

Abstract
A major challenge of cancer immunotherapy is the persistence and outgrowth of subpopulations that lose expression of the target antigen. IL-15 is a potent cytokine that can promote organ-specific autoimmunity when up-regulated on tissue cells. Here we report that T cells eradicated 2-wk-old solid tumors that expressed IL-15, eliminating antigen-negative cells. In contrast, control tumors that lacked IL-15 expression consistently relapsed. Interestingly, even tumors lacking expression of cognate antigen were rejected when expressing IL-15, indicating that rejection after adoptive T-cell transfer was independent of cognate antigen expression. Nevertheless, the T-cell receptor of the transferred T cells influenced the outcome, consistent with the notion that T-cell receptor activation and effector status determine whether IL-15 can confer lymphokine killer activity-like properties to T cells. The effect was limited to the microenvironment of tumors expressing IL-15; there were no noticeable effects on contralateral tumors lacking IL-15. Taken together, these results indicate that expression of IL-15 in the tumor microenvironment may prevent the escape of antigen loss variants and subsequent tumor recurrence by enabling T cells to eliminate cancer cells lacking cognate antigen expression in a locally restricted manner.
Keywords: NKG2D, NK receptors
The relapse rate among cancer patients remains high, despite initial success with various treatments, owing to outgrowth of resistant cancer variants. Adoptively transferred cytotoxic T cells have the advantage of potentially achieving sterilizing immunity, killing cancerous cells much like infected cells. In support of this strategy, CD8+ T-cell infiltration into tumors is associated with a better prognosis (1–4). Furthermore, in mouse models, tumor-specific CD8+ T cells prevent growth and can be used to treat well-established tumors in adoptive therapy (5). However, cancer may relapse through the escape of antigen loss variants that are insensitive to antigen-specific CD8+ T cells (6–8), impeding efforts of adoptive T-cell therapy.
IL-15 has multiple functions in the immune system and signals through a heterodimer composed of IL-2 receptor beta (IL-2Rβ) and the common gamma chain (γC) (9, 10). The unique high-affinity receptor alpha chain (IL-15Rα) presents IL-15 in trans to IL-2Rβ/γC on neighboring cells. Importantly, IL-15 is commonly found in the inflamed tissues of patients with autoimmune disorders and celiac disease, where it may promote tissue damage (11, 12), either by serving as a costimulatory molecule for the T-cell receptor (TCR) (13–15) or by endowing T cells through the licensing of natural killer group 2D receptor (NKG2D) to exert lymphokine-activated killer (LAK) activity (13, 15–17). LAK activity by cytotoxic T cells, previously dismissed as an in vitro artifact, has been correlated with IL-15 expression by intestinal cells in individuals with celiac disease (13, 15, 18, 19). However, previous studies in humans were correlative in nature and could not determine whether killing of epithelial cells in a noncognate manner involves low-affinity TCR recognition of self or microbial antigens.
Antitumor activity of IL-15 in vivo has been reported in two types of regimens. In the first type, IL-15 was added to cultures during activation of tumor-specific T cells in vitro before adoptive transfer (20–22); in the second, IL-15 was given systemically (23–25). These reports examined the effects of IL-15 in cancer models, although treatments either were given before tumors had been established or produced only partial responses. Other studies examining the effects of IL-15 expression by cancer cells have suggested that IL-15 can prevent tumor outgrowth and/or metastasis (26), and our laboratories have recently shown the eradication of established IL-15–expressing tumors by densely granulated natural killer (NK) cells (27).
Based on accumulating evidence that IL-15 requires cell contact to function (27–29) and that it promotes organ-specific autoimmunity when expressed by tissue cells (30), we postulated that if cancerous cells expressed IL-15, then they could endow cytotoxic T cells with the ability to reject large established tumors and even prevent relapse. To test this idea, we adoptively transferred CD8+ T cells into mice bearing well-established tumors expressing IL-15 and evaluated tumor regression and regrowth. Our results show that IL-15 elicits a powerful response against established solid tumors and may be a more powerful costimulatory molecule for the TCR than previously thought, in that it could even endow the TCR with the ability to mediate cytolysis of tumors lacking expression of cognate antigens.
Results
We previously reported that cancer cells expressing low antigen levels relapse after treatment with specific CD8+ T cells, whereas tumors expressing high levels of antigens are completely rejected (31). We wanted to determine whether IL-15 in the tumor microenvironment would endow antigen-specific cytotoxic T cells with the ability to prevent tumor escape despite low levels of antigen expression in the same tumor model. To this effect, we transduced the fibrosarcoma mesenchymal cell line MC57 to express low levels of a fusion protein of an SIYRYYGL (SIY) peptide trimer and EGFP with either IL-15 (32) in an enhanced cyan fluorescent protein (ECFP) vector (M-SIY-IL15) or the empty vector (M-SIY) (Fig. 1A and Table S1). M-SIY and M-SIY-IL15 have similar EGFP and ECFP fluorescence (Fig. 1B), have been shown to secrete IL-15 (27), and exhibit comparable growth before treatment (Fig. 1C and Fig. S1).
