

Abstract
Genome-wide association studies (GWAS) are a powerful means of identifying genes with disease-associated common variants, but they are not well-suited to detecting genes with disease-associated rare and low-frequency variants. In the current study of Behçet disease (BD), nonsynonymous variants (NSVs) identified by deep exonic resequencing of 10 genes found by GWAS (IL10, IL23R, CCR1, STAT4, KLRK1, KLRC1, KLRC2, KLRC3, KLRC4, and ERAP1) and 11 genes selected for their role in innate immunity (IL1B, IL1R1, IL1RN, NLRP3, MEFV, TNFRSF1A, PSTPIP1, CASP1, PYCARD, NOD2, and TLR4) were evaluated for BD association. A differential distribution of the rare and low-frequency NSVs of a gene in 2,461 BD cases compared with 2,458 controls indicated their collective association with disease. By stringent criteria requiring at least a single burden test with study-wide significance and a corroborating test with at least nominal significance, rare and low-frequency NSVs in one GWAS-identified gene, IL23R (P = 6.9 × 10−5), and one gene involved in innate immunity, TLR4 (P = 8.0 × 10−4), were associated with BD. In addition, damaging or rare damaging NOD2 variants were nominally significant across all three burden tests applied (P = 0.0063–0.045). Furthermore, carriage of the familial Mediterranean fever gene (MEFV) mutation Met694Val, which is known to cause recessively inherited familial Mediterranean fever, conferred BD risk in the Turkish population (OR, 2.65; P = 1.8 × 10−12). The disease-associated NSVs in MEFV and TLR4 implicate innate immune and bacterial sensing mechanisms in BD pathogenesis.
Keywords: complex genetics, genetic association
Behçet disease (BD) is a multisystem inflammatory disease characterized mainly by recurrent orogenital ulcers, uveitis, and skin inflammation (1). Genetic factors are involved in the pathogenesis of the disease. In addition to the human leukocyte antigen gene encoding B*51 (HLA-B*51), genome-wide association studies (GWAS) have identified common variants in regions encompassing the major histocompatibility complex class I genes, IL10, and IL23R-IL12RB2 associated with BD in both the Turkish and Japanese populations (2, 3). Moreover, a GWAS with imputed genotypes identified the C-C chemokine receptor 1 gene (CCR1), STAT4, a region encompassing five genes encoding the killer cell lectin-like receptor family members, K1, C1, C2, C3, and C4 in the natural killer gene complex (KLRK1, KLRC1, KLRC2, KLRC3, and KLRC4), and the endoplasmic reticulum aminopeptidase gene (ERAP1) associated with BD (4). Although the GWAS is a powerful means for identifying common variants associated with complex disease, it is generally ineffective in identifying associations resulting from rare (minor allele frequency [MAF] < 1%) and low-frequency (MAF = 1–5%) variants.
It has long been debated whether the innate immune system is involved in the pathogenesis of BD (1). Clinical manifestations such as episodic inflammation and neutrophil recruitment to the sites of inflammation strongly suggest that the innate immune response is a major contributor, although genetic evidence to support this hypothesis is lacking.
Recent advances in sequencing technology are allowing investigators to resequence selected genes to discover variants in large collections of patients with complex genetic traits and genetically matched control participants (5–9). The strength of this strategy is that rare and low-frequency coding variants are likely to influence protein function (6, 10), and therefore influence disease risk. To determine whether rare and low-frequency variants are associated with BD, we performed deep resequencing of 10 GWAS-identified genes (IL23R, IL10, CCR1, STAT4, KLRK1, KLRC1, KLRC2, KLRC3, KLRC4, and ERAP1) and 11 genes known to have roles in innate immunity [IL1B, IL1R1, IL1RN, the nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin domain containing protein-3 gene (NLRP3), the familial Mediterranean fever gene (MEFV), the TNF receptor super family 1A gene (TNFRSF1A), the proline-serine-threonine phosphatase interacting protein gene (PSTPIP1), the caspase 1 gene (CASP1), the PYD and CARD domain gene (PYCARD), the nucleotide-binding oligomerization domain 2 gene (NOD2), and the toll-like receptor 4 gene (TLR4)].
Results
Discovery and Validation of Rare and Low-Frequency Nonsynonymous Variants in Targeted Genes in Japanese Samples.
In the first stage, 382 Japanese case participants and 384 matched control participants from the previously reported GWAS (3) were used for rare and low-frequency variant discovery by resequencing (Fig. S1). These samples were evenly divided into 16 DNA pools, with up to 48 samples per pool. Exons of the targeted genes (45.7 kb of targeted regions total) were amplified by PCR performed on each DNA pool. The PCR products from a single DNA pool were combined and sequenced on a Genome Analyzer IIx (Illumina). We analyzed the sequence data with CASAVA 1.6 software (Illumina) and identified putative low-frequency variants (Materials and Methods). To evaluate the sensitivity of this method, Sanger sequencing was performed as the gold standard on the IL23R exons of 94 Japanese individuals (46 case participants and 48 control participants) from 2 of the pools. We detected all of the validated rare and low-frequency variants that we found by resequencing these pools, and no additional variants were identified.
