

Abstract
Neural activity enhances adult neurogenesis, enabling experience to influence the construction of new circuits. GABAA receptor-mediated depolarization of newborn neurons in the adult and developing brain promotes glutamatergic synaptic integration since chronic reduction of GABA depolarization impairs morphological maturation and formation of glutamatergic synapses. Here we demonstrate an acute role of GABA depolarization in glutamatergic synaptic integration. Using proopiomelanocortin enhanced–green fluorescent protein reporter mice, we identify a developmental stage when adult-generated neurons have glutamatergic synaptic transmission mediated solely by NMDA receptors (NMDARs), representing the initial silent synapses before AMPA receptor (AMPAR)-mediated functional transmission. We show that pairing synaptic stimulation with postsynaptic depolarization results in synapse unsilencing that requires NMDAR activation. GABA synaptic depolarization enables activation of NMDARs in the absence of AMPAR-mediated transmission and is required for synapse unsilencing induced by synaptic activity in vitro as well as a brief exposure to an enriched environment in vivo. The rapid appearance of AMPAR-mediated EPSCs and the lack of maturational changes show that GABA depolarization acutely allows NMDAR activation required for initial synapse unsilencing. Together, these results also reveal that adult-generated neurons in a critical period for survival use GABA signaling to rapidly initiate functional glutamate-mediated transmission in response to experience.
Introduction
Adult-generated dentate granule cells (GCs) must survive and integrate into the existing network to contribute to hippocampal function. Only a fraction of newly generated GCs survive, whereas most undergo apoptosis within the first few weeks of maturation (Biebl et al., 2000; Dayer et al., 2003; Sierra et al., 2010). Interestingly, environmental enrichment (EE), learning tasks, and electrical stimulation selectively increase survival of newborn GCs that are 1–3 weeks postmitotic (Kee et al., 2007; Tashiro et al., 2007; Kitamura et al., 2010; Anderson et al., 2011), indicating this is a critical period when an animal's experience controls the long-term fate of each newborn cell. Single-cell knockdown studies show that cell-autonomous synaptic mechanisms can enable precise cell- and information-specific control of survival. For example, blocking GABAergic depolarization (Jagasia et al., 2009) or deleting NMDA receptors (NMDARs) (Tashiro et al., 2006) enhances the death of critical-period GCs. Thus, cell survival is associated with both GABAergic and glutamatergic signaling, but it is not clear how experience affects synaptic activation of new neurons in this critical period or how GABA and glutamatergic mechanisms interact to promote initial synaptic integration.
A well-established model of synaptic integration involves activity-dependent incorporation of AMPA receptors (AMPARs) into nascent “silent” NMDAR-only-containing synapses (Isaac et al., 1995; Liao et al., 1995; Durand et al., 1996; Wu et al., 1996; Isaac et al., 1997). AMPAR incorporation (“synapse unsilencing”) requires depolarization to relieve Mg2+ block of NMDARs. In developing neonatal neurons, inadequate glutamate receptor-mediated depolarization limits activity-dependent AMPAR incorporation (Liao and Malinow, 1996), suggesting that another source of depolarization is needed for initial synapse unsilencing. Because of the high intracellular [Cl−] maintained by the early expression of the NKCC1 chloride importer, GABAA receptor (GABAR)-mediated responses in developing neurons are depolarizing (Represa and Ben-Ari, 2005). Ben-Ari et al. (1997) proposed that GABA could provide the depolarization necessary for initial synapse unsilencing, because synaptic GABAR-mediated depolarization can allow NMDAR activation (Leinekugel et al., 1997; Wang and Kriegstein, 2008). However, the many trophic actions of GABA on neural maturation before excitatory synaptogenesis (Represa and Ben-Ari, 2005; Ge et al., 2007a) and the difficulty of identifying neurons at the appropriate developmental stage has precluded the direct demonstration of the acute role of GABA in NMDAR-mediated synapse unsilencing.
Here we establish the critical role of GABA depolarization in AMPAR incorporation at the first excitatory synapses on adult-generated neurons. We identify a population of newborn GCs that have glutamatergic transmission mediated solely by silent synapses. We show that synapse unsilencing produced by neural activity in vitro requires GABAR-mediated depolarization as well as NMDAR activation. Housing mice in EE for 2 weeks, known to increase survival of newborn neurons, leads to the appearance of AMPAR EPSCs without a change in morphology or intrinsic properties. Intriguingly, merely 2 h of EE is sufficient to generate synapse unsilencing in newborn GCs that is dependent on GABAR-mediated depolarization. Our results demonstrate that GABA depolarization is required for the unsilencing of initial glutamatergic synapses on developing neurons and allows rapid functional integration of critical-period neurons in response to experience.
Materials and Methods
Animals
Adult (8–12 weeks old) female hemizygous proopiomelanocortin enhanced–green fluorescent protein (POMC–GFP) transgenic mice (Cowley et al., 2001; Overstreet et al., 2004) were maintained on a C57BL/6J background. All animal procedures followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee. Mice were housed in standard cages (12 × 7.5 inches) until 8–9 weeks of age when some were moved to EE cages (23 × 14 inches) consisting of 8–14 mice with a variety of toys and shelters that were exchanged twice per week (no running wheels). For 2 h EE experiments, animals were given an intraperitoneal injection of vehicle, 30 mg/kg bumetanide or 10 mg/kg tiagabine and 15 min later moved from their home cage to EE. To test locomotion and activity levels, a separate group of mice were injected with vehicle, bumetanide or tiagabine and after 15 min were placed in a 16 × 16 × 15 inches transparent box with a video monitoring system. All mice were killed at the same time of day (early morning) to avoid potential confounds from circadian cycles.
Electrophysiology
Anesthetized mice were perfused intracardially with ice-cold cutting solution containing the following (in mm): 110 choline chloride, 25 d-glucose, 2.5 MgCl2, 2.5 KCl, 1.25 Na2PO4, 0.5 CaCl2, 1.3 Na-ascorbate, 3 Na-pyruvate, and 25 NaHCO3 (bubbled with 95% O2/5% CO2). The brain was quickly removed, and 300 μm horizontal slices were prepared using a Vibratome 3000EP and the same cutting solution. Transverse slices were then incubated at 37°C for 30 min in artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 Na2PO4, 2 CaCl2, 1 MgCl2, 25 NaHCO3, and 25 d-glucose (bubbled with 95% O2/5% CO2). They were then transferred to room temperature in the same solution and incubated at least 30 min before use.