Fig. 1.

Expression of IL-15 by cancer cells prevents relapse after treatment with tumor-specific T cells. (A) Schematic diagram of the transgenes used in this study. SIY-EGFP, (SIY)3 triple SIYRYYGL-AAY repeat, fused to EGFP; IL-15-ECFP, IL-2SP (signal peptide) fused to IL-15 and coupled to ECFP by the internal ribosomal entry site (IRES); control, IRES ECFP alone. (B) M-parental, M-SIY, and M-SIY-IL15 were analyzed by flow cytometry for expression of SIY (EGFP) and IL-15 or control plasmid (ECFP). (C) Rag−/−γC−/− mice were injected s.c. with M-SIY or M-SIY-IL15 cells, followed 2 wk later by 2C splenocytes i.v. or no further treatment. Lines represent individual tumors compiled from three individual experiments. The incidence of relapse of M-SIY tumors compared with M-SIY-IL15 tumors was statistically significant (P < 0.05). (D) Escaping M-SIY tumors were reisolated from treated Rag−/−γC−/− mice and analyzed for EGFP (SIY antigen) and ECFP (control vector) fluorescence.
We opted to use Rag−/−γC−/− mice as hosts because they are incapable of responding to IL-15, thus permitting uninhibited establishment of tumors. Either M-SIY or M-SIY-IL15 cells were injected s.c. into Rag−/−γC−/− mice. After 2 wk, when tumors were clinical size (∼400–500 mm3), splenocytes from mice transgenic for the 2C TCR, which recognizes SIY presented by the MHC class I molecule Kb, were transferred to treat tumor-bearing animals. M-SIY tumors expressing only antigen shrank initially, but eventually escaped (Fig. 1C). Cancer cells from escaping tumors were readapted to culture, and analysis revealed that although they expressed EGFP before inoculation (Fig. 1B), they no longer did so after tumor outgrowth, and thus the relapsing tumor was composed of antigen-negative cells (Fig. 1D). ECFP expression was maintained, confirming no selection for loss of the empty vector. In contrast, tumors expressing SIY and IL-15 conjointly were completely rejected without relapse (Fig. 1C). (In this study, we defined relapse as progressive regrowth, after regression, by day 65.) This effect was dependent on treatment, as demonstrated by the fact that M-SIY-IL15 tumors grew progressively when no T cells were transferred. Taken together, these results indicate that IL-15 expression in the tumor microenvironment induced 2C T cells to prevent escape of antigen-negative cells, thereby preventing tumor relapse.
To further characterize how IL-15 functions in preventing tumor relapse, we wanted to know whether the effects of IL-15 were systemic, given the consistent evidence suggesting that IL-15 functions in a cell contact-dependent manner (28, 29). To investigate this possibility, we first measured the percentage of circulating 2C T cells after transfer into Rag−/−γC−/− mice bearing either the M-SIY or M-SIY-IL15 tumors, and found that mice bearing M-SIY-IL15 tumors indeed had more 2C T cells than those bearing M-SIY tumors in the peripheral blood (Fig. 2A). This increase in peripheral T cells raised the likely possibility that the effect of IL-15 may be systemic. To determine whether this increase in T cells was sufficient to prevent tumor relapse, we challenged single hosts with M-SIY cells on one flank and with M-SIY-IL15 cells on the opposite flank. Two weeks later, 2C T cells were transferred, and tumor growth was monitored. Interestingly, the presence of the M-SIY-IL15 tumor did not prevent the contralateral M-SIY tumor from relapsing (Fig. 2B). Thus, although there was a systemic increase in 2C T cells, this effect was insufficient to prevent tumor relapse. These data suggest that the ability of IL-15 to enhance T-cell functions is limited to the local microenvironment where IL-15 is expressed.
Fig. 2.

Prevention of relapse by IL-15 is restricted to the tumor where it is expressed. (A) Rag−/−γC−/− mice were injected s.c. with M-SIY (blue) or M-SIY-IL15 (red) cells and then 2 wk later with 2C splenocytes i.v. Peripheral blood specimens were obtained at various times and analyzed for Vβ8 and CD8 expression by flow cytometry. Mean ± SEM percentages of Vβ8+CD8+ cells gated on live lymphoctes from at least three animals per bar are shown. (B) Rag−/−γC−/− mice were injected s.c. on opposite flanks with M-SIY (blue) and M-SIY-IL15 (red) cells and then 2 wk later with 2C splenocytes i.v. Data are compiled from three individual experiments. The incidence of relapse of M-SIY tumors compared with M-SIY-IL15 tumors was statistically significant (P < 0.05).
