni and Taphozous sp.) and one species from Bangladesh (Pteropus giganteus), suggesting the potential for recombination to occur. Three coinfections involved two distinct pegiviruses from clades G or K whereas, in the fourth case, a bat was infected with both a hepacivirus (clade C) and a pegivirus (clade K) (SI Appendix, Table S4).
Fig. 1.
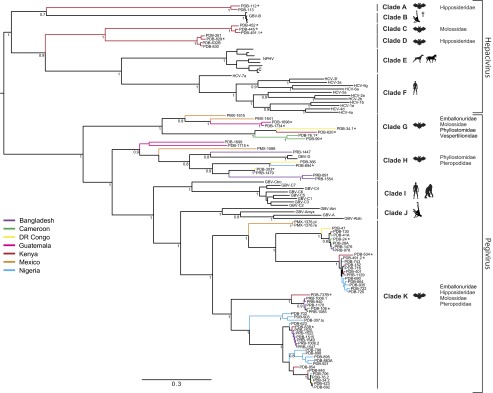
Bayesian phylogenetic tree of a 300-nt conserved region of the RdRp gene of the bat-derived viruses and selected members of the Hepacivirus and Pegivirus genera. The 83 bat-derived viruses identified in this study are shown along with the clade designations (A to K); an asterisk indicates viruses for which the near full-length genome was obtained. The countries of origin for the sequences generated in this study are indicated by the branch color; the host and the bat families associated with each clade are indicated next to the clade name. Bayesian posterior probabilities are shown only for nodes with significant support (>0.7). The scale bar indicates the average number of nucleotide substitutions per site. The virus names corresponding to the abbreviations and GenBank accession numbers are provided in SI Appendix, Table S10. C, canine hepacivirus; †, experimentally infected New World monkeys.
Genomic Organization of Bat-Derived Hepaciviruses and Pegiviruses.
Twenty near full-length genome sequences were generated, representing all six clades in which the bat hepaciviruses (BHVs) and bat pegiviruses (BPgVs) were identified (SI Appendix, Table S7). Similar to all other members of the Flaviviridae, the BHVs and BPgVs have a positive-sense, single-stranded RNA genome that contains a single large ORF encoding a polyprotein precursor (26). The ORF is flanked at the 5′ and 3′ end by nontranslated regions (NTRs). The BHV and BPgV genome sequences comprise at least 8,916 nt and encode precursor polyproteins of 2,842–3,469 aa in length (Fig. 2, SI Appendix, Table S7). Conserved flavivirus protein domains consistent with structural and nonstructural proteins were recognized in the BHV and BPgV predicted polyproteins, enabling accurate prediction of the location and function of the encoded proteins (SI Appendix, Tables S8 and S9). A comparison of predicted protein products indicated that the envelope (E) and the nonstructural proteins NS2 and NS5A were most variable, with less than 34.5%, 33.5%, and 20.4% (hepaciviruses) and 48.3%, 59.2%, and 37.7% (pegiviruses) aa sequence identities, respectively. In contrast, NS3 and NS5B were the most conserved with aa sequence identities in the range of 39.2–53.3% (NS3) and 35.3–46% (NS5B) for hepaciviruses and 43.8–78.7% (NS3) and 39.5–71.1% (NS5B) for pegiviruses (Datasets S1 and S2).
Fig. 2.

Comparison of the genome organization and putative proteomic map of the bat-derived viruses with representative members of the Hepacivirus and Pegivirus genera. The putative genomic organization of the bat-derived viruses is shown for each clade and was predicted using sequence comparisons with known hepaciviruses and pegiviruses. The HCV genomic RNA contains a unique ORF that encodes a precursor polyprotein that is cleaved by viral and cellular proteases into structural proteins [core (C), envelope glycoproteins (E1, E2)], and nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B). The HCV ARFPs are translated in an alternate reading frame overlapping C and represented in orange. The structural and nonstructural proteins are shown in green and red, respectively; the region between E2 and NS2 is shown in light blue, and the variable region (VR) identified in viruses from clade G is represented by the dark blue jagged box. Arrowheads indicate putative host peptidase (purple) and viral NS2-NS3 (white) and NS3-NS4A (yellow) peptidase cleavage sites. A predicted protein of 7–28 kDa is present between E2 and NS2 in the BHVs and BPgVs. *, HCV p7 (41, 42) and GBV-B p13 (43); **, predicted proteins of 21 kDa for GBV-A and 6 kDa for GBV-C (19); ***, predicted proteins of 27–28 kDa; NTR, nontranslated region; ‡, signal peptidase cleavage site predicted for GBV-C (19).