Slices were bathed in oxygenated 32°C ACSF at a rate of ∼2 ml/min during recordings. Whole-cell recording pipettes contained the following (in mm): 120 Cs-gluconate, 17.5 CsCl, 4 MgCl2, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 7 phosphocreatine, and 10 EGTA, pH 7.2 (310 mOsm). For pairing experiments, the same Cs-gluconate internal solution was used, but the EGTA was reduced to 0.1 mm (see Fig. 2). For perforated patch experiments (see Fig. 3A), pipettes contained 135 mm KCl, 4 mm MgCl2, 10 mm HEPES, and 200 μg/ml gramicidin, with the pipette tip containing the same solution without gramicidin and voltages were corrected for series resistance offline (average Rs = 99 ± 40 MΩ). For experiments with physiological intracellular [Cl−], the CsCl was increased to 30 mm and Cs gluconate decreased to 97 mm (see Fig. 3B,C). To examine the intrinsic properties (see Fig. 5D), patch pipettes contained the following (in mm): 120 K-gluconate, 17.5 KCl, 4 MgCl2, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 7 phosphocreatine, and 0.1 EGTA. Synaptic stimulations were delivered in the inner molecular layer using a monopolar stimulating electrode filled with ACSF and constant-current stimulator (Digitimer). Unless noted otherwise, for each newborn GC the minimum stimulus intensity to evoke a postsynaptic response was found (typically a GABA PSC), and then the intensity was increased to four times that threshold intensity for the remainder of the experiment. The probability of failures in response to synaptic stimulation was measured during 10–100 synaptic stimulations. Series resistance was uncompensated (8–35 MΩ), and experiments were discarded if substantial changes (>20%) were observed. Junction potentials were not corrected. Recordings were filtered at 2 kHz and sampled at 10 kHz (MultiClamp 700B; Molecular Devices). Data were acquired with AxoGraph X acquisition software (Axograph Scientific) and analyzed with Axograph X and Prism (GraphPad Software).
Figure 2.
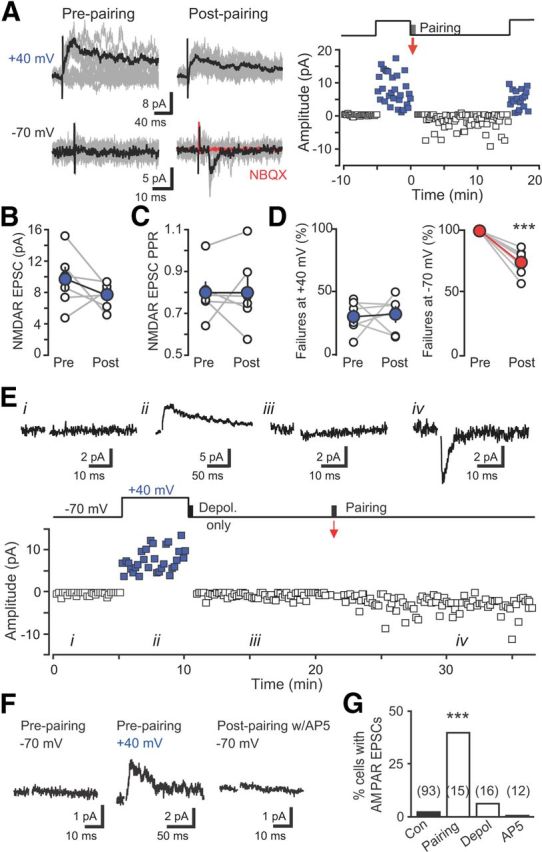
Initial synapse unsilencing requires NMDAR activation. A, Left, EPSCs before and after pairing reveal rapid incorporation of AMPARs. Ten traces overlaid (gray) with averages (black). Right, Plot of EPSC amplitude versus time at −70 mV (black) and +40 mV (blue). Pairing protocol (red arrow) consisted of postsynaptic depolarization to 0 mV for 300 ms with presynaptic stimulation repeated at 1 Hz for 30 s. Baseline noise was subtracted. B, C, Neither the NMDAR EPSC amplitude (B) nor paired-pulse ratio (C) was altered by pairing. D, Failures of the NMDAR EPSC were unaffected by the pairing protocol (measured as the number of successes/total number of stimulations), whereas failures of the AMPAR EPSC were reduced. ***p < 0.001, paired t tests. E, Example EPSCs from the indicated times (above) and amplitude time course (below) showing that depolarization alone did not induce AMPA EPSCs that subsequently were induced by pairing depolarization with presynaptic stimulation. Artifacts blanked for clarity. F, Synapse unsilencing by the pairing protocol was blocked by AP5 (50 μm). G, Summary graph showing the percentage of newborn GCs with AMPAR EPSCs in each condition. The number of cells tested is shown in parentheses. ***p < 0.001, χ2 test compared with control. Con, Control; Depol, Depolarization.
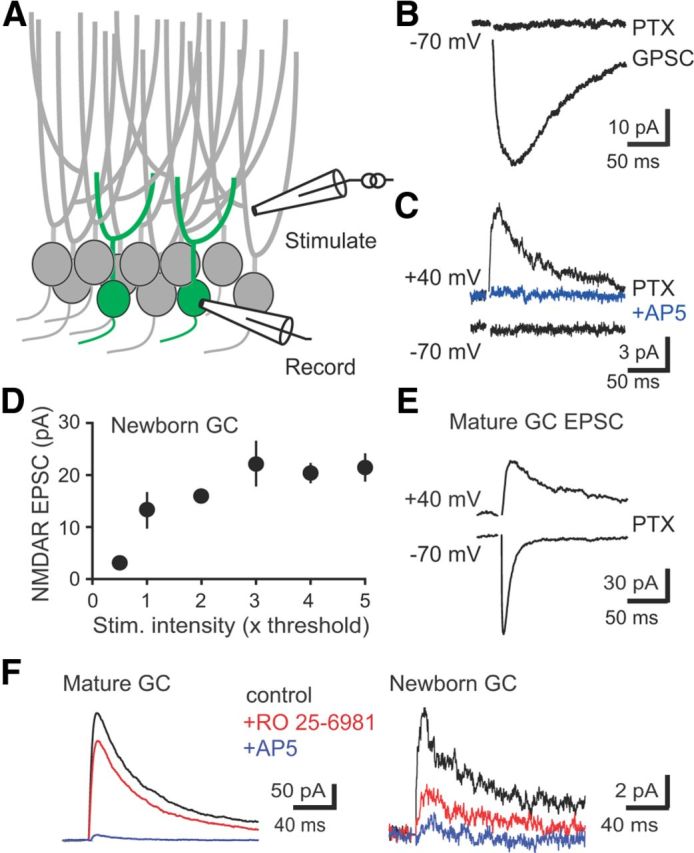
Figure 3.

Synaptic GABAergic depolarization allows activation of NMDARs. A, Left, Examples of GABAergic PSCs at holding potentials between −70 and 0 mV in gramicidin perforated patch recordings from newborn GCs in control and bumetanide-treated slices. NBQX (5 μm) and AP5 (50 μm) were used to isolate GPSCs. Right, Bumetanide (10 μm) hyperpolarized the reversal potential of GPSCs. B, Left, PSPs measured at −70 mV were partially blocked by AP5, indicating an NMDAR-mediated component. Right, PTX blocked PSP at −70 mV, although NMDAR EPSPs measured near EGABA (approximately −34 mV) were unaffected by PTX (insets; calibration: 2 mV, 100 ms). C, Right, Quantification of the AP5-sensitive component of PSPs recorded in control ACSF (n = 4) and PTX (n = 5) at each membrane potential. ***p < 0.001; ns, not significant; t test. Con, Control.
Figure 5.