The finding that antigen-negative cells did not grow out in tumors expressing IL-15 raised the question of whether IL-15 enables T cells to kill cancer cells lacking cognate antigen. To better characterize this property, we challenged Rag−/−γC−/− mice with MC57 expressing IL-15 (M-IL15) or MC57 expressing the empty ECFP vector (M-control), both lacking the cognate SIY antigen (Table S1). After 2 wk, once the tumors were established, we transferred 2C splenocytes and monitored tumor growth. To our surprise, 2C T cells eradicated MC57 tumors in the absence of cognate antigen expression when they expressed IL-15 (in 13 of 14 mice; Fig. 3 and Table 1). We excluded the possibility that this effect was linked to expression of non-2C T-cell receptors on 2C T cells by reproducing the effect using 2C/Rag−/− splenocytes (Fig. 3). Anti-CD8 antibody treatment at the time of T-cell transfer protected the tumors from rejection, thereby affirming a requirement for CD8+ T cells in the IL-15–mediated antitumor effect (Fig. 3). In contrast to the M-IL15 tumors, all M-control tumors lacking IL-15 expression relapsed after various intervals. Taken together, these results suggest that IL-15 expression in the tumor can lead to eradication of large established tumors in the absence of cognate antigen expression.
Fig. 3.
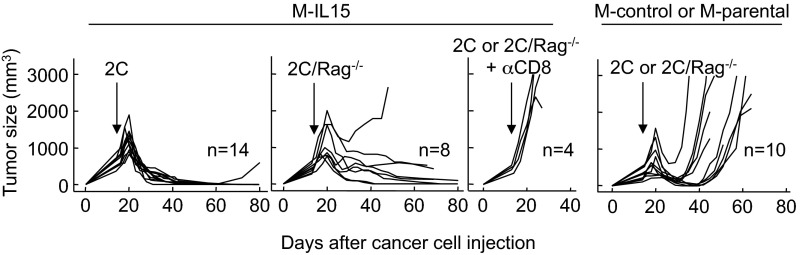
IL-15 can induce T-cell–dependent complete eradication of tumors that lack cognate antigen. Rag−/−γC−/− mice were injected s.c. with M-IL15 or M-control (or M-parental) cancer cells, all lacking SIY expression, and then 2 wk later with 2C or 2C/Rag−/− splenocytes i.v. The indicated mice also received anti-CD8 antibody to deplete T cells. Details on numbers of experiments and mice, as well as statistical significance, are provided in Table 1.
Table 1.
Effect of IL-15 on CD8+ T cells in rejection of tumors lacking cognate antigen
| Treatment | Outcome | |||
| Tumor | Ab | T cells | Regression | relapse |
| M-control or M-parental | — | OT-1 or OT-1/Rag−/− | 1/5A | 1/1 |
| M-SIY-IL15 or M-IL15 | — | OT-1 | 2/2B | 1/2 |
| — | OT-1/Rag−/− | 5/5C* | 5/5 | |
| — | OT1-2C | 5/5D* | 5/5 | |
| αCD8 | OT-1/Rag−/− | 0/5D | N/A | |
| M-control or M-parental | — | 2C/Rag−/− | 10/10A | 10/10 |
| M-IL15 | — | 2C | 14/14A | 1/14† |
| — | 2C/Rag−/− | 12/12E | 1/12† | |
| — | 2C/Prf−/− | 6/6C | 4/6 | |
| αCD8 | 2C | 0/2B | N/A | |
| αCD8 | 2C/Rag−/− | 0/2B | N/A | |
| 9604-parental | — | OT-1 or OT-1/Rag−/− | 0/4D | N/A |
| 9604-IL15 | — | OT-1/Rag−/− | 5/5D* | 3/5 |
| 9604-IL15-Kb | — | OT-1/Rag−/− | 4/5D* | 4/4 |
N/A, non applicable.
Data compiled from four individual experiments.
Data from one experiment.
Data compiled from three individual experiments.
Data compiled from two individual experiments.
Data compiled from six individual experiments.
P < 0.05 compared with control/parental tumors.
P < 0.005 compared with control/parental tumors.
We next wanted to determine how IL-15 mediates its antitumor effects. Having found that in the presence of IL-15, tumors isolated from Rag−/−γC−/− hosts after 2C/Rag−/− T-cell treatment displayed higher levels of IFN-γ in total tissue homogenates (Fig. S2A), we wanted to ascertain the role of IFN-γ. We found that neutralization with anti–IFN-γ antibody prevented tumor regression in presence or absence of IL-15 expression by MC57 tumors (Fig. S2B). Next, knowing that IL-15 up-regulates the expression of cytolytic granules in CD8+ T cells (16), we evaluated the frequency of perforin-positive 2C T cells, and found a significantly higher frequency in M-IL15 tumors compared with M-control tumors (Fig. 4A). Using perforin-deficient 2C T cells, we found that tumors regressed in absence of perforin, but that IL-15 could no longer prevent tumor relapse in four of six mice (Fig. 4B), indicating that induction of perforin is critical to the antitumor properties of IL-15.