Among the 20 BHVs and BPgVs with complete polyprotein-coding sequences, 14 presented a genomic organization similar to that of hepaciviruses or pegiviruses (Fig. 2, SI Appendix, Table S7). Resembling HCV (27–30), the genome of the BHV PDB-829 in clade D contains a potential alternate reading frame (ARF) that overlaps the core protein-coding gene (SI Appendix, Fig. S2). The HCV ARF proteins (ARFPs) are derived either through ribosomal frameshift or through internal translation initiation (27, 30). The putative BHV PDB-829 ARF begins at the alternative GUG start codon located 41 nt upstream of the main C ORF and has a coding capacity of 156 aa (31). In contrast to HCV, there is no in-frame stop codon that terminates the BHV PDB-829 ARF at the start of the main C ORF (27, 31). Interestingly, the ARF identified in BHV PDB-829 is absent in the BHVs from clade C. Genomes of viruses from clade G presented a distinct genomic feature, a region of variable length referred to as the variable region (VR, 273–843 nt in length) located upstream of the predicted E1 (Fig. 3). No sequence homology to this region is observed in members of the Flaviviridae or in any other viral sequence present in the GenBank/European Molecular Biology Laboratory (EMBL) databases. The predicted aa coding sequences deduced from the VRs are basic (pI = 8.49–9.22) with two to four transmembrane regions. Predicted signal peptidase cleavage sites are identified 18–21 aa downstream of the putative initiation codon (Fig.3). Further studies are necessary to determine whether the VR is part of E1 or encodes for a separate protein. The translated envelope protein sequences (E1, E2) of the BHVs and BPgVs contain 5–9 potential N-linked glycosylation sites, compared with 10–16 and 3–7 sites in hepaciviruses and pegiviruses, respectively (SI Appendix, Tables S8, S9 and Fig. S3A). The BHVs contain 8–11 of the 18 E2 cysteine residues strictly conserved among HCV genotypes and shown to form nine disulfide bridges essential for proper folding of the envelope glycoprotein (SI Appendix, Fig. S3 A and B) (32).
Fig. 3.

Schematic representation of the genomic organization of viruses from clade G. (A) Overall genomic organization and expanded diagram of the predicted variable region (VR). The putative gene products after polyprotein cleavage are indicated in green for the structural proteins, red for the nonstructural proteins, light blue for the region analogous to the HCV-p7, and dark blue for the variable region (VR). ***, predicted proteins of 27–28 kDa. (B) Amino acid sequence alignment of the predicted VR of viruses from clade G. Putative AUG initiator codons were chosen based on the presence of predicted signal peptidase cleavage sites identified 18–21 aa downstream as indicated in red open box. Predicted signal peptidase cleavage sites between VR and E1 are shown in blue open box. Strictly conserved and similar aa are indicated by black or gray shades, respectively.
Discussion
Our study suggests that bats are a major natural reservoir for both hepaciviruses and pegiviruses. Hepaciviruses, formerly detected only in primates, were recently identified in horses and dogs (21–23). Pegiviruses have been found only in primates and in one species of Old World frugivorous bat (P. giganteus) (24). The viruses characterized in this study showed an unprecedented viral biodiversity and represent the most diverse repertoire of hepaciviruses and pegiviruses described to date (Fig. 1 and SI Appendix, Fig. S1). In addition, our phylogenic analyses suggest that the bat-derived viruses are likely to occupy a basal position relative to the previously described hepaciviruses and pegiviruses although a wider sampling of various mammalian species is required to determine whether bats represent the ultimate reservoir host for these viruses. The BHVs and BPgVs are widely distributed, are present on several continents of the New and Old World, and are identified in both suborders of bats (Yinpterochiroptera and Yangochiroptera) from 21 species, 20 genera, and six families, indicating a widespread circulation (Fig. 4). Nearly five percent of the bats studied here were infected with BHVs or BPgVs (0.6% and 4%, respectively; 0.4% were undetermined). All bats collected were apparently healthy despite the high levels of viremia detected, suggesting that BHVs and BPVs may not be pathogenic in their hosts.
Fig. 4.