All of the nonsynonymous-coding rare and low-frequency variants were genotyped in the individual samples of the sequenced pools by TOF-MS genotyping (iPLEX, Sequenom) or TaqMan genotyping assays (Applied Biosystems) and were also genotyped in the remaining Japanese BD collection samples. We also genotyped the additional low-frequency (1–5% MAF) nonsynonymous variants (NSVs) found in the 1000 Genomes HapMap Japanese in Tokyo, Japan (JPT) samples (n = 100 samples; http://browser.1000genomes.org) but not in our sequence data. Because there is not a well-accepted standard method used to identify genes harboring disease-associated rare and low-frequency NSVs, we applied three different burden tests to evaluate the collection of NSVs found for each of the 21 genes tested. Disease association was determined by stringent criteria requiring significant association (P < 0.05 adjusted for 21 genes tested; P < 0.0024) by at least one burden test and a corroborating test with at least nominal significance (P < 0.05).
In the first stage, 82 rare and low-frequency NSVs (MAF ≤ 5% either in cases or controls) were validated (Tables S1 and S2). Call rates for variants were higher than 98%. After removing one variant in MEFV because of linkage disequilibrium with another variant, we applied the three burden tests to the collections of independent NSVs found in each of the 21 genes tested. Permutation was performed 1,000,000 times to test for significance. By the C-alpha test, a statistical test developed to account for a mixture of risk, protective, and neutral effects within a set of rare variants (11), we found significant association (P < 0.0024) for IL23R (5 NSVs, permuted P = 3.9 × 10−4) and nominally significant associations (P < 0.05) in IL1R1 (3 NSVs, permuted P = 0.021) and TLR4 (6 NSVs, permuted P = 0.043) in Japanese BD (Table 1). Both the data-adaptive sum test (12) and the step-up test (13), which are alternative approaches to test for associations of collective rare and low-frequency variant effects (Materials and Methods), showed significant association (P < 0.0024) of the IL23R NSVs and nominal significance (P < 0.05) of the IL1R1 NSVs (Table 1).
Table 1.
Association of nonsynonymous coding low-frequency and rare variants identified in Japanese and Turkish populations
| Gene | C-alpha test | aSum test | Step-Up test | ||||||
| Alla | Damagingb | Rare damagingc | Alla | Damagingb | Rare damagingc | Alla | Damagingb | Rare damagingc | |
| Japanese | |||||||||
| IL23R | 0.00039** | 0.00035** | 0.06 | 2 × 10E-5** | 0.00022** | NA | 0.00044** | 0.00026** | NA |
| IL10 | 0.36 | 0.36 | 0.36 | 0.22 | 0.22 | 0.22 | 0.19 | 0.19 | 0.19 |
| NLRP3 | 0.14 | 0.056 | 0.056 | 0.022* | 0.032* | 0.032* | 0.10 | 0.10 | 0.10 |
| IL1R1 | 0.021* | 0.38 | 0.38 | 0.0033* | NA | NA | 0.031* | NA | NA |
| IL1B | 0.23 | 0.23 | 0.23 | NA | NA | NA | NA | NA | NA |
| IL1RN | 0.64 | NA | NA | 0.9822 | NA | NA | 0.81 | NA | NA |
| STAT4 | 0.35 | 0.50 | 0.50 | 0.34 | 0.51 | 0.51 | 1 | 1 | 1 |
| CCR1 | 0.50 | 0.50 | 0.50 | 0.23 | 0.23 | 0.23 | 0.51 | 0.51 | 0.51 |
| ERAP1 | 0.84 | 0.84 | 0.84 | 0.39 | 0.39 | 0.39 | 0.75 | 0.75 | 0.75 |
| TLR4 | 0.043* | 0.22 | 0.22 | 0.21 | 0.64 | 0.64 | 0.068 | 0.068 | 0.068 |
| CASP1 | 0.51 | NA | NA | NA | NA | NA | NA | NA | NA |
| TNFRSF1A | 0.64 | NA | NA | 0.71 | NA | NA | 0.77 | NA | NA |
| KLRK1 | 0.25 | 0.25 | 0.46 | 0.44 | 0.44 | 0.59 | 0.55 | 0.55 | 0.55 |
| KLRC3 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| KLRC1 | 0.22 | 0.20 | 0.20 | 0.41 | 0.53 | 0.53 | 0.53 | 0.53 | 0.53 |
| PSTPIP1 | 0.15 | 0.15 | 0.15 | 0.026* | 0.063 | 0.063 | 0.16 | 0.16 | 0.16 |
| MEFV | 0.41 | 0.41 | 0.83 | 0.82 | 0.95 | 0.62 | 0.78 | 0.78 | 0.78 |
| NOD2 | 0.21 | 0.036* | 0.036* | 0.23 | 0.092 | 0.092 | 0.10 | 0.10 | 0.10 |
| Turkish | |||||||||
| IL23R | 0.0014** | 0.00099** | 0.90 | 0.073 | 0.004* | 0.73 | 0.019* | 0.019* | 0.90 |
| IL10 | 0.26 | 0.26 | 0.26 | 0.42 | 0.42 | 0.42 | 0.39 | 0.39 | 0.39 |
| IL1R1 | 0.42 | 0.48 | 0.48 | 0.81 | NA | NA | 0.46 | NA | NA |
| STAT4 | 0.24 | 0.24 | 0.24 | 0.61 | 0.61 | 0.61 | 0.39 | 0.39 | 0.39 |
| CCR1 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| ERAP1 | 0.53 | 0.65 | 0.24 | 0.61 | 0.93 | 0.12 | 0.62 | 0.62 | 0.62 |
| TLR4 | 0.0029* | 0.0028* | 0.048* | 0.0024* | 0.001** | 0.024* | 0.034* | 0.034* | 0.034* |
| KLRK1 | 0.16 | 0.15 | 0.15 | 0.037* | 0.024* | 0.024* | 0.15 | 0.15 | 0.15 |
| KLRC1 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| MEFV | <1 × 10E-6** | <1 × 10E-6** | 0.29 | 4 × 10E-6** | 4 × 10E-6** | 0.63 | 0.58 | 0.58 | 0.58 |
| MEFV w/o M694V | 0.14 | 0.39 | 0.29 | 0.097 | 0.57 | 0.63 | 0.58 | 0.58 | 0.58 |