Fig. 4.

Perforin is required for complete eradication of tumors that lack cognate antigen. (A) Rag−/−γC−/− mice were injected s.c. with M-IL15 or M-control cancer cells, followed 2 wk later with 2C or 2C/Rag−/− splenocytes i.v. Five days later, tumors were isolated and tumor-infiltrating lymphocytes analyzed for perforin (PRF) and granzyme B (GRZB) expression. Dot plots show representative results of six mice per group analyzed in two experiments. Data of all mice are pooled in the graph. (B) Rag−/−γC−/− mice were injected s.c. with M-IL15 cancer cells, followed 2 wk later with 2C/Prf−/− or 2C/Rag−/− splenocytes i.v. Data are compiled from two independent experiments.
After finding that IL-15 endows 2C T cells with potent antitumor properties in the absence of cognate antigen expressed by cancer cells, we wanted to examine the role of the TCR in this process. This was prompted by the finding that NKG2D is not required for the IL-15–mediated antitumor effects on MC57 (Fig. S3). We had investigated the role of NKG2D, because MC57 tumors expressed the NKG2D ligand RNA export 1 (Rae-1) (33) (Fig. S3A), and NKG2D was up-regulated on 2C T cells after transfer into tumor-bearing mice (Fig. S3B). Furthermore, NKG2D has been shown to mediate LAK activity in human CD8+ T cells activated by IL-15 (13, 15, 18). To further evaluate the role of the TCR, we assessed how IL-15 affected the ability of T cells with a polyconal TCR repertoire to reject and prevent relapse of large established tumors. Interestingly, in contrast to 2C T cells, which were able to shrink tumors independently of IL-15, polyclonal T cells were able to shrink established tumors only in the presence of IL-15 (Fig. S4). Furthermore, they prevented tumor relapse, similar to 2C T cells (Fig. S4).
To examine the involvement of the TCR more specifically, we treated tumors with splenocytes from mice transgenic for the OT-1 TCR, specific for an ovalbumin peptide presented by Kb. Our first striking finding was that OT-1 T cells, in contrast to 2C T cells, could not shrink established MC57 tumors in the absence of IL-15, suggesting that 2C and OT-1 TCR transgenic T cells have different intrinsic abilities to recognize and/or eliminate MC57 tumors. Importantly, IL-15 did enhance the antitumor properties of OT-1 T cells, as demonstrated by the ability of OT-1 T cells to shrink large established MC57 tumors expressing IL-15 in the absence of cognate antigen (Fig. 5A and Table 1). However, OT-1 in contrast to 2C T cells were unable to prevent relapse (Fig. 5A and Table 1). Of note, 2C and OT-1 T cells displayed similar proliferative capacities when transferred into Rag−/−γC−/− hosts (Fig. S5). Altogether, these observations suggest that T cells vary in their capacities to eradicate tumors in presence of IL-15 due to differences in their TCR, and this despite the lack of requirement for cognate antigen.
Fig. 5.

IL-15–induced eradication of tumors that lack cognate antigen is TCR-dependent. (A) Rag−/−γC−/− mice were injected s.c. with indicated cancer cells, followed 2 wk later with OT-1 or OT-1/Rag−/− splenocytes i.v. The indicated mice also received anti-CD8 antibody. Details of the numbers of experiments and mice, as well as statistical significance, are provided in Table 1. (B) (Left) Flow cytometry analysis of the transduction rate of OT-1 T cells expressing the 2C TCR (OT1-2C). (Center) OT1 cells transduced to express the 2C TCR were used to treat established M-IL15 tumors in Rag−/−γC−/− hosts. (Right) Mean ± SEM tumor sizes at the time of treatment (open bars) and at the start of regression (black bars) are shown after treatment with OT-1/Rag-/−, 2C/Rag−/−, or OT1-2C cells. (C) Rag−/−γC−/− mice were injected s.c. with indicated 9604 cancer cells and 2 wk later were given OT-1/Rag−/− (or OT-1) splenocytes i.v. Data are compiled from four independent experiments. Relapsing 9604-IL15-Kb tumors were adapted to culture and analyzed for Kb expression. (Left) Representative comparison of reisolates from a treated mouse and a nontreated mouse (one of four reisolated lines shown for each) compared with 9404-IL-15-Kb and 9604-IL15 cultured in vitro. (Right) Quantification of the decrease in Kb expression on reisolated cancer lines of treated mice versus nontreated mice.
To directly test this hypothesis, we transduced OT-1/Rag−/− splenocytes to express the 2C TCR (OT1-2C). Conspicuously, although only 25% of T cells were transduced, we observed a difference in tumor regression caused by OT-1 and OT1-2C T cells. In particular, OT1-2C T cells rejected the M-IL15 tumor with kinetics similar to 2C T cells and decreased the tumor burden as observed with nontransduced OT-1 T cells (Fig. 5B); nevertheless, tumor relapse occurred. Thus, it appears that the TCR is likely involved in the rejection process of M-IL15 tumors by 2C and OT-1 T cells in the absence of cognate antigen recognition, possibly by permitting recognition of low-avidity self or tumor antigens presented by the MHC class I molecule Kb.