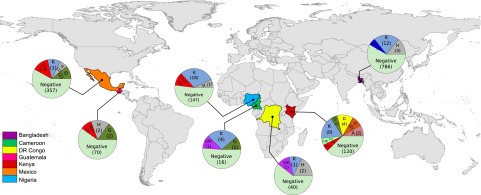
Geographic distribution of the bat-derived hepaciviruses and pegiviruses from specimens collected between 2007–2011. The proportion of the different clades of bat-derived hepaciviruses and pegiviruses is represented in pie charts for each country. Numbers of samples were log converted. The number of samples tested is indicated in parentheses. Map and pie charts were produced using the statistical software R (44).
A long evolutionary history of these viruses in bats is further supported by the distribution across the different taxonomic groups of bats and geographic areas in which the viruses were found. The three distinct clades of BHVs that were all identified in Kenyan specimens were each associated with common insectivorous African bats (O. martiensseni and H. vittatus) of the distantly related families Molossidae and Hipposideridae (Fig. 1). An additional BHV of undetermined clade was also identified in an insectivorous Scotoecus sp. (Family Vespertilionidae) bat from Kenya (SI Appendix, Table S4). The BPgVs clustered in three distinct clades, all of which were paraphyletic, indicating a long history of diversification and cross-species transmission. Whereas the viruses from clades G and H were separated by deep branches associated with distinct bat species (with the exception of PDB-303 and PDB-1479), the clusters in clade K were associated with multiple bat species from different genera and families, representing Yinpterochiroptera and Yangochiroptera bats. Moreover, within each cluster, the closely related BPgVs originated from different countries or even continents as well as from taxonomically distinct bat hosts. Transmission of viruses between bats may occur through shedding in excreta, as suggested by the identification of BPgV in feces, via mating, or during gestation or parturition. However, the mechanism by which these viruses (or their ancestors) moved between continents remains to be elucidated. Further studies, especially in Asian bats, are likely to provide additional information on the evolutionary history and geographical distribution of these viruses.
If bats are currently or historically implicated in transmission of hepaci- and pegiviruses to other species, potential routes for transmission include consumption of fresh bat bodies, contamination of food with bat excreta, or direct exposure to bat blood or excreta or infection via intermediate hosts. Such mechanisms were identified for Nipah virus (NiV), where humans become exposed through contact with infected pigs, consumption of date palm sap contaminated with infected bat excreta, or through direct exposure to NiV-infected blood/excreta from the Indian flying fox, P. giganteus (33). In the case of SARS, the emergence of SARS-CoV in the human population probably occurred from bats to humans via palm civets present in Chinese wet markets (34–36). Whatever the mechanism of interspecies transmission, the distinct bat-derived pegivirus lineages identified in six bat families from Africa, Asia, and Central America and their widespread distribution indicate a much older association of pegiviruses with bats than with any human or nonhuman primate host. Similarly, whereas earlier studies have suggested that the evolution of both GBV-A and GBV-C is compatible with virus–host codivergence within primates (37–39), our phylogenetic analysis reveals multiple cross-species transmissions—both within bats and likely among other mammalian species—over an unknown time-scale. Therefore, the evolutionary history of the hepaciviruses and pegiviruses is clearly more complex than previously thought.
Taken together, our data indicate that bats are a major natural reservoir for hepaciviruses and pegiviruses. Although our current knowledge does not allow us to assess the true prevalence, host range, or geographical distributions of BHVs and BPgVs, it does reveal that these viruses are highly genetically diverse and are circulating in major lineages of Chiroptera in both the Old and New World. As such, our findings shed light on the possible evolutionary history of HCV and human GB viruses, which in turn opens avenues for investigating cross-species transfer and zoonotic potential. Additional field surveillance for these hepaci- and pegiviruses in other mammalian taxa will be necessary to unveil the diversity, biogeography, epizootiology, and natural history of these viruses, as well as the mechanisms that drive spill-over infections and host shifts.
Materials and Methods
Bat Sample Collection.