| NOD2 | 0.023* | 0.019* | 0.019* | 0.152 | 0.019* | 0.019* | 1 | 0.081 | 0.081 |
| Meta-analysis (Stouffer's z) | |||||||||
| IL23R | 6.9 × 10E-5** | 4.4 × 10E-5** | 0.79 | 0.0054* | 0.0002** | NA | 0.0018** | 0.0015** | NA |
| TLR4 | 0.0008** | 0.002** | 0.0348* | 0.0017** | 0.0021** | 0.037* | 0.015* | 0.015* | 0.015* |
| NOD2 | 0.016* | 0.0063* | 0.0063* | 0.12 | 0.0092* | 0.0092* | 1 | 0.045* | 0.045* |
Association of combined rare and low frequency nonsynonymous variants from targeted genes in 528 Japanese BD cases and 586 controls, and 1,933 Turkish BD cases and 1,872 controls. Genes with no rare or low frequency NSVs are not shown. *P < 0.05 (nominal significance) and **P < 0.0024 (significance correcting for 21 genes) after permutation 1,000,000 times for C-alpha test and 10,000 times for adaptive sum and step-up tests. NA, not applicable due to lack of variants in either cases or controls, an absence of predicted damaging variants, or detection of a single variant, which cannot be evaluated by the adaptive sum or step-up tests. Meta-analyses (Stouffer’s weighted Z-method) of the Japanese and Turkish association data for IL23R, TLR4, and NOD2 are shown.
Includes all validated nonsynonymous variants with MAF ≤ 5% in either cases or controls.
Indicates P values after permutation including only variants predicted to be damaging.
Includes damaging variants with MAF < 1% in either cases or controls.
To account for the predicted effects on proteins of the variants as a priori information in the statistical analysis, we used computational protein prediction programs (SIFT (http://sift.jcvi.org) or PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) to obtain prediction categorization of the variants and limited the analysis to variants predicted to be damaging (Materials and Methods). By the C-alpha test, in addition to IL23R, which retained significance after the filtering (with 2 damaging variants, permuted P = 3.5 × 10−4), NOD2 was also nominally significant (6 damaging variants, permuted P = 0.036). All 6 of the damaging NSVs of NOD2 were also rare (MAF < 1% in either case or control participants), and therefore showed the same significance for disease association (Table 1).
Discovery and Validation of Rare and Low-Frequency NSVs in Targeted Genes in Turkish Samples.
In stage 2, rare and low-frequency variants in the 10 GWAS genes (IL23R, IL10, CCR1, STAT4, KLRK1, KLRC1, KLRC2, KLRC3, KLRC4, and ERAP1), three autoinflammatory pathway genes implicated from stage 1 (IL1R1, NOD2, TLR4), and MEFV were selected for resequencing in the Turkish population (384 case participants and 384 control participants). We applied the same method used in stage 1 (Fig. S1). Ninety-seven rare and low-frequency NSVs from the resequencing were subsequently validated by TOF-MS or TaqMan assays (Tables S1 and S3). After removing 4 variants because of linkage disequilibrium, the results of C-alpha testing in 1,933 patients with BD and 1,872 control participants were significant (P < 0.0024) for IL23R (11 NSVs, permuted P = 0.0014) and nominally significant (P < 0.05) for TLR4 (11 NSVs, permuted P = 0.0029) and NOD2 (26 NSVs, permuted P = 0.023) (Table 1). To support the findings by C-alpha test, we performed two additional burden tests (the adaptive sum and step-up tests), as we did for the Japanese samples. TLR4 and IL23R showed P < 0.05 in the adaptive sum and/or the step-up tests, supporting their associations identified by the C-alpha test (Table 1).
Limiting the analysis to damaging variants improved significance for IL23R (7 damaging NSVs, P = 9.9 × 10−4), NOD2 (19 damaging NSVs, P = 0.019), and TLR4 (5 damaging NSVs, P = 0.0028). Furthermore, rare damaging variants in TLR4 (4 rare damaging NSVs, P = 0.048) and NOD2 (19 rare damaging NSVs, P = 0.019) also showed nominal significance.
A meta-analysis combining the C-alpha test results from the Japanese and Turkish populations for IL23R, TLR4, and NOD2 showed that low-frequency and rare NSVs in IL23R (P = 6.9 × 10−5) and TLR4 (P = 8.0 × 10−4) were significantly associated with BD (P < 0.0024) (Table 1). These significant associations were additionally supported (all with P ≤ 0.015) by both the adaptive sum and step-up tests (Table 1). Predicted damaging and rare damaging variants in NOD2 were found to be nominally associated with BD (P < 0.05) in the C-alpha test meta-analysis and were further supported with nominal significance by the adaptive sum and step-up test meta-analyses. Positions of the low-frequency and rare NSVs we identified in IL23R, TLR4, and NOD2 are shown in Fig. 1.
Fig. 1.