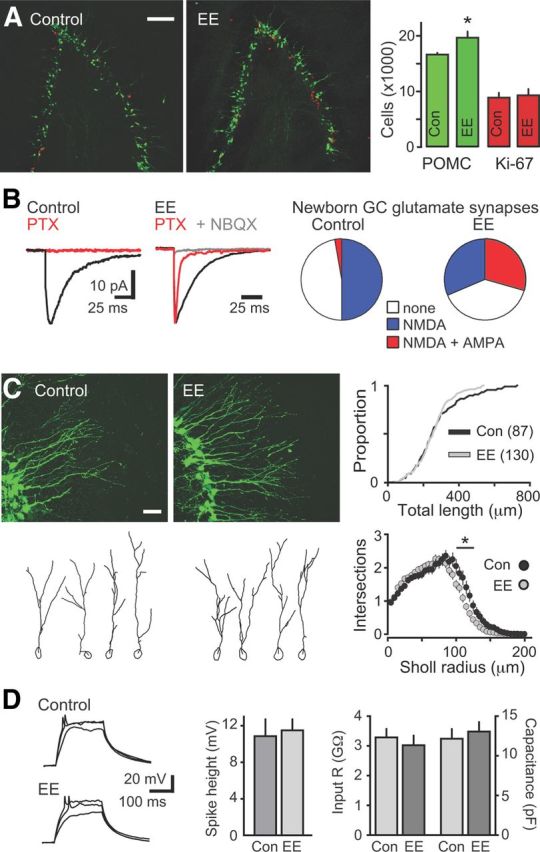
EE enhances newborn GC survival and promotes and initial synapse unsilencing in vivo. A, Left, POMC–GFP+ (green) and Ki67+ (red) expression identify newborn GCs and proliferative cells, respectively, in the DG of mice housed in standard environment (control; Con) and EE. Scale bar, 100 μm. Right, EE increased the number of GFP+ cells (n = 7 control and EE mice, p < 0.05, Mann–Whitney U test) without changing the number of Ki67+ cells (n = 3 control and EE mice, p = 0.78). B, PTX (red) blocked synaptic currents in newborn GCs from control mice (left), whereas NBQX-sensitive AMPAR EPSCs were present in newborn GCs from EE mice (middle). Right, EE increased the percentage of newborn GCs with functional AMPAR-containing synapses (p < 0.0001) and decreased the percentage of newborn GCs with no response (p < 0.05, χ2 test, n = 93 newborn GCs in control and 147 in EE). C, Left, Representative confocal images (top) and dendrite tracings (bottom) of newborn GCs from control and EE mice. Scale bar, 20 μm. Right top, The cumulative distribution of TDLs was not altered by EE, and there was no difference in the TDL (281 ± 15 vs 261 ± 9 μm), farthest extent of the dendrites (119 ± 4 vs 113 ± 2 μm), or number of nodes (5.8 ± 0.4 vs 5.5 ± 0.3; p > 0.05, unpaired t tests). Right bottom, There was a slight reduction in intersections at a distance of 95–120 μm from the soma in newborn GCs after EE (*p < 0.05). D, Left, Examples of current injections (40–60 pA) in newborn GCs from control and EE mice. Middle, EE did not alter the percentage of cells that fired action potentials (p = 0.12, χ2 test, n = 19 control and 25 EE) or the amplitude of spikes (0.76, unpaired t test). Right, The input resistance (p = 0.54) and capacitance (p = 0.59) were not different between newborn GCs in control and EE mice, indicating that newborn GCs were at the same developmental stage.
Immunohistochemistry
Mice were intracardially perfused with 0.1 m PBS, followed by 4% PFA. Brains were collected and postfixed overnight in 4% PFA. Fifty micrometer horizontal brain slices were prepared on a Vibratome 1000. Tissue was processed as free-floating sections and mounted in Vectashield (Vector Laboratories).
GFP staining.
Tissue was rinsed three times in 0.1 m PBS for 10 min, blocked in PBS blocking buffer (10% normal goat serum, 3% BSA, and 0.4% Triton X-100 in 0.1 m PBS) for 90 min at room temperature, and then incubated with 1:1000 dilution of anti-GFP antibody (catalog #A-21311, anti-GFP conjugated to Alexa Fluor 488; Invitrogen) in PBS blocking buffer overnight at 4°C with gentle shaking. Tissue was then rinsed in PBS three times for 10 min.
Ki67 staining.
Sections were permeabilized and blocked in a TBS buffer containing Triton X-100 (0.4%), BSA (3%), glycine (1%), and normal goat serum (10%) (blocking buffer). Sections were incubated with a rabbit anti-Ki67 antiserum (10 μg/ml, ab15580; Abcam) in blocking buffer (overnight, 4°C). After three washes in TBS, sections were incubated in a goat anti-rabbit antibody coupled to Alexa Fluor 568 (1:200 in blocking buffer, overnight, 4°C; Invitrogen).
Stereology
The number of GFP-expressing (GFP+) or Ki67+ cells was determined using stereological counts as described previously (Pugh et al., 2011). The number of cells was counted on every sixth section for the entire left dentate gyrus (DG), allowing for an estimated total for each hippocampus using StereoInvestigator (MicroBrightField).
Dendrite analyses
Sections immunostained for GFP were imaged with an Olympus FluoView 300 confocal microscope using a 60× oil-immersion objective with a Z step of 0.25 μm. Neuronal morphology was traced from confocal image stacks using Neurolucida (version 9; MicroBrightField). In all cases, dendrites were drawn and analyzed by an investigator naive to treatment conditions. Cells with obvious truncations were excluded from analysis. Measurements included total dendrite length (TDL) and Sholl analyses of length, nodes and intersections at 5 μm intervals. The furthest extent of dendritic projections was determined by the furthest Sholl radius containing measurable dendrite length (i.e., rounded to the nearest 5 μm). TDL, nodes, and dendritic extents were compared by two-sample unpaired t tests. Sholl analyses were compared using a two-way ANOVA with Bonferroni's post hoc tests (Prism).
Statistical analyses
Statistical analyses were performed using paired and unpaired Student's t tests, ANOVA with Bonferroni's post hoc tests, or χ2 test as indicated. Data were tested for normality and variance before analyses with t tests and ANOVA. When data were not normally distributed, a Mann–Whitney U test was performed. When variance was unequal between multiple groups, a Kruskall–Wallis test with post hoc Dunn's comparison was used. Statistical analyses were performed using Prism software (GraphPad Software).