To further test the role of peptide/MHC–TCR interactions in the IL-15–dependent antitumor response, we took advantage of the MHC class I-deficient cancer line 9604. We transduced this cell line with the IL-15 plasmid described in Fig. 1A (Fig. S6A) both alone and in association with a vector expressing Kb. Rag−/−γC−/− mice receiving 9604-IL15 or 9604-IL15-Kb cancer cells developed solid tumors. Both type of tumors regressed with similar kinetics after the transfer of OT-1/Rag−/− splenocytes (Fig. 5C). Furthermore, as observed for M-IL15 tumors treated with OT-1 T cells, 9604-IL15-Kb and 9604-IL15 cancers relapsed. However, analysis of relapsed 9604-IL15-Kb tumors for Kb expression showed a striking loss of MHC class I expression in all cells, suggesting that MHC class I-positive cancer cells were preferentially eliminated by OT-1 T cells. These results, in combination with data showing that the nature of the TCR influences IL-15–driven rejection of tumors, indicate that even though the antitumor properties of IL-15 do not require expression of cognate antigens by tumors, they do involve peptide/MHC–TCR interactions. Interestingly, T cells in these treated mice exhibited up-regulated stimulatory NK receptors (Fig. S6B), suggesting that these receptors may be implicated along with the TCR in the tumor rejection process.
Discussion
This study has revealed that IL-15 expression in the tumor environment has a dramatic effect on T-cell–mediated rejection of established tumors. We showed that the presence of IL-15 could prevent the relapse of loss variants after treatment with tumor-specific T cells. Furthermore, we found that IL-15 enhances the antitumor properties of T cells in a noncognate, yet TCR- and perforin-dependent, mechanism. The antitumor effects of IL-15 observed in this study are likely underestimated, given our previous findings that NK cells are empowered to reject established tumors in the presence of IL-15 (27), and the fact that the experimental systems used here excluded the effect of NK cells. It remains to be deternined whether the combined effect of IL-15 on NK cells and T cells can prevent tumor relapse under suboptimal conditions (e.g., after transfer of OT-1 cells).
Our data suggest that IL-15 promotes regression and even elimination of established solid tumors lacking expression of cognate antigens by endowing CD8+ T cells with potent LAK activity in vivo. Given that the nature of the TCR and MHC class I expression influences the elimination of tumor cells, acquisition of LAK activity by CD8+ T cells likely involves reduction of the TCR activation threshold, leading to recognition of self or tumor peptides that otherwise would be inconsequential. IL-15 could costimulate the TCR directly by activating phosphoinositide 3-kinase or indirectly by promoting the expression and/or activation of costimulatory NK receptors, such as NKG2D (13, 15). Although we found NKG2D to be dispensable for tumor rejection (Fig. S3C), we have not ruled out the possible involvement of NK receptors with stimulatory properties (it is possible that there is a redundancy between activating NK receptors). In fact, tumors lacking MHC class I expression were significantly reduced in size after treatment, and activating NK receptors, including natural killer cell group 2E and 2C (NKG2E/C), and the MHC class I specific C-type lectin NK receptor Ly49D, were up-regulated on CD8+ T cells transferred into IL-15 tumor-bearing mice (Fig. S6B). These activating NK receptors may have overlapping functions with the TCR and/or play a role in reducing the TCR activation threshold. Thus, in contrast to our original thinking, we now argue that acquisition of LAK activity by CD8+ T cells in the presence of IL-15 might involve both the TCR and activating NK receptors. The use of redundant effector mechanisms in the elimination of tumors is now a well-established concept. For instance, it has been shown that the knockdown of effector molecules, such as IFN-γ and TNF-α, in T cells does abrogate full tumor rejection, but does not prevent regression of tumors expressing the cognate antigen (31, 34, 35).
These results have important implications for the design of IL-15 immunotherapy. They demonstrate that cognate antigens are not required for the antitumor properties of T cells in the presence of IL-15. However, they also indicate that the extent to which IL-15 can exert its antitumor effect, and in particular prevent relapse of large established tumors, will rely on the selection of T cells used for immunotherapy. Here we show that certain T-cell clones (e.g., 2C) may exert stronger antitumor effects than others (e.g., OT-1). Importantly, however, polyclonal T cells with a WT repertoire fully reject IL-15–secreting tumors (i.e., there is no tumor relapse in presence of IL-15), suggesting that the use of polyclonal T cells in combination with IL-15 in cancer therapy may have advantages over the use of clonal T cells, which would need to be evaluated individually for their antitumor effects. Our findings further imply that therapeutic strategies should aim to bring IL-15 to the tumor.