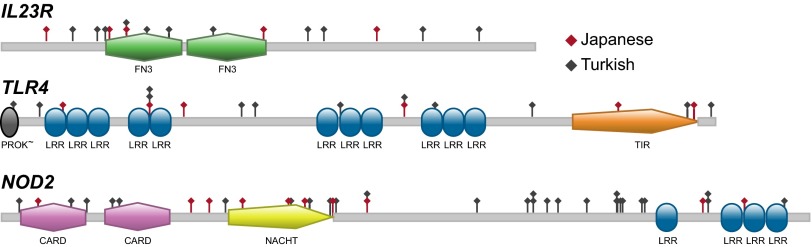
Positions of low-frequency and rare variants in IL23R, TLR4, and NOD2 detected by targeted resequencing. Red and gray symbols indicate variants identified in the Japanese and Turkish samples, respectively. The variant identifications can be found in Tables S2 and S3. Protein domains: CARD, caspase activation and recruitment domain; FN3, Fibronectin tenth type III domain; LRR, leucine-rich repeat domain; NACHT, nucleoside triphosphatase domain identified in NAIP, CIITA, HET-E, and TP1 proteins; PROK∼, prokaryotic membrane lipoprotein lipid attachment domain; TIR, Toll/interleukin 1 receptor domain. Figures were created with Prosite (http://prosite.expasy.org).
MEFV M694V Associated with BD in the Turkish Population.
MEFV rare and low-frequency variants (25 NSVs) also exhibited significant association with BD in the Turkish population (C-alpha permuted P < 1.0 × 10−6 and adaptive sum permuted P = 4.0 × 10−6). Mutations in this gene are responsible for the recessive autoinflammatory disease familial Mediterranean fever (FMF), for which a high carrier rate has been found in Mediterranean populations, suggesting a selective advantage of the FMF-associated alleles (14). FMF-associated variants have previously been suggested to play a role in BD (15, 16). A closer look at the MEFV variants showed that the significance was primarily derived from a single FMF mutation, p.Met694Val (M694V) (without M694V, C-alpha permuted P = 0.14), indicating that variants such as p.Met680Ile and p.Val726Ala, which are two other mutations commonly responsible for FMF in this population (14), do not confer strong risk for BD in the heterozygous state (Table 1 and Table S3). M694V has been recognized as a highly penetrant FMF variant that is associated with more severe inflammatory manifestations of FMF, including increased risk for amyloid deposition (14). Furthermore, this same allele has been identified as a risk factor for ankylosing spondylitis (17) and inflammatory bowel disease (18) in the Turkish population, suggesting that MEFV M694V predisposes or contributes to particular inflammatory conditions.
This variant was not found in the Japanese population, nor was an association of MEFV rare and low-frequency variants found in Japanese BD. In the Turkish samples, the MEFV M694V allele by itself achieved genome-wide significance (allelic test P = 1.79 × 10−12; odds ratio [OR], 2.65), which was not identified by GWAS (2, 4), presumably because this mutation was not represented by the GWAS single-nucleotide polymorphisms (SNPs). Furthermore, heterozygous carriage of the allele without a second exon 10 mutation was associated with significant BD risk (genotypic test, P = 2.79 × 10−12; OR, 2.73; 178/1,933 patients with BD and 67/1,872 control participants carried the M694V allele without another exon 10 mutation).
Discussion
Identifying disease-associated rare or low-frequency variants is hindered by the low power of such analyses in conventional single-variant association tests. We therefore used three different burden tests developed to increase power by combining the rare and low-frequency NSVs of a gene and evaluating their collective effect. The C-alpha test was recently developed by Neale et al. (11) to robustly evaluate genes containing both risk and protective alleles by testing for a nonrandom distribution of the set of variants in the case and control participants. A nonrandom distribution indicates that the set of rare and low-frequency variants contributes to disease risk. Two other burden tests using a regression model with grouped variants allowing both deleterious and protective alleles, the adaptive sum test (12) and the step-up test (13), were also applied. The adaptive sum test is a data-driven approach to determining the direction of variants for a subsequent sum test. The step-up test incorporates the weights of the inverse of the MAF in control participants and employs an optimal selection strategy for aggregating variants.
No single-burden test is recognized as a gold standard method for testing the association of rare variants. Because the three different methods use different information from the data, we sought consistent results across two or more burden tests, requiring a minimum P < 0.0024 in one test and P < 0.05 in the second test, to provide robust evidence for association in our study. Using these criteria, we find robust evidence for the association of rare and low-frequency NSVs in IL23R and TLR4 with BD. We also found a strong association of a single NSV, the M694V FMF mutation of MEFV, with BD in the Turkish population.
Although our study includes the largest collection of patients with BD yet reported for a genetic analysis, our ability to identify genes with disease-associated NSVs among the group of genes tested was limited by the number of samples used for variant discovery (382 Japanese case participants, 384 Japanese control participants, 384 Turkish case participants, and 384 Turkish control participants) and by the total number of case and control participants genotyped (2,461 case participants and 2,458 control participants). Thus, some of the other genes we sequenced may also have disease-influencing variants, but despite applying burden tests, our study may be underpowered to detect their effects.
The association of rare and low-frequency NSVs in IL23R, one of the genes identified by GWAS, further supports its role in BD. Among the IL23R NSVs, most of the significance was derived from two low-frequency variants with protective effects (higher frequency in control participants than in case participants): p.Gly149Arg (G149R) in the Japanese population [allelic test OR, 0.54; 95% confidence interval (CI), 0.39–0.75] and p.Arg381Gln (R381Q) in the Turkish population (allelic test OR, 0.68; 95% CI, 0.54–0.85). These variants are also protective for Crohn disease (Table 2) (7), suggesting that BD and Crohn disease may share some pathogenic cytokine response pathways.