Results
Glutamatergic transmission mediated by NMDAR2B-containing silent synapses
We used POMC–GFP reporter mice that identify newborn GCs at a well-defined developmental stage when GABA is depolarizing and AMPAR-mediated transmission is not yet established (Overstreet et al., 2004; Overstreet Wadiche et al., 2005). In adult mice, this stage is typically achieved at 10–12 d after mitosis (Overstreet et al., 2004; Overstreet-Wadiche et al., 2006), coinciding with a critical period for activity-dependent survival (Tashiro et al., 2007; Jagasia et al., 2009; Sierra et al., 2010). We made whole-cell recordings from POMC–GFP-labeled newborn GCs in acute hippocampal slices. Consistent with the timing and sequence of synaptic integration established by retroviral labeling that shows GABA synaptic input develops before glutamatergic input (Espósito et al., 2005; Ge et al., 2006), focal stimulation near the dendrites of newborn GCs held at −70 mV evoked GABAAR-mediated postsynaptic currents (GPSCs) that were blocked by picrotoxin (PTX; Fig. 1A,B; 91 of 93 cells). Surprisingly, subsequent depolarization to +40 mV revealed an AP5-sensitive NMDAR-mediated EPSC (NMDAR EPSC) in ∼50% of newborn GCs (n = 46 of 93 cells; Fig. 1C). Importantly, minimal stimulation was not required to identify NMDAR-only synapses in POMC–GFP-labeled neurons. Increasing stimulation intensity generated only small NMDAR-only EPSCs (silent synapses) in newborn GCs (Fig. 1D), whereas all neighboring mature GCs displayed dual AMPAR/NMDAR EPSCs at similar intensities (Fig. 1E). NMDAR EPSCs in newborn GCs were blocked by the selective NMDAR2B receptor antagonist Ro 25-6981 [R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol] (1 μm; 76 ± 6% block, n = 9) to a greater extent compared with NMDAR EPSCs recorded in mature GCs (23 ± 8%, n = 4; p < 0.0001, unpaired t test; Fig. 1F), consistent with inclusion of NMDAR2B receptors at immature synapses (Tovar and Westbrook, 1999; Ge et al., 2007b). Thus, POMC–GFP expression identifies a developmental stage when glutamatergic signaling is mediated solely by silent synapses that have a large NMDAR2B receptor component. The appearance of NMDAR-only synapses in only ∼50% of cells and the inability to substantially increase the NMDAR EPSC amplitude with increasing stimulation intensities suggests that these silent synapses represent the initial glutamatergic contacts.
Figure 1.
Newborn GCs have silent NMDAR-only synapses. A, Diagram of recording configuration. B, Synaptic stimulation near newborn GC dendrites evoked GPSCs blocked by PTX (100 μm; −70 mV holding potential). C, A typical silent synapse in PTX, with an NMDAR EPSC at +40 mV but no AMPAR EPSC at −70 mV. NMDAR EPSCs were blocked by AP5. D, The amplitude of NMDAR EPSCs in newborn GCs did not increase substantially with increased stimulation intensity (n = 5 cells with silent synapses), suggesting that very few synapses were present. Stimulus intensity was normalized to the intensity at which the NMDAR EPSC was just detectable (1× threshold). E, Similar stimulation intensities evoked dual AMPAR/NMDAR EPSCs in all neighboring mature GCs (n = 15). F, Differential block of NMDAR EPSCs in mature and immature GCs by Ro 25-6981. NMDAR EPSCs in mature GCs were recorded in NBQX.
NMDAR-dependent synapse unsilencing
We tested whether activity-dependent incorporation of AMPARs at the first synapses on adult-born neurons is similar to synapse unsilencing during early brain development (Isaac et al., 1995; Liao et al., 1995; Durand et al., 1996). First, we used low-frequency synaptic stimulation (0.1 Hz) in the presence of PTX to identify newborn GCs with NMDAR-only silent synapses, confirming glutamate release onto the recorded cell. Then we paired synaptic stimulation with postsynaptic depolarization to 0 mV to test for rapid incorporation of AMPARs. In 6 of 15 cells, AMPAR EPSCs developed over minutes after return to low-frequency stimulation (Fig. 2A). The appearance of AMPAR EPSCS was not accompanied by changes in the NMDAR EPSC amplitude or paired-pulse ratio (Fig. 2B,C) or the percentage of failures at +40 mV (Fig. 2D), ruling out presynaptic alterations in glutamate release by the pairing protocol. Thus, coincident postsynaptic depolarization with synaptic stimulation is sufficient for incorporation of AMPARs at the first silent synapses, resulting in rapid functional integration of adult-generated GCs into the hippocampal circuit.
To test whether Ca2+ influx during postsynaptic depolarization of newborn GCs (Stocca et al., 2008) was sufficient for synapse unsilencing, we repeated the pairing protocol in the absence of synaptic stimulation. However, postsynaptic depolarization alone was unable to induce AMPAR EPSCs in 15 of 16 cells, although subsequent pairing of synaptic stimulation with depolarization was successful in two of four attempts (Fig. 2E). Because depolarization alone was not sufficient for synapse unsilencing, we tested whether synaptic stimulation was required to active NMDARs. After first confirming the presence of NMDAR EPSCs at +40 mV, inclusion of AP5 (50 μm) during the pairing protocol prevented the appearance of AMPAR EPSCs (0 of 12 cells; Fig. 2F,G), even with increased frequency of depolarization (5 Hz, n = 6; data not shown). Thus, similar to silent synapses in the developing brain (Isaac et al., 1995; Liao et al., 1995; Durand et al., 1996), NMDAR activation is required for AMPAR incorporation at the first glutamatergic synapses on adult-generated neurons.
Synapse unsilencing by GABAR- and NMDAR-mediated synaptic activity
We next sought to directly test a long-standing postulate that synaptic GABA depolarization allows initial synapse unsilencing during patterned neural network activity (Ben-Ari et al., 1997). The presence of silent synapses along with the lack of AMPAR-containing synapses and the depolarized reversal potential for exogenous GABA-activated currents (Overstreet Wadiche et al., 2005) make POMC–GFP-labeled newborn GCs valuable for addressing this question.
First we confirmed that synaptic GABAR activation depolarizes newborn GCs. The GABA reversal potential (EGABA) measured with gramicidin perforated patch recordings was −34 ± 3 mV (n = 6; Fig. 3A), more depolarized than the measured resting membrane potential (RMP) of −50.1 ± 2.0 mV (p < 0.01; n = 5) and much more depolarized than EGABA in mature cells (data not shown; −80 ± 4 mV, n = 4; p < 0.001). Note that the very high input resistance of newborn GCs (Overstreet et al., 2004; Overstreet Wadiche et al., 2005) results in an artificially depolarized RMP attributable to the shunt through the recording seal; therefore, the actual RMP is likely more hyperpolarized than −50 mV. GABA depolarizes newly generated GCs due to activity of the NKCC1 chloride importer (Ge et al., 2006) that is blocked by the diuretic bumetanide. Bumetanide (10 μm) caused a negative shift in EGABA (−49 ± 4 mV, n = 9; p < 0.05; Fig. 3A), confirming that POMC–GFP cells have depolarizing GABAergic signaling attributable to high expression of NKCC1. Conversely, bumetanide did not affect EGABA in mature GCs (−86 ± 5 mV, n = 5; p = 0.11; data not shown), and thus bumetanide provides a tool to hyperpolarize EGABA in newborn GCs.
To test whether synaptic GABA depolarization removed the voltage-dependent Mg2+ block from NMDARs at silent synapses, we recorded postsynaptic potentials (PSPs) in newborn GCs with silent synapses using the calculated native intracellular [Cl−]i. PSPs measured at −70 mV were reduced by AP5, indicating the presence of a NMDAR-mediated component (Fig. 3B,C, left). However, the NMDA component was absent when GABAARs were first blocked with PTX (Fig. 3B, right). Furthermore, NMDAR EPSPs measured at a depolarized potential near EGABA (approximately −34 mV) in the same cells were unaffected by PTX (Fig. 3B,C, insets). Together, these results show that synaptic GABAR-mediated depolarization relieves the Mg2+ block of synaptic NMDARs at the first silent synapses on newborn GCs.