Bats were captured at multiple sites in seven countries (SI Appendix, Table S1). Animals were collected with mist netting or hoop nets in caves and around human dwellings or from roost locations and foraging sites. All bats captured were apparently healthy. The species of each animal was identified by field biologists and recorded, as well as the sex and morphometric measurements (forearm, body lengths, and weight). Bats captured in Bangladesh and Mexico were anesthetized using isoflurane or manually restrained, respectively. Animals were safely released at the site of capture following sample collection. Oral and rectal swabs and blood were collected (when possible) from each animal. Bats sampled in Africa and Guatemala were anesthetized by intramuscular inoculation with ketamine hydrochloride (0.05–0.1 mg/g of body weight) and euthanized under sedation by intracardiac exsanguination and cervical dislocation. Animals were necropsied by aseptic technique for tissue collection and immediately stored on dry ice or liquid nitrogen in the field, for final storage at −80 °C. One hundred microliters to 3 mL of blood were collected and placed immediately into serum separator tubes (BD vacutainer or microtainer; Becton Dickinson). Serum was kept on ice separated by centrifugation and immediately frozen on dry ice or liquid nitrogen. The capture and sample collection protocols were approved by the Institutional Animal Care and Use Committee of the Centers for Disease Control and Prevention (Atlanta, GA), the University of California (Davis, CA; protocol 16048), and the Tufts New England Medical Center (Boston, MA; protocol G2907) as well as by local authorities in the countries of sampling.
Samples and High-Throughput Sequencing.
Aliquots of sera from bat specimens collected in Africa and Guatemala were combined into 43 pools for nucleic acid extraction using the NucliSENS easyMAG (bioMerieux). Total nucleic acid was treated with DNase-I before random amplification and pyrosequencing (25). After assembly (Newbler v2.3; 454 Life Sciences), reads were clustered and assembled into contiguous fragments for comparison with the GenBank database of nucleic acids and proteins via the BLAST (40).
Screening and Quantitative PCR.
All bat specimens were initially screened for viral genetic material in pools of 8–10 specimens. BHV and BPgV screening was performed using four nested PCR assays amplifying a 300-nt conserved fragment of the RdRp gene (NS5B) (corresponding to the nt positions 8127–8483 in the HCV-1a genome, GenBank accession number M62321). Consensus primers (SI Appendix, Table S11) were designed by multiple alignments of NS5B flavivirus nucleotide sequences identified in the unbiased high-throughput sequencing (UHTS) data. Thereafter, individual specimens from each positive pool were screened using the same set of primers. Additionally, specific primers were designed for all sera pools for which flavivirus sequences were identified in the UHTS data but were negative by NS5B consensus PCRs. Reverse transcription was performed using SuperScript II RT (Invitrogen). PCR primers were applied at 0.2 µM concentrations with 1.5 µL of cDNA (diluted 1:5) and HotStar polymerase (Qiagen). First round cycle conditions were: 95 °C for 7 min; 15 cycles at 95 °C for 30 s, 65 °C for 35 s (−1 °C per cycle), and 72 °C for 40 s; 35 cycles at 95 °C for 30 s, 48 °C for 30 s, and 72 °C for 40 s; and 1 cycle at 72 °C for 7 min. Second round cycling conditions were: 95 °C for 7 min; 35 cycles at 95 °C for 30 s, 48 °C for 30 s, and 72 °C for 40 s; and 1 cycle at 72 °C for 7 min. All PCR products were sequenced in both directions to confirm the presence of viral genetic material in samples. Quantitative SYBR green PCR assays were developed to determine the BHV or BPgV genome copy number. Standards were prepared for each assay by cloning the NS5B amplicon from representative viruses (see SI Appendix, Table S12 for primer sequences). Quantitative PCR (qPCR) reactions were cycled on the 7500 Fast Real-Time PCR System (Applied Biosystems) under the following conditions: 50 °C for 2 min, 95 °C for 10 min, then 45 cycles of 95 °C for 15 s and 60 °C for 60 s.
Genome Sequencing and Phylogenetic and Evolutionary Analyses.
Details are provided in SI Appendix, SI Materials and Methods.
GenBank accession numbers for the viruses identified in this study are provided in SI Appendix, Tables S6 and S7.