Table 2.
Risk effects of the rare and low frequency nonsynonymous variants associated with Behçet disease (BD) and Crohn disease (CD)
| Gene | Variant | BD | CD | Function |
| IL23R | G149R | Protective | Protective | Unknown |
| R381Q | Protective | Protective | Reduces interleukin 23 dependent interleukin 17 production | |
| TLR4 | D299G | Protective | Risk | Reduces response to LPS |
| T399I | Protective | Risk | Reduces response to LPS | |
| NOD2 | R702W | Protective | Risk | Reduces response to MDP |
| G908R | Protective | Risk | Reduces response to MDP | |
| L1007fs | Protective | Risk | Reduces response to MDP | |
| MEFV | M694V | Risk | Risk | Increases response to LPS |
Conditional analysis and linkage disequilibrium showed that association of the IL23R low-frequency coding G149R NSV was independent of the previously reported disease-associated common variant rs1495965 in the Japanese samples (455 Japanese case participants and 596 control participants: r2, 0.01; D′, 0.33), but R381Q partially explained the protective effect of the minor allele variant of the reported rs924080 association in Turkish samples (1151 Turkish case participants and 1260 control participants: R381Q and rs924080 have r2, 0.07 and D′, 0.90; the P value for association of rs924080 was reduced from 3.9 × 10−6 to 1.6 × 10−4 by conditioning on R381Q).
In this study, we found that rare and low-frequency NSVs in two genes of innate immunity, MEFV and TLR4, were associated with BD, suggesting that innate immune responses directed against bacterial components are involved in the pathogenesis of BD. TLR4 is a sensor for the bacterial component lipopolysaccharide (LPS) (19). On LPS stimulation, neutrophils and monocytes produce inflammatory cytokines such as TNF-α, which is a recognized target for the treatment of BD. Two of the TLR4 variants, p.Asp299Gly (D299G) and p.Thr399Ile (T399I), that we found in the Turkish population have previously been associated with hyporesponsiveness to endotoxin (20). Although these hyporesponsive variants have previously been associated with risk for Crohn disease (21), they were apparently protective for BD [D299G: OR, 0.64 (95% CI, 0.47–0.86); T399I: OR, 0.82 (95% CI 0.63–1.06); Table 2].
Although NOD2 did not meet our stringent criteria for robust disease association, analyses limited to the damaging or the rare damaging NSVs were nominally significant in our meta-analysis across all three burden tests (Table 1). Three coding variants in NOD2, p.Arg702Trp (R702W), p.Gly908Arg (G908R), and p.Leu1007fsinsC (L1007fs), that are associated with risk for Crohn disease were present in the Turkish samples, but not in the Japanese samples. These Crohn disease–associated alleles, previously shown to reduce responses to MDP (22), showed BD protective effects (OR, 0.40, 0.66, and 0.38, for R702W, G908R, and L1007fs, respectively; Table 2), which is also consistent with three previous reports in which these Crohn disease–associated alleles were reported at a lower frequency in patients with BD than in healthy control patients (23–25). These data suggest that some inflammatory processes seen in Crohn disease are distinct from those seen in BD. Interestingly, the locations of the Japanese and the Turkish NOD2 NSVs differ: the Japanese variants cluster around the nucleoside triphosphatase NACHT domain, whereas the Turkish variants cluster at the leucine-rich repeat end of the protein (Fig. 1), perhaps reflecting the differences in the frequencies of colitis in BD observed in these populations, which is low among Turkish patients but high among Japanese patients with BD. Functional analyses of the uncharacterized NOD2 variants will be required to better understand their possible roles in disease, which may be BD-specific and different from Crohn disease.
The role of MEFV in human disease is not fully understood; however, current evidence suggests that FMF-associated MEFV variants are gain-of-function mutations leading to increased responsiveness to bacterial products (26). Mice bearing the FMF mutation MEFV M694V activate caspase 1 more efficiently than wild-type mice, which results in overproduction of interleukin 1β when stimulated by LPS alone (26). The strong association we found of MEFV M694V with BD risk links innate immunity to BD.
The BD risk-associated coding alleles with known functions of TLR4, NOD2, and MEFV are all associated with greater responsiveness to bacterial products than the disease-protective alleles, suggesting that robust host responses to microbes contribute to the inflammation of BD. We recently found a BD-associated common SNP near CCR1, the C–C motif chemokine receptor 1, a receptor that is important for the recruitment of effector immune cells to sites of inflammation. Unlike the functionally characterized TLR4, NOD2, and MEFV BD-associated coding alleles, the BD-associated noncoding CCR1 allele is associated with reduced gene function (4), which could impair microbial clearance. Taken together, the genetic data suggest that both impaired microbial clearance and exuberant host responses contribute to BD susceptibility.
Although BD has features of innate immune response dysregulation, the previous GWAS failed to identify disease-associated common variants in genes of innate immunity. Deep resequencing of targeted genes in BD, however, led us to identify disease-associated rare and low-frequency NSVs in genes involved in innate immunity, as well as in adaptive immunity, suggesting that both innate and acquired immune interactions between host and microbes play important roles in the pathogenesis of BD.
Materials and Methods
Patients.