Directly testing the role of synaptic GABA depolarization in synapse unsilencing is difficult because silent synapses cannot be identified with GPSCs intact, and AMPAR EPSCs in newborn GCs were too small to be reliably resolved at the GABA reversal potential. Thus, we tested whether driving synaptic activity before recording was sufficient for AMPAR incorporation that we quantified as the percentage of newborn GCs with AMPAR EPSCs. We induced synaptic network activity in acute slices with the potassium channel blocker 4-aminopyridine (4-AP; 100 μm; Michelson and Wong, 1994). 4-AP generates rhythmic low-frequency GABA release onto newborn GCs as well as glutamate release that can be detected at +40 mV (Markwardt et al., 2009, 2011; Fig. 4A). After pretreating slices for 2 h in 4-AP, we tested synaptic input to newborn GCs after 4-AP washout (confirmed by the absence of rhythmic synaptic activity). Intriguingly, the percentage of newborn GCs with AMPAR EPSCs was increased to 34% (11 of 32 cells; Fig. 4B,F) compared with newborn GCs from untreated slices (2%; 2 of 93 cells; p < 0.0001, χ2 test), demonstrating that 4-AP-evoked synaptic activity recapitulates synapse unsilencing by postsynaptic depolarization paired with synaptic glutamate release (Fig. 1).
Figure 4.
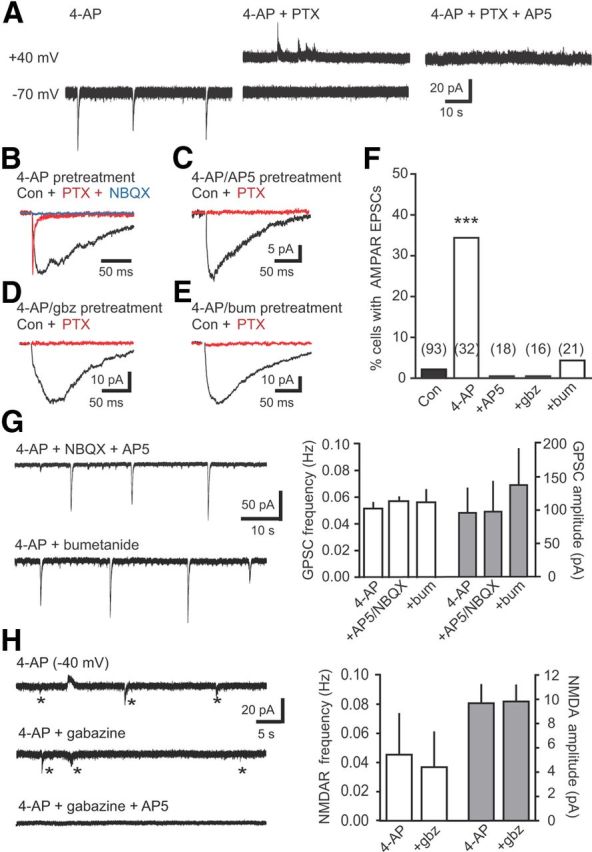
GABA depolarization is required for initial synapse unsilencing by synaptic activity. A, 4-AP (100 μm) induced rhythmic GABAR activation blocked by PTX in newborn GCs held at −70 mV. Subsequent depolarization to +40 mV revealed NMDAR EPSCs blocked by AP5 (50 μm). B, Example synaptic currents in a newborn GC after washout of 4-AP after 2 h incubation. Addition of PTX revealed an AMPAR EPSC (red; normalized to peak of GPSC) that was blocked by NBQX (10 μm; blue). C–E, Examples of synaptic currents in newborn GCs after incubation in 4-AP plus AP5 (50 μm; C), 4-AP plus gabazine (5 μm; D), or 4-AP plus bumetanide (10 μm; E). F, Summary of the percentage of newborn GCs with AMPAR EPSCs in control ASCF (black) or after 4-AP incubation with inclusion of the indicated antagonists (white). The number of cells tested is shown in parentheses. ***p < 0.001, χ2 test, compared with control. G, Left, Examples of rhythmic GABA release induced by 4-AP in the presence of blockers that prevent AMPAR incorporation. Right, The NKCC1 antagonist bumetanide (10 μm; n = 4) and the NMDAR and AMPAR antagonists AP5/NBQX (50 and 5 μm, respectively; n = 4) did not alter the frequency and amplitude of rhythmic GABAR PSCs (ANOVA). H, Left, Example of NMDAR-mediated EPSCs (asterisks) in a newborn GC held near the GABA reversal potential (small GPSCs are outward) in the indicated conditions. Right, Gabazine did not alter the frequency or amplitude of NMDAR EPSCs (n = 4, paired t test). bum, Bumetanide; Con, control; gbz, gabazine.
Synapse unsilencing by synaptic network activity was likewise dependent on NMDARs because inclusion of AP5 during the 4-AP pretreatment completely prevented the appearance of AMPAR EPSCs (Fig. 4C,F). We next tested the role of GABARs in synapse unsilencing by including the GABAR antagonist gabazine during 4-AP treatment and found that gabazine blocked the appearance of AMPAR EPSCs (Fig. 4D,F). To test whether GABAR-mediated depolarization was necessary for synapse unsilencing, we included bumetanide during 4-AP treatment and found that it also reduced the percentage of newborn GCs with AMPAR EPSCs back to control levels (Fig. 4E,F). These results are consistent with the idea that synapse unsilencing by synaptic activity requires GABAR-mediated depolarization to allow NMDAR activation. In separate control experiments, we tested whether each antagonist that blocked synapse unsilencing affected 4-AP-induced network activity. However, neither bumetanide nor AP5/NBQX affected the frequency and amplitude of rhythmic GABA PSCs in newborn GCs (Fig. 4G), consistent with a large literature showing that 4-AP-induced network activity in the DG is maintained in blockers of synaptic transmission (Müller and Misgeld, 1991; Michelson and Wong, 1994; Bijak and Misgeld, 1996; Markwardt et al., 2009; Gonzalez-Sulser et al., 2011). Additionally, the frequency and amplitude of NMDAR-mediated activity in 4-AP was unaffected by gabazine (measured at −40 mV; Fig. 4H), and robust rhythmic activity in mature GCs persisted in gabazine (0.077 ± 0.012 Hz, n = 4; data not shown). Although each antagonist could alter other aspects of network activity, the lack of effect of each blocker on GABA and glutamate release to newborn GCs suggests that blockade of AMPAR incorporation is attributable to postsynaptic actions. Together, these results show that GABA depolarization is necessary to trigger incorporation of synaptic AMPARs on newborn GCs in response to network activity in vitro.