We believe that the major effects of IL-15 that we have identified (i.e., its ability to promote regression and even prevent relapse of established tumors) are associated with our decision to have IL-15 expressed by the cancer cells instead of administering it systemically or in T-cell cultures before treatment (20–25). Nevertheless, this does have the potential for clinical application through the use of advanced molecular engineering to target tumors. The expression of IL-15Rα on cancer and stromal cells, along with local IL-15 levels, are important for the activation of both T and NK cells for effective tumor eradication. Not all cancer cells express Rα, and soluble IL-15:IL-15Rα conjugates have been shown to be functional in vivo (36). Coupling these conjugates to tumor-specific receptors, such as antibodies, single-chain variable fragments (scFv), and single-chain soluble TCRs, should enable the effective delivery of these conjugates to tumors. The use of receptor-redirected T cells as delivery vehicles has been studied as well (37). We predict that once a proper method of administration is established, IL-15 will advance efforts in preventing relapse caused by the persistence of cells lacking cognate antigen expression, which continues to hinder cancer treatment.
Methods
Antibodies and Mice.
Anti-Vβ8 (F23.1), anti-Vα2 (B20.1), anti-CD8 (53-6.7), anti-CD16/CD32 (2.4G2), anti-Ly49A (A1), anti-Ly49D (4E5), anti-Ly49F (HBF-719), and anti-Kb (AF6-88.5) antibodies were purchased from BD Pharmingen; anti-CD94 (18d3), anti-CD122 (TM-b1), anti-NK1.1 (PK136), anti-NKG2AB6 (16a11), anti-NKG2D (CX5), and anti-perforin (eBioOMAK-D), from eBioscience; anti-human granzyme B (GB11), from Invitrogen; and polyclonal antibodies anti–panRae-1-PE and anti–IL15Rα-biotin, from R&D Systems. The Rag−/−γC−/− mice were purchased from Taconic Farms; the C57BL/6 mice, from Jackson Laboratory. The 2C mice were provided by J. Chen (Massachusetts Institute of Technology, Cambridge, MA) and subsequently crossed to C57BL/6-Rag−/− (B6.129S7-Rag1tm1Mom/J) or Prf−/− (C57BL/6-Prf1tm1Sdz/J; both from Jackson Laboratory). OT-1 mice were provided by M. Mescher (University of Minnesota, Minneapolis). OT-1/Rag−/− mice were provided by A. Ma (University of San Francisco), and Kb−/−Db−/− mice were provided by A. Chervonsky (University of Chicago) (38). All animal experiments were approved by the University of Chicago’s Institutional Animal Care and Use Committee.
Cell Lines.
All cells were cultured in DMEM containing 5% FCS. P. Ohashi (University of Toronto, Toronto, Canada), with permission of H. Hengartner (University Hospital, Zurich, Switzerland), provided the MC57G cells. The KbDb-deficient cancer cell line 9604 has been described previously (27). The IL-15 cDNA sequence containing the IL-2 signal peptide (IL-2SP) was kindly provided by M. Caligiuri (Ohio State University, Columbus, OH) (32). Primers (5′) 5′ GGGGTCTCGCATGTACAGCAT GCAGCTCG 3′ and (3′) 5′ GGCTCGAGCTATTTGTCATCGTCGTCCTTG 3′ (Integrated DNA Technologies) were used for cloning into the pMFG-IRES-ECFP plasmid (IL-15-ECFP). The plasmid pMFG-IL15Rα has been described previously (27). pLXIN-Kb was generated by cloning Kb into pLXIN (Clontech). Viral supernatants from calcium phosphate-transfected packaging cells were used to transduce the cancer cells. ECFP+ MC57 cells were sorted on a FACSAria cell sorter (BD Biosciences) at the University of Chicago’s Flow Cytometry Facility and designated M-IL15 and M-control cells. These cell lines were also transduced with pMFG-(SIY)3-EGFP using the same method, and the lowest 5% of EGFP+ cells were sorted (designated M-SIY-IL15 and M-SIY). Sorted ECFP+ 9604 cells were designated 9604-IL15. The 9604-IL15-Kb cell line was also transduced and sorted to express Kb; both cell lines were also transduced to express IL15Rα.
In Vivo Tumor Experiments.
On day 0, cancer cells were trypsinized and washed twice with PBS. Mice were injected s.c. with 2–5 × 106 cells in PBS. For depletion, 200 μg of anti-CD8 antibody (2.43.1) was injected i.p. every 3–4 d, beginning at the time of transfer and continuing until termination. For NK depletion, mice were treated with 100 μg of anti-NK1.1 (PK136; BioXCell). NK cell depletion was confirmed by flow cytometry of peripheral blood samples with anti-NK1.1 and anti-CD122. Blocking was done with anti-NKG2D (CX5, 50 μg biweekly; generously provided by L. Lanier, University of San Francisco) or anti–IFN-γ (XMG1.2, 0.5 mg biweekly; BioXCell). Tumor volume was measured with calipers along three orthogonal axes and calculated using the ABC/2 method. The duration of treatment varied from 13 d to 15 d after cancer cell inoculation, depending on tumor size. Splenocytes from donor mice (one donor per one to two hosts) were treated with Tris-NH4Cl, washed, and injected i.v. in the retro-ortibal plexus in PBS. For TCR transduction experiments, OT-1/Rag−/− splenocytes were activated with 1 μM SIINFEKL and transduced with viral supernatants containing 2C TCR or a control plasmid using the retronectin spin technique (39).