The study consisted of stages 1 and 2, as shown in the diagram in Fig. S1. In stage 1, Japanese BD and control samples included in the previously reported GWAS were used (3). All of the Japanese patients with BD fulfilled Japanese BD diagnostic criteria (3). For stage 2, both the Turkish patients with BD and control participants enrolled in the previously reported GWAS (2) and an additional similarly collected 815 case participants and 627 control participants were included (4). Turkish patients with BD fulfilled the International Study Group Diagnostic Criteria for BD (27). We excluded individuals who also met the Tel-Hashomer clinical criteria for the diagnosis of FMF (28) from the Turkish GWAS collection (n = 6). All study participants provided written informed consent, and the study was approved by the ethics committees of all of the investigative institutions.
Pooled DNA.
Pooled DNA was prepared as previously described, with modifications (5). Concentrations of genomic DNA were measured by Quant-IT dsDNA high-sensitivity kit (Invitrogen) in triplicate and then adjusted to 10 ng/μL. Eight DNA pools from patients with BD (n = 382 or 384) and 8 pools from healthy control participants (n = 384) were prepared for both the Japanese and Turkish collections. Each pool consisted of up to 48 equally represented samples, resulting in up to 96 alleles per pool.
PCR of Exons from Targeted Genes.
For stage 1, IL10, IL23R, CCR1, STAT4, KLRK1, KLRC1, KLRC2, KLRC3, KLRC4, and ERAP1, identified by previous GWAS, and genes involved in autoinflammatory diseases or innate immunity (MEFV, NLRP3, PSTPIP1, NOD2, TNFRSF1A, TLR4, IL1B, IL1RN, IL1R1, CASP1, and PYCARD) were selected for deep resequencing. Primers, PCR conditions, and DNA polymerases (LA Taq, Takara; Advantage GC-rich LA kit, Clontech; and AmpliTaq Gold, Applied Biosystems) used in the study are listed in Table S4. PCR was performed using pooled DNA as the template. All of the PCR amplicons were visualized by agarose gel electrophoresis [E-Gel 96, 2% (wt/vol) agarose, Invitrogen]. Estimated equimolar amounts of all of the PCR amplicons from a single DNA pool were combined, purified by QIAquick PCR Purification Kit (Qiagen), and adjusted to 2.5 μg DNA in 125 μL TE for fragmentation. In stage 2, IL10, IL23R, CCR1, STAT4, KLRK1, KLRC1, KLRC2, KLRC3, KLRC4, ERAP1, TLR4, NOD2, IL1R1, and MEFV were selected for resequencing in the Turkish BD collection DNA pools.
DNA Fragmentation.
Combined PCR amplicons were sheared with the S2 Adaptive Focused Acoustics 200-bp protocol (Covaris). Fragmented DNAs were further purified by QIAquick PCR Purification Kit (Qiagen) and analyzed with high-sensitivity DNA chips on a 2100 Bioanalyzer (Agilent Technologies).
Library Preparation.
Adapters were ligated to the fragmented PCR products with a ChIP-Seq DNA sample prep kit (Illumina) according to manufacturer’s protocol with modification. Fragmented PCR amplicons (100 ng) were treated with T4 and Klenow polymerase (20°C for 30 min) to make blunt-end DNA. After the addition of adenine to 3′ ends (37°C for 30 min), adapters were added and incubated at room temperature overnight. After purification with Ampure XP (Agencourt Bioscience), the ligated products were amplified by 14 cycles of PCR. After purification by Ampure XP, samples were loaded onto 2% (wt/vol) agarose gels, and DNA fragments of ∼220 bp were extracted by MinElute Gel Extraction Kit (Qiagen). DNA was quantified by the 2100 Bioanalyzer and adjusted to 1.5 μM in 0.1% Tween in TE.
Deep Sequencing and Data Quality Control.
Resequencing was performed by Genome Analyzer IIx (Illumina) with a single-end 36-cycle sequencing kit V4 (Illumina). Data were aligned and SNPs detected by CASAVA 1.6 software, allowing 4 mismatches in 36-bp reads. We also used Wheeler Aligner (http://bio-bwa.sourceforge.net) for the alignment, the Genome Analysis Toolkit (www.broadinstitute.org/gsa/wiki/index.php/Main_Page) for realignment and recalibration, and Samtools (http://samtools.sourceforge.net) to analyze per base coverage. Quality of resequencing was evaluated by FastQC software (Babraham Bioinformatics). Depth of coverage for each pool is shown in Fig. S2. All of the pools achieved mean coverage >2500× (>50× per sample and >25× per allele). A >500× coverage was achieved for 84% and 96% of the targeted nucleotides in the Japanese pools and Turkish pools, respectively.
We first randomly selected 177 of 275 variants called in the Japanese pooled DNA libraries and validated them by TOF-MS (iPLEX, Sequenom) in all of our Japanese samples (528 case participants and 586 control participants), including the variant discovery samples. All samples had call rates higher than 90%, and the call rates of all validated SNPs were higher than 98%. Among the validated variants, we compared the MAF estimated by resequencing and based on the individual genotypes in each pool. The results showed good agreement between the two methods, indicating that pooled DNA preparation and resequencing gave good estimates of the actual allele frequencies (Fig. S3).
We found a substantial number of false-positives from the initial analysis (93/177; 52.5%). Many of the false-positive SNP calls were from nucleotides with low coverage (<500×), from low-quality pools with a quality score lower than 20 at any bp position, or from variants that were common (MAF > 4%) in any of the pools but not found in the 1000 Genomes project data.