Synapse unsilencing in vivo by EE
Our results suggest that POMC–GFP-labeled newborn GCs are primed to undergo functional integration via AMPAR incorporation into nascent silent synapses. We next sought to test whether synapse unsilencing can also occur in vivo with increased network activity. We housed mice in EE for 2 weeks to promote exploration and novelty-induced neural activity in DG (Nitz and McNaughton, 2004). As expected, EE enhanced the survival of newborn GCs measured by an increase the number of POMC–GFP GCs without a change in progenitor proliferation (Fig. 5A), consistent with the reported timing of a critical period for survival that occurs during the time window of POMC–GFP expression (Overstreet-Wadiche et al., 2006; Tashiro et al., 2007). We then compared newborn GC synaptic activity in acute slices from EE and control mice. Approximately 40% of newborn GCs after EE had NMDAR-only silent synapses (31 of 78 cells), similar to the percentage of newborn GCs with silent synapses in control mice. NMDAR EPSCs after EE were likewise sensitive to the selective NMDAR2B antagonist Ro 25-6981 (controls, 76 ± 6%, n = 9; EE, 74 ± 6%, n = 12; p = 0.8; data not shown). Remarkably, there was a large increase in the percentage of newborn GCs with AMPAR EPSCs after EE (Fig. 5B; 32%, 47 of 149 cells), consistent with synapse unsilencing in vivo, together with a decrease in the percentage of newborn GCs with no responses (Fig. 5B, right).
EE-induced synapse unsilencing is not associated with morphological maturation
The appearance of AMPAR EPSCs after 2 weeks of EE could result from accelerated maturation, because GABA-induced synaptic integration of developing neurons is typically associated with morphological maturation and dendrite outgrowth (Ge et al., 2006; Cancedda et al., 2007; Wang and Kriegstein, 2008; Duveau et al., 2011). However, we did not detect enhanced morphological maturation after EE. Sholl analysis revealed that the TDL, number of nodes, and extent into the molecular layer was unaltered by EE, and there was a slight decrease in dendrite complexity in the inner molecular layer (Fig. 5C). Furthermore, intrinsic measures of GC maturation, including input resistance and capacitance, were unaltered, and there was no change in the percentage of newborn GCs that fired action potentials or the amplitude of action potentials (Fig. 5D). Thus, POMC–GFP-labeled newborn GCs are at the same developmental stage in control and EE mice, despite the enhancement in glutamatergic synaptic input. However, neural activity induced by EE could accelerate the rate of GC maturation (Piatti et al., 2011), resulting in a more rapid progression of newborn GCs through the POMC–GFP-expressing stage, as occurs in the early postnatal period when the POMC–GFP stage is achieved a few days earlier (8–10 d after mitosis; Overstreet-Wadiche et al., 2006). Regardless, the lack of morphological and intrinsic alterations demonstrates that enhanced network activity induced by EE can promote glutamatergic synapse unsilencing independent of other measures of maturation.
Short exposure to EE is sufficient for synapse unsilencing in vivo
We used a shorter enrichment paradigm to test the role of GABAergic depolarization in synapse unsilencing in vivo because chronic interference with GABAergic depolarization impairs dendrite maturation and potentially the establishment of silent synapses (Ge et al., 2006; Wang and Kriegstein, 2008). Intriguingly, both a 24 h and a 2 h exposure to EE were equally effective in inducing AMPAR EPSCs in newborn GCs compared with the 2 week treatment (Fig. 6A,B; 24 h EE, 5 of 16 cells, p < 0.001 compared with control, p = 1.0 compared to 2 week EE; 2 h EE, 9 of 32 cells, p < 0.001 compared with control; p = 0.7 compared with 2 week EE; χ2 test). Thus, the appearance of AMPAR EPSCs likely results from acute activity-dependent synapse unsilencing rather than chronic nonspecific effects of EE. The sufficiency of 2 h is in accordance with the rapid time course of activity-dependent synapse unsilencing in vitro (Figs. 2, 4). Furthermore, the amplitude of AMPAR EPSCs induced by 2 weeks or 2 h of EE, pairing, and 4-AP incubation were similar (Fig. 6C), and there were no differences in the rise and decay times (data not shown; p = 0.99 and p = 0.53, respectively). The AMPA/NMDA ratio in newborn GCs was also similar across conditions (Fig. 6D,F) but lower than the AMPA/NMDA ratio in mature GCs from EE mice (Fig. 6E,F; n = 15; p < 0.05). There was a trend toward larger AMPAR EPSCs after in vitro 4-AP treatment that did not reach statistical significance, potentially implying that it was the strongest stimulus paradigm. Regardless, the similar frequency and properties of AMPA EPSCs across all conditions support the idea that synapse unsilencing by pairing or synaptic activity in vitro can also be achieved by the experience of EE in vivo.
Figure 6.
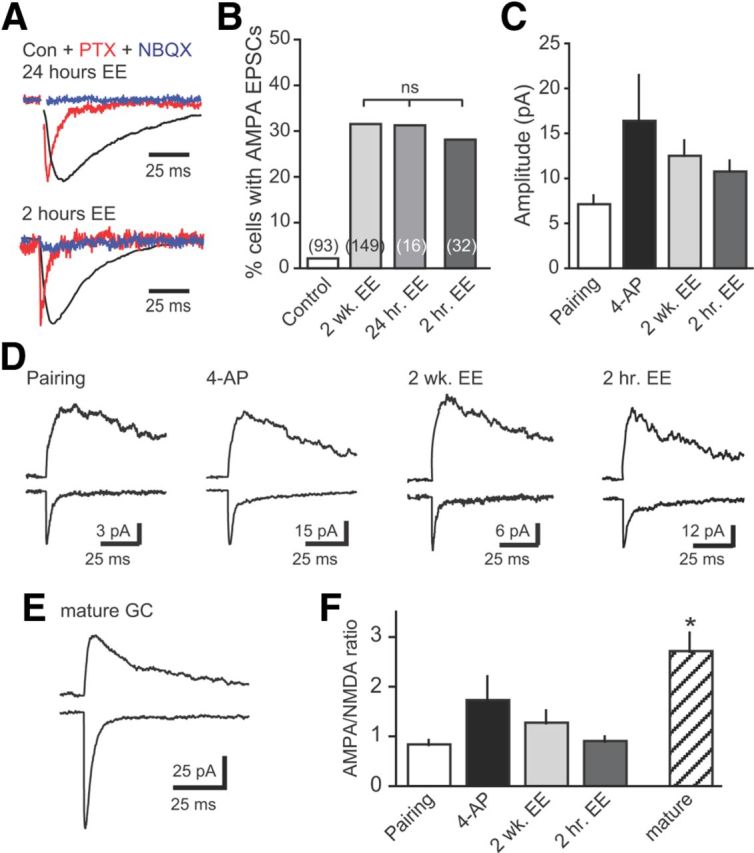
Short exposure to EE is sufficient for unsilencing in vivo. A, Examples of synaptic currents in newborn GCs after 24 or 2 h of EE, demonstrating that a short exposure to EE is sufficient for synapse unsilencing. EPSCs insensitive to PTX (red; normalized to peak of GPSC) were blocked by NBQX (5 μm; blue). B, Summary of the percentage of newborn GCs with AMPA EPSCs after exposure to EE for 2 weeks (data from Fig. 5), 24 h, or 2 h. C, The amplitude of AMPA EPSCs induced by pairing, 4-AP-driven synaptic activity, and EE was similar (p = 0.22, one-way ANOVA). D, Examples of AMPAR and NMDAR EPSCs in newborn GCs, measured at −70 mV and at +40 mV, respectively. E, Example of AMPAR and NMDAR EPSCs in a neighboring mature GC. F, The AMPA/NMDA ratio was similar in newborn GCs across all conditions (p = 0.16, ANOVA) but lower than in mature GCs (n = 15; *p < 0.01, t test of all newborn GCs compared with mature GCs). Con, Control.