Supplementary Material
Acknowledgments
We thank the King and Chiefs of the Idanre community, Ondo State, Nigeria; the Federal Ministry of Health, Abuja, Nigeria; S. Wuyah, M. Lawal, M. Ari, A. Mohammed, I. Onaja, and G. Kia; the Faculty of Veterinary Medicine, Ahmadu Bello University (ABU), Zaria, Nigeria; and the Vice Chancellor and Management of ABU. We thank Luis Escobar, Alejandra Estevez, María Reneé López, Ramon Medrano, Maria E. Morales, and María Luisa Muller (Center for Health Studies, Universidad del Valle de Guatemala) and Julio Martinez (Guatemala Ministry of Agriculture-Animal Health Department). We thank Sophronia Yu, Edwin Danga, Evelyne Mulama, Solomon Gikundi, Leonard Nderitu, and Eric Ogola (Centers for Disease Control-Kenya), Rafael Ciraiz (Guatemala Ministry of Public Health and Social Assistance), and Arif Islam (from Bangladesh) for excellent technical assistance and logistics. This work was supported by National Institutes of Health Grants AI051292 and AI57158 (Northeast Biodefense Center) (to W.I.L.), National Institute of Allergy and Infectious Diseases Grant 5R01AI079231-02, US Agency for International Development PREDICT Grant GHNA 0009 0001 000, and an award from the US Department of Defense. This study was also supported by the Emerging Pandemic Threats Program of the US Agency for International Development, the National Center for Emerging and Zoonotic Infectious Diseases, the Centers for Disease Control and Prevention (Atlanta, GA), and Technical Support Corps funds from the Global Disease Detection Program of the Centers for Disease Control and Prevention (Atlanta, GA).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database. For a list of accession numbers for the viruses identified in this study, see SI Appendix, Tables S6 and S7.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303037110/-/DCSupplemental.
References
- 1.Jones KE, et al. Global trends in emerging infectious diseases. Nature. 2008;451(7181):990–993. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teeling EC, et al. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science. 2005;307(5709):580–584. doi: 10.1126/science.1105113. [DOI] [PubMed] [Google Scholar]
- 3.Zhang G, et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science. 2013;339(6118):456–460. doi: 10.1126/science.1230835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newman SH, Field HE, de Jong CE, Epstein JH, editors. eds (2011) Investigating the Role of Bats in Emerging Zoonoses: Balancing Ecology, Conservation and Public Health Interests (FAO Animal Production and Health Manual No. 12) (Food and Agriculture Organization of the United Nations, Rome)
- 5.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: Important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19(3):531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chua KB, et al. Nipah virus: A recently emergent deadly paramyxovirus. Science. 2000;288(5470):1432–1435. doi: 10.1126/science.288.5470.1432. [DOI] [PubMed] [Google Scholar]
- 7.Drexler JF, et al. Bats host major mammalian paramyxoviruses. Nat Commun. 2012;3:796. doi: 10.1038/ncomms1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lau SK, et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci USA. 2005;102(39):14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leroy EM, et al. Fruit bats as reservoirs of Ebola virus. Nature. 2005;438(7068):575–576. doi: 10.1038/438575a. [DOI] [PubMed] [Google Scholar]
- 10.Li W, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310(5748):676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 11.Luis AD, et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: Are bats special? Proc R Soc B. 2013;280(1756):20122753. doi: 10.1098/rspb.2012.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray K, et al. A morbillivirus that caused fatal disease in horses and humans. Science. 1995;268(5207):94–97. doi: 10.1126/science.7701348. [DOI] [PubMed] [Google Scholar]
- 13.Yob JM, et al. Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerg Infect Dis. 2001;7(3):439–441. doi: 10.3201/eid0703.010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alter HJ. Discovery of the non-A, non-B hepatitis virus: The end of the beginning or the beginning of the end. Transfus Med Rev. 1989;3(2):77–81. doi: 10.1016/s0887-7963(89)70071-7. [DOI] [PubMed] [Google Scholar]
- 15.Choo QL, et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244(4902):359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 16.Ray Kim W. Global epidemiology and burden of hepatitis C. Microbes Infect. 2002;4(12):1219–1225. doi: 10.1016/s1286-4579(02)01649-0. [DOI] [PubMed] [Google Scholar]
- 17.Simmonds P. Genetic diversity and evolution of hepatitis C virus—15 years on. J Gen Virol. 2004;85(Pt 11):3173–3188. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 18.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5(6):453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 19.Stapleton JT, Foung S, Muerhoff AS, Bukh J, Simmonds P. The GB viruses: A review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J Gen Virol. 2011;92(Pt 2):233–246. doi: 10.1099/vir.0.027490-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bukh J, Apgar CL, Govindarajan S, Purcell RH. Host range studies of GB virus-B hepatitis agent, the closest relative of hepatitis C virus, in New World monkeys and chimpanzees. J Med Virol. 2001;65(4):694–697. doi: 10.1002/jmv.2092. [DOI] [PubMed] [Google Scholar]
- 21.Burbelo PD, et al. Serology-enabled discovery of genetically diverse hepaciviruses in a new host. J Virol. 2012;86(11):6171–6178. doi: 10.1128/JVI.00250-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lyons S, et al. Nonprimate hepaciviruses in domestic horses, United kingdom. Emerg Infect Dis. 2012;18(12):1976–1982. doi: 10.3201/eid1812.120498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapoor A, et al. Characterization of a canine homolog of hepatitis C virus. Proc Natl Acad Sci USA. 2011;108(28):11608–11613. doi: 10.1073/pnas.1101794108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Epstein JH, et al. Identification of GBV-D, a novel GB-like flavivirus from old world frugivorous bats (Pteropus giganteus) in Bangladesh. PLoS Pathog. 2010;6:e1000972. doi: 10.1371/journal.ppat.1000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palacios G, et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med. 2008;358(10):991–998. doi: 10.1056/NEJMoa073785. [DOI] [PubMed] [Google Scholar]
- 26.Simmonds P, et al. (2012) in Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses, eds King AMQ, Lefkowitz E, Adams MJ, Carstens EB (Academic, New York), pp 1003-1020.