On the basis of this experience and our interest in the rare and low-frequency variants, we applied the following criteria to select subsequent variants for validation: the variant call must come from a nucleotide with sequence coverage >500× (i.e., >5×/allele in a pool of 48 DNAs) and must have an estimated MAF >1% in at least one pooled DNA; the call or calls must come from pools with high-quality scores (resequencing was repeated for low-quality pools; 2 pools from stage 1 were repeated); variants with an estimated MAF > 4% in any of the DNA pools, but not found in 1000 Genomes data or the Infevers database (for MEFV, NOD2, PSTPIP1, and TNFRSF1A, http://fmf.igh.cnrs.fr/ISSAID/infevers), were omitted; and variants for validation were limited to nonsynonymous coding variants with pooled DNA estimated average MAF < 10%. After filtering the variant calls for the first three requirements, the transition/transversion ratio from resequencing the Japanese pools was 1.8, and from the Turkish pools it was 3.0.
Rare and low-frequency, nonsynonymous coding variants within the targeted gene exons fulfilling these criteria were selected for validation with TOF-MS. We also tested any additional low-frequency (1–5% MAF) coding NSVs within our targeted genes from HapMap 1000 Genomes data (HapMap JPT for the Japanese collection and CEU for the Turkish collection). All of the validated rare variants (MAF < 1% in a sample collection) were evaluated to determine whether they were found by individual genotyping in one of the DNA samples from the pool in which the variant was called. If the pool did not contain a sample with the variant, the TOF-MS of any DNA sample with a missing genotype was repeated. Thus, for all of the validated rare variants, every pool containing the variant by resequencing contained at least one sample with the variant by MS-TOF. In addition, if a rare variant was found in a DNA specimen from a pool in which the resequencing failed to detect a variant, Sanger sequencing was performed to determine whether the MS-TOF result was a false-positive. A comparison of the number of pools containing a variant identified by resequencing and by TOF-MS for all of the validated rare variants (in genes IL10, IL23R, IL1B, IL1R1, IL1RN, NLRP3, MEFV, TNFRSF1A, PSTPIP1, CASP1, PYCARD, NOD2, and TLR4) also agreed well (Fig. S3 D and E).
Validation and Additional Genotyping.
All of the called SNPs from resequencing were validated with iPLEX assays (Sequenom), as previously described (2), by genotyping the samples from the DNA pools and were further analyzed by genotyping additional samples. The number of samples genotyped was as follows: stage 1 (528 Japanese case participants and 586 Japanese control participants from previous GWAS) and stage 2 (1116 Turkish case participants and 1249 Turkish control participants enrolled in previous GWAS and an additional 817 Turkish case participants and 623 Turkish control participants). For all of the variants that failed TOF-MS design or reaction, TaqMan genotyping was performed (Applied Biosystems). Genotyping was performed in an unbiased fashion by masking the phenotype of the samples.
Statistical analysis.
All statistical analyses were performed in each population separately. Calculation of odds ratios, principal component (PC) analysis to evaluate whether the low-frequency variants clustered in a genetically identifiable subset of the population, and LD calculations among validated variants were performed by SVS v7.5.2 software (Golden Helix). For coding variants in linkage disequilibrium (r2 > 0.8 and/or D′ > 0.8), we included only one in the gene-based burden tests, selecting the one predicted to be damaging (Tables S2 and S3). For TLR4 D299G/T399I, we included the lower-frequency D299G variant. The individuals with the low-frequency alleles were distributed throughout the population cluster by their locations on PC1 and PC2, showing that these alleles were not limited to a genetically defined subset of the population (Fig. S4). The C-alpha test and permutations were performed with an R script (kindly provided by Benjamin Neale, The Broad Institute of MIT and Harvard, Cambridge, MA) (11).
Singleton variants (found in only one sample) within a gene were combined for the analysis, as described (11). C-alpha test statistic P values < 0.05 after 1,000,000 permutations were considered nominally significant. We also performed two other burden tests based on a regression model, the adaptive sum test (12) and the step-up test (13), to corroborate the results from C-alpha test. These two methods were designed to take into consideration opposite direction effects (protective and deleterious) when combining the variants. For the adaptive sum test, we preselected 0.1 for the alpha value, which is the marginal regression P value cutoff for the decision to flip the coding of opposite directional variant genotypes to generate one direction for all variants, as suggested by previous research (12), for the subsequent sum test per gene. The step-up test incorporated three aspects in the generalized regression model for testing association: the direction of variant effects, the weights of the inverse of the minor allele frequencies in control participants, and the selection of the best combination of rare variants included in a single aggregated group in a manner similar to forward stepwise regression. We used 10,000 or 50,000 permutations for obtaining P values in both tests and performed tests using R v2.14.1 with the script released by Han and Pan (12) for the adaptive sum test and using R package “thgenetics” v0.3 for the step-up test. For each gene, we tested association between disease and the gene, using subsets of validated variants classified as “Damaging variants” or “Rare damaging variants.” Damaging variants were those encoding premature stop codon and frameshift mutations and those missense changes predicted to be deleterious by either the computational protein prediction program, SIFT (either “damaging with low confidence” or “damaging”) (29) or PolyPhen2 (either “possibly damaging” or “probably damaging”) (30). Rare damaging variants were the subset of damaging variants with MAF < 1% in either case or control participants from the appropriate population.