GABA depolarization is required for synapse unsilencing in vivo
To examine the role of GABAergic depolarization in synapse unsilencing in vivo, we injected mice with bumetanide (30 mg/kg, i.p.) before the 2 h exposure to EE. Similar to its effects in vitro, bumetanide reduced synapse unsilencing in response to network activity induced by EE (Fig. 7A,B; 2 of 23 cells; p < 0.05 compared with vehicle control, χ2 test). To further test whether the effect of bumetanide resulted from impaired GABA depolarization of newborn GCs rather than altered inhibition within the network, we also manipulated GABA signaling by blocking neuronal GABA transporters (GAT1). Phasic GABA signaling to newborn GCs is robustly enhanced by blockade of GAT1, whereas phasic signaling to mature GCs is less affected (Markwardt et al., 2009). Whereas bumetanide selectively reduces GABA depolarization, tiagabine enhances GABA signaling regardless of the polarity of the GABAR response. Accordingly, tiagabine treatment (10 mg/kg, i.p.) before EE did not block synapse unsilencing in newborn GCs (Fig. 7A,B). Although there was a trend toward a higher percentage of newborn GCs with AMPA EPSCs after EE plus tiagabine (38 vs 28% in vehicle control), the difference did not achieve statistical significance (8 of 21 cells; p = 0.5, χ2 test). In separate experiments, treatment of mice with either bumetanide or tiagabine did not alter overall activity levels monitored in an open field, showing that altered behavior was unlikely to influence these results (Fig. 7C,D). Consistent with its previously reported anxiolytic effect (Thoeringer et al., 2010), tiagabine treatment enhanced the time spent in the center of the field (Fig. 7E), indicating that this dose was sufficient for CNS effects. Together, the selective block of synapse unsilencing by bumetanide but not tiagabine demonstrates that GABA depolarization is needed to activate the first silent synapses on adult-born GCs in response to a brief experience of EE.
Figure 7.
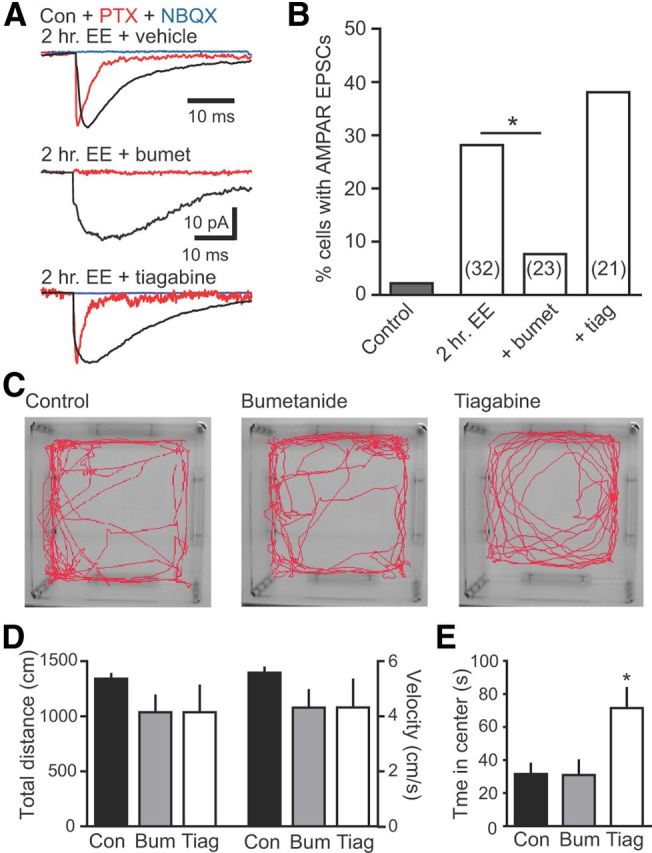
GABAergic depolarization is necessary for synapse unsilencing by EE. A, Examples of synaptic currents in newborn GCs with vehicle, bumetanide (30 mg/kg, i.p.; middle) or tiagabine (10 mg/kg, i.p.) treatment before 2 h of EE. B, Summary of the percentage of newborn GCs with AMPAR EPSCs in each condition. Bumetanide reduced the percentage of newborn GCs with EE-induced AMPA EPSCs, whereas tiagabine had no significant effect. The number of cells tested is shown in parentheses. *p < 0.05, χ2 test. There was no difference in the percentage of newborn GCs with AMPAR EPSCs in the 2 h EE uninjected and vehicle-injected control groups (4 of 15 and 5 of 17, respectively; p = 1.0, χ2 test), so they were combined (n = 32). C, A separate group of mice were given vehicle (control; n = 7), bumetanide (30 mg/kg; n = 7), or tiagabine (10 mg/kg; n = 6) before exploring a novel open field for 4 min. D, E, Neither bumetanide nor tiagabine altered the total distance traveled and velocity (D; distance, p = 0.3; velocity, p = 0.3, ANOVA), but tiagabine increased the time spent in the center (E; p < 0.05). bumet/Bum, Bumetanide; Con, control; tiag/Tiag, tiagabine.
Discussion
Experience-dependent synaptic modification is a well-established mechanism for neural plasticity in the adult brain. In the adult DG, experience can modify neural circuitry not only at the level of single synapses but also at the cellular level by controlling the long-term survival of newborn GCs. Here we show that EE, an experience that promotes survival, elicits AMPA EPSCs at the first silent NMDAR-only-containing synapses on newborn GCs. Using in vitro paradigms, we show that depolarization paired with synaptic stimulation drives rapid AMPAR incorporation at silent synapses in an NMDAR-dependent manner and that synaptic GABA depolarization relieves the voltage-dependent Mg2+ block of NMDARs at silent synapses. GABA depolarization and NMDAR activation work synergistically to initiate functional glutamatergic synaptic transmission, since rhythmic synaptic activity induced by 4-AP in vitro generates synapse unsilencing that requires both NMDAR activation and GABAergic depolarization. Furthermore, we show that in vivo synapse unsilencing in response to a brief experience of EE also requires GABAergic depolarization. Interestingly, we found a similar percentage of newborn GCs with functional glutamatergic transmission after a variety of activity paradigms in vivo and in vitro, likely representing a pool of newborn GCs primed to respond to correlated GABA depolarization and glutamate release. Together, these results illustrate that GABAergic synaptic input to critical-period neurons is required for rapid synapse unsilencing, and thus functional integration, in response to experience.