Isolation of Cancer and Stromal Cells.
For the analysis of cancer and stromal cells ex vivo, tumors were surgically excised and placed in PBS, minced into 1-mm3 pieces, and incubated for 30 min in 2 mg/mL collagenase D and 100 U/mL DNase I (both Roche Diagnostics). Tumor fragments were pipetted up and down for 2 min after the addition of prewarmed trypsin (0.025% final concentration; Gibco). Suspensions were spun down, washed, and filtered through a 70-μm nylon mesh, resulting in a single-cell suspension, which was used for flow cytometry analysis (for perforin and granzyme) or readaptation to culture in vitro. Alternatively, needle biopsy specimens from escaping tumors were collected and cultured before analysis.
FACS Analysis.
Samples were Fc-blocked and stained with antibodies at 4 °C. Peripheral blood was first treated with Tris/NH4Cl. FACS data were collected with a FACScalibur or LSRII flow cytometer (BD Biosciences) and analyzed using FloJo software (Tree Star).
IFN-γ ELISA.
Tumors were harvested, weighed, and immediately frozen at −80 °C. Samples were later thawed in 1 mL PBS and subjected to homogenization with a glass tissue grinder, then assayed with an IFN-γ ELISA Kit (eBioscience).
Statistics.
Tumor regression and relapse rates were compared using Fisher’s exact test for two-tailed P values. Blood samples and average tumor sizes were compared using an unpaired Student t test.
Supplementary Material
Acknowledgments
We thank Michael Caligiuri and Lewis Lanier for reagents and Ainhoa Arina, David Binder, Jennifer Stone, and David Kranz for technical support and fruitful discussions. This research was supported by National Institutes of Health Grants P01-CA97296, R01-CA22677, R01-CA37156 (to H.S.), R01-DK067180-06 (to B.J.), and University of Chicago Digestive Disease Research Core Center (DK42086) and Committee on Immunology Training Grant T32 AI 0709.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1301022110/-/DCSupplemental.
References
- 1.Naito Y, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58(16):3491–3494. [PubMed] [Google Scholar]
- 2.Zhang L, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 3.Pagès F, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353(25):2654–2666. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 4.Sato E, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005;102(51):18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spiotto MT, et al. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 2002;17(6):737–747. doi: 10.1016/s1074-7613(02)00480-6. [DOI] [PubMed] [Google Scholar]
- 6.Urban JL, Burton RC, Holland JM, Kripke ML, Schreiber H. Mechanisms of syngeneic tumor rejection: Susceptibility of host-selected progressor variants to various immunological effector cells. J Exp Med. 1982;155(2):557–573. doi: 10.1084/jem.155.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uyttenhove C, Maryanski J, Boon T. Escape of mouse mastocytoma P815 after nearly complete rejection is due to antigen-loss variants rather than immunosuppression. J Exp Med. 1983;157(3):1040–1052. doi: 10.1084/jem.157.3.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yee C, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99(25):16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giri JG, et al. IL-15, a novel T cell growth factor that shares activities and receptor components with IL-2. J Leukoc Biol. 1995;57(5):763–766. doi: 10.1002/jlb.57.5.763. [DOI] [PubMed] [Google Scholar]
- 10.Carson WE, et al. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J Exp Med. 1994;180(4):1395–1403. doi: 10.1084/jem.180.4.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waldmann TA. Targeting the interleukin-15/interleukin-15 receptor system in inflammatory autoimmune diseases. Arthritis Res Ther. 2004;6(4):174–177. doi: 10.1186/ar1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol. 2009;9(12):858–870. doi: 10.1038/nri2670. [DOI] [PubMed] [Google Scholar]
- 13.Roberts AI, et al. NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J Immunol. 2001;167(10):5527–5530. doi: 10.4049/jimmunol.167.10.5527. [DOI] [PubMed] [Google Scholar]
- 14.Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci USA. 2003;100(16):9452–9457. doi: 10.1073/pnas.1632807100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meresse B, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. 2004;21(3):357–366. doi: 10.1016/j.immuni.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 16.Gamero AM, Ussery D, Reintgen DS, Puleo CA, Djeu JY. Interleukin 15 induction of lymphokine-activated killer cell function against autologous tumor cells in melanoma patient lymphocytes by a CD18-dependent, perforin-related mechanism. Cancer Res. 1995;55(21):4988–4994. [PubMed] [Google Scholar]