Significant association (correcting for 21 genes) had P value < 0.0024. For genes with at least nominal significance (P < 0.05) in each population, we performed a meta-analysis to combine the data from the two populations using Stouffer's weighted Z-method (31). Genes with robust evidence for disease-associated rare and low-frequency variants were those with significant association (P < 0.0024) by at least one burden test and that also had one or more corroborating test or tests with at least nominal significance (P < 0.05) for the same variant collection, either within a single population or in the combined population meta-analysis.
Supplementary Material
Acknowledgments
We thank Dr. Benjamin Neale (Broad Institute of MIT and Harvard) for providing the C-alpha test script and Dr. Ivona Aksentijevich (National Human Genome Research Institute, National Institutes of Health) for comments on the manuscript. This research was supported by the Intramural Research Programs of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Human Genome Research Institute and by the Center for Human Immunology, Autoimmunity, and Inflammation of the NIH; by the Istanbul University Research Fund; and by Research on Specific Disease of the Health Science Research Grants from the Japanese Ministry of Health, Labor, and Welfare and the Japan Rheumatism Foundation.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306352110/-/DCSupplemental.
References
- 1.Sakane T, Takeno M, Suzuki N, Inaba G. Behcet's disease. N Engl J Med. 1999;341:1284–1291. doi: 10.1056/NEJM199910213411707. [DOI] [PubMed] [Google Scholar]
- 2.Remmers EF, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet's disease. Nat Genet. 2010;42:698–702. doi: 10.1038/ng.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizuki N, et al. Genome-wide association studies identify IL23R -IL12RB2 and IL10 as Behcet's disease susceptibility loci. Nat Genet. 2010;42:703–706. doi: 10.1038/ng.624. [DOI] [PubMed] [Google Scholar]
- 4.Kirino Y, et al. Genome-wide association analysis identifies new susceptibility loci for Behcet's disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. 2013;45:202–207. doi: 10.1038/ng.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raychaudhuri S. Mapping rare and common causal alleles for complex human diseases. Cell. 2011;147:57–69. doi: 10.1016/j.cell.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Momozawa Y, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat Genet. 2010;43:43–47. doi: 10.1038/ng.733. [DOI] [PubMed] [Google Scholar]
- 8.Rivas MA, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diogo D, et al. Rare, low-frequency, and common variants in the protein-coding sequence of biological candidate genes from GWASs contribute to risk of rheumatoid arthritis. Am J Hum Genet. 2013;92:15–27. doi: 10.1016/j.ajhg.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marth GT, et al. The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neale BM, et al. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han F, Pan W. A data-adaptive sum test for disease association with multiple common or rare variants. Hum Hered. 2010;70:42–54. doi: 10.1159/000288704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann TJ, Marini NJ, Witte JS. Comprehensive approach to analyzing rare genetic variants. PLoS ONE. 2010;5:e13584. doi: 10.1371/journal.pone.0013584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: The molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27:621–668. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Touitou I, et al. MEFV mutations in Behcet's disease. Hum Mutat. 2000;16:271–272. doi: 10.1002/1098-1004(200009)16:3<271::AID-HUMU16>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 16.Livneh A, et al. A single mutated MEFV allele in Israeli patients suffering from familial Mediterranean fever and Behcet's disease (FMF-BD) Eur J Hum Genet. 2001;9:191–196. doi: 10.1038/sj.ejhg.5200608. [DOI] [PubMed] [Google Scholar]
- 17.Cosan F, et al. Association of familial Mediterranean fever-related MEFV variations with ankylosing spondylitis. Arthritis Rheum. 2010;62:3232–3236. doi: 10.1002/art.27683. [DOI] [PubMed] [Google Scholar]
- 18.Akyuz F, et al. Association of the MEFV gene variations with inflammatory bowel disease in Turkey. J Clin Gastroenterol. 2013;47:e23–e27. doi: 10.1097/MCG.0b013e3182597992. [DOI] [PubMed] [Google Scholar]
- 19.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 20.Arbour NC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 21.Shen X, et al. The Toll-like receptor 4 D299G and T399I polymorphisms are associated with Crohn's disease and ulcerative colitis: A meta-analysis. Digestion. 2010;81:69–77. doi: 10.1159/000260417. [DOI] [PubMed] [Google Scholar]
- 22.Girardin SE, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 23.Kappen JH, et al. Low prevalence of NOD2 SNPs in Behcet's disease suggests protective association in Caucasians. Rheumatology (Oxford) 2009;48:1375–1377. doi: 10.1093/rheumatology/kep292. [DOI] [PubMed] [Google Scholar]
- 24.Ahmad T, et al. CARD15 polymorphisms in Behcet's disease. Scand J Rheumatol. 2005;34:233–237. doi: 10.1080/03009740510018714. [DOI] [PubMed] [Google Scholar]
- 25.Uyar FA, Saruhan-Direskeneli G, Gul A. Common Crohn's disease-predisposing variants of the CARD15/NOD2 gene are not associated with Behcet's disease in Turkey. Clin Exp Rheumatol. 2004;22:S50–S52. [PubMed] [Google Scholar]
- 26.Chae JJ, et al. Gain-of-function pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34:755–768. doi: 10.1016/j.immuni.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.International Study Group for Behcet's Disease Criteria for diagnosis of Behcet's disease. Lancet. 1990;335:1078–1080. [PubMed] [Google Scholar]
- 28.Livneh A, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40:1879–1885. doi: 10.1002/art.1780401023. [DOI] [PubMed] [Google Scholar]
- 29.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitlock MC. Combining probability from independent tests: The weighted Z-method is superior to Fisher's approach. J Evol Biol. 2005;18:1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.