- 27.Branch AD, Stump DD, Gutierrez JA, Eng F, Walewski JL. The hepatitis C virus alternate reading frame (ARF) and its family of novel products: The alternate reading frame protein/F-protein, the double-frameshift protein, and others. Semin Liver Dis. 2005;25(1):105–117. doi: 10.1055/s-2005-864786. [DOI] [PubMed] [Google Scholar]
- 28.Walewski JL, Keller TR, Stump DD, Branch AD. Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA. 2001;7(5):710–721. doi: 10.1017/s1355838201010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Z, et al. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J. 2001;20(14):3840–3848. doi: 10.1093/emboj/20.14.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vassilaki N, Mavromara P. The HCV ARFP/F/core+1 protein: Production and functional analysis of an unconventional viral product. IUBMB Life. 2009;61(7):739–752. doi: 10.1002/iub.201. [DOI] [PubMed] [Google Scholar]
- 31.Ina Y, Mizokami M, Ohba K, Gojobori T. Reduction of synonymous substitutions in the core protein gene of hepatitis C virus. J Mol Evol. 1994;38(1):50–56. doi: 10.1007/BF00175495. [DOI] [PubMed] [Google Scholar]
- 32.Krey T, et al. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 2010;6(2):e1000762. doi: 10.1371/journal.ppat.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luby SP, et al. Foodborne transmission of Nipah virus, Bangladesh. Emerg Infect Dis. 2006;12(12):1888–1894. doi: 10.3201/eid1212.060732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guan Y, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302(5643):276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 35.Lau SK, et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J Virol. 2010;84(6):2808–2819. doi: 10.1128/JVI.02219-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song HD, et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci USA. 2005;102(7):2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Charrel RN, De Micco P, de Lamballerie X. Phylogenetic analysis of GB viruses A and C: Evidence for cospeciation between virus isolates and their primate hosts. J Gen Virol. 1999;80(Pt 9):2329–2335. doi: 10.1099/0022-1317-80-9-2329. [DOI] [PubMed] [Google Scholar]
- 38.Patel MR, Loo YM, Horner SM, Gale M, Jr, Malik HS. Convergent evolution of escape from hepaciviral antagonism in primates. PLoS Biol. 2012;10(3):e1001282. doi: 10.1371/journal.pbio.1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharp PM, Simmonds P. Evaluating the evidence for virus/host co-evolution. Curr Opin Virol. 2011;1(5):436–441. doi: 10.1016/j.coviro.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 40.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 41.Griffin SD, et al. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003;535(1-3):34–38. doi: 10.1016/s0014-5793(02)03851-6. [DOI] [PubMed] [Google Scholar]
- 42.Lin C, Lindenbach BD, Prágai BM, McCourt DW, Rice CM. Processing in the hepatitis C virus E2-NS2 region: Identification of p7 and two distinct E2-specific products with different C termini. J Virol. 1994;68(8):5063–5073. doi: 10.1128/jvi.68.8.5063-5073.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takikawa S, et al. Functional analyses of GB virus B p13 protein: Development of a recombinant GB virus B hepatitis virus with a p7 protein. Proc Natl Acad Sci USA. 2006;103(9):3345–3350. doi: 10.1073/pnas.0511297103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. Available at www.R-project.org/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.