- 17.Tang F, et al. Cytosolic PLA2 is required for CTL-mediated immunopathology of celiac disease via NKG2D and IL-15. J Exp Med. 2009;206(3):707–719. doi: 10.1084/jem.20071887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green PH, Jabri B. Coeliac disease. Lancet. 2003;362(9381):383–391. doi: 10.1016/S0140-6736(03)14027-5. [DOI] [PubMed] [Google Scholar]
- 19.Hüe S, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21(3):367–377. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 20.Klebanoff CA, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA. 2004;101(7):1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teague RM, et al. Interleukin-15 rescues tolerant CD8+ T cells for use in adoptive immunotherapy of established tumors. Nat Med. 2006;12(3):335–341. doi: 10.1038/nm1359. [DOI] [PubMed] [Google Scholar]
- 22.Mueller K, Schweier O, Pircher H. Efficacy of IL-2- versus IL-15-stimulated CD8 T cells in adoptive immunotherapy. Eur J Immunol. 2008;38(10):2874–2885. doi: 10.1002/eji.200838426. [DOI] [PubMed] [Google Scholar]
- 23.Roychowdhury S, et al. Failed adoptive immunotherapy with tumor-specific T cells: Reversal with low-dose interleukin 15 but not low-dose interleukin 2. Cancer Res. 2004;64(21):8062–8067. doi: 10.1158/0008-5472.CAN-04-1860. [DOI] [PubMed] [Google Scholar]
- 24.Dubois S, Patel HJ, Zhang M, Waldmann TA, Müller JR. Preassociation of IL-15 with IL-15R α-IgG1-Fc enhances its activity on proliferation of NK and CD8+/CD44high T cells and its antitumor action. J Immunol. 2008;180(4):2099–2106. doi: 10.4049/jimmunol.180.4.2099. [DOI] [PubMed] [Google Scholar]
- 25.Epardaud M, et al. Interleukin-15/interleukin-15R alpha complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer Res. 2008;68(8):2972–2983. doi: 10.1158/0008-5472.CAN-08-0045. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi H, et al. Role of trans-cellular IL-15 presentation in the activation of NK cell-mediated killing, which leads to enhanced tumor immunosurveillance. Blood. 2005;105(2):721–727. doi: 10.1182/blood-2003-12-4187. [DOI] [PubMed] [Google Scholar]
- 27.Liu RB, et al. Densely granulated murine NK cells eradicate large solid tumors. Cancer Res. 2012;72(8):1964–1974. doi: 10.1158/0008-5472.CAN-11-3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato N, Patel HJ, Waldmann TA, Tagaya Y. The IL-15/IL-15Ralpha on cell surfaces enables sustained IL-15 activity and contributes to the long survival of CD8 memory T cells. Proc Natl Acad Sci USA. 2007;104(2):588–593. doi: 10.1073/pnas.0610115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mortier E, Woo T, Advincula R, Gozalo S, Ma A. IL-15Ralpha chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J Exp Med. 2008;205(5):1213–1225. doi: 10.1084/jem.20071913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Budagian V, Bulanova E, Paus R, Bulfone-Paus S. IL-15/IL-15 receptor biology: A guided tour through an expanding universe. Cytokine Growth Factor Rev. 2006;17(4):259–280. doi: 10.1016/j.cytogfr.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Spiotto MT, Rowley DA, Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nat Med. 2004;10(3):294–298. doi: 10.1038/nm999. [DOI] [PubMed] [Google Scholar]
- 32.Fehniger TA, et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med. 2001;193(2):219–231. doi: 10.1084/jem.193.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerwenka A, et al. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12(6):721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 34.Zhang B, Karrison T, Rowley DA, Schreiber H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Invest. 2008;118(4):1398–1404. doi: 10.1172/JCI33522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Hernandez MdeL, et al. Adoptive transfer of tumor-specific Tc17 effector T cells controls the growth of B16 melanoma in mice. J Immunol. 2010;184(8):4215–4227. doi: 10.4049/jimmunol.0902995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kroemer A, et al. The innate NK cells, allograft rejection, and a key role for IL-15. J Immunol. 2008;180(12):7818–7826. doi: 10.4049/jimmunol.180.12.7818. [DOI] [PubMed] [Google Scholar]
- 37.Chmielewski M, Abken H. CAR T cells transform to trucks: Chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother. 2012;61(8):1269–1277. doi: 10.1007/s00262-012-1202-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vugmeyster Y, et al. Major histocompatibility complex (MHC) class I KbDb−/− deficient mice possess functional CD8+ T cells and natural killer cells. Proc Natl Acad Sci USA. 1998;95(21):12492–12497. doi: 10.1073/pnas.95.21.12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sommermeyer D, et al. Designer T cells by T cell receptor replacement. Eur J Immunol. 2006;36(11):3052–3059. doi: 10.1002/eji.200636539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.