Defining the critical period during adult neurogenesis
Tashiro et al. (2007) described a critical period occurring 1–3 weeks after cell birth when the experience of EE enhances the survival of new GCs. Learning tasks and direct electrical stimulation of the perforant path likewise rescue cell death during this time (Kitamura et al., 2010, Anderson et al., 2011). The first 3 weeks of GC maturation is a dynamic phase of growth and synaptogenesis when new neurons transition from GABA-only signaling to functional integration that is defined by glutamatergic synapse formation with the perforant path (Espósito et al., 2005; Ge et al., 2006; Mongiat et al., 2009). Because the tempo of this transition is variable and subject to activity-dependent modification even at the single-cell level (Espósito et al., 2005; Ambrogini et al., 2010; Piatti et al., 2011), defining the precise functional stage corresponding to the critical period is challenging. The 1–3 week time window likely represents the duration of this critical period for the cohort of cells rather than the duration for an individual cell, similar to POMC–GFP expression that identifies a functional developmental stage that the majority of newborn cells achieve at 10–12 d after mitosis but the entire cohort achieves between ∼3 and 24 d after cell birth (Overstreet-Wadiche et al., 2006). Thus, the time window when GABAergic depolarization plays a major role in cell survival and synapse unsilencing may be brief, but heterogeneous maturation of cells within the cohort desynchronizes the timing across 3 weeks. We speculate that the critical period closes when sufficient glutamatergic innervation and NMDAR activation ensures transcriptional suppression of the intrinsic apoptotic pathway (Tashiro et al., 2006; Leveille et al., 2010).
GABA depolarization drives maturation and survival
The importance of GABA depolarization in neuronal development was clearly illustrated by single-cell knock-out studies showing that deletion of the chloride importer NKCC1 reduces newborn neuron dendrite outgrowth and synaptic integration (Ge et al., 2006; Wang and Kriegstein, 2008). Similarly, deletion of specific GABAA subunits alters the number and maturation of newborn GCs (Duveau et al., 2011; Song et al., 2012). Survival of 1- to 2-week-old GCs is specifically associated with GABA-induced CREB activation (Jagasia et al., 2009). Consistent with a role of CREB signaling in survival, ∼70% of POMC–GFP newborn GCs express pCREB, with a similar percentage exhibiting GABAR-induced spontaneous Ca2+ transients (Overstreet-Wadiche et al., 2006). CREB is upregulated by EE (Huang et al., 2006) and coordinates expression of widespread genes involved in survival and integration of adult-generated neurons (Merz et al., 2011), including the microRNA miR-132 that regulates excitatory synaptogenesis (Luikart et al., 2011). GABA-mediated depolarization presumably contributes to many Ca2+-dependent molecular cascades essential for proliferation, survival, and growth (Ge et al., 2007a), enabling GABA signaling to dynamically regulate adult neurogenesis across multiple stages of GC maturation (Dieni et al., 2012).
GABAergic depolarization drives AMPAR insertion
Nascent excitatory synapses in the developing brain contain exclusively NMDARs that become functional by activity-dependent trafficking of AMPARs (Durand et al., 1996), analogous to the postsynaptic mechanism of long-term potentiation (Isaac et al., 1995; Liao et al., 1995). AMPAR insertion requires activation of NMDARs that is typically generated by AMPAR-mediated depolarization at neighboring functional synapses. However, the source of depolarization at early developmental stages that lack AMPAR-containing synapses has not been directly addressed. It was proposed years ago that GABAergic synaptic activity provides the depolarization required for AMPAR insertion (Ben-Ari et al., 1997), and subsequent studies have confirmed a role of GABAergic depolarization in synaptic integration of developing neurons (Akerman and Cline, 2006; Ge et al., 2006; Wang and Kriegstein, 2008). However, impaired GABA depolarization delays dendrite development (Cancedda et al., 2007), confounding identification of the specific role of GABA depolarization in acute synapse unsilencing. Our results provide direct experimental verification of the longstanding idea that GABAergic synapses supply the acute depolarization necessary for rapid AMPAR insertion at NMDA-only-containing synapses on developing neurons. POMC–GFP-labeled GCs are particularly useful for this purpose because the lack of AMPAR-containing synapses negates the necessity of minimal stimulation paradigms and provides an all-or-none response.
Experience-dependent modification of dentate circuitry
Experience-dependent control of survival is proposed to sculpt the formation of new circuits by selecting individual critical-period cells that receive sufficient activation to support long-term integration into the network (Tashiro et al., 2006). This cell and information-specific process could potentially tune surviving cells to respond to future reinstatement of those experiences (Kee et al., 2007; Tashiro et al., 2007; but see Alme et al., 2010). Our data show that EE rapidly initiates functional excitatory integration, although the consequences for future activation of “rescued” GCs requires additional testing. Importantly, our results illustrate one specific mechanism by which GABA synaptic signaling enables glutamatergic synaptic integration in critical-period neurons that could contribute to impaired maturation and survival after cell-specific blockade of GABAergic depolarization (Ge et al., 2006; Tashiro et al., 2006; Jagasia et al., 2009; Duveau et al., 2011). GABA-mediated excitatory synapse unsilencing may represent a general mechanism whereby GABA signaling contributes to experience-dependent regulation of neurodevelopment in both the adult and developing brain.
Footnotes
This work was supported by National Institutes of Health Grants NS061788 and NS078887 (J.H.C.), NS064025 (L.S.O.-W.), and NS065920 (J.I.W.), University of Alabama at Birmingham (UAB) Intellectual and Developmental Disabilities Research Center Grant P30-HD38985, and UAB Neuroscience Blueprint Cores Grants P30-NS47466 and P30-NS57098. We thank members of the Wadiche laboratories and Dr. Anastasios Tzingounis for discussions throughout this project and Nick Piazza for participating in pilot experiments.
The authors have no competing financial interests.
References
- Akerman CJ, Cline HT. Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J Neurosci. 2006;26:5117–5130. doi: 10.1523/JNEUROSCI.0319-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alme CB, Buzzetti RA, Marrone DF, Leutgeb JK, Chawla MK, Schaner MJ, Bohanick JD, Khoboko T, Leutgeb S, Moser EI, Moser MB, McNaughton BL, Barnes CA. Hippocampal granule cells opt for early retirement. Hippocampus. 2010;20:1109–1123. doi: 10.1002/hipo.20810. [DOI] [PubMed] [Google Scholar]
- Ambrogini P, Cuppini R, Lattanzi D, Ciuffoli S, Frontini A, Fanelli M. Synaptogenesis in adult-generated hippocampal granule cells is affected by behavioral experiences. Hippocampus. 2010;20:799–810. doi: 10.1002/hipo.20679. [DOI] [PubMed] [Google Scholar]
- Anderson ML, Sisti HM, Curlik DM, 2nd, Shors TJ. Associative learning increases adult neurogenesis during a critical period. Eur J Neurosci. 2011;33:175–181. doi: 10.1111/j.1460-9568.2010.07486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaiarsa JL. GABAA, NMDA and AMPA receptors: a developmentally regulated “menage a trois.”. Trends Neurosci. 1997;20:523–529. doi: 10.1016/S0166-2236(97)01147-8. [DOI] [PubMed] [Google Scholar]
- Biebl M, Cooper CM, Winkler J, Kuhn HG. Analysis of neurogenesis and programmed cell death reveals a self-renewing capacity in the adult rat brain. Neurosci Lett. 2000;291:17–20. doi: 10.1016/S0304-3940(00)01368-9. [DOI] [PubMed] [Google Scholar]
- Bijak M, Misgeld U. Suppression by GABAB receptors of 4-aminopyridine-induced hyperactivity in guinea-pig dentate neurons. Neurosci Lett. 1996;205:49–52. doi: 10.1016/0304-3940(96)12368-5. [DOI] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Dayer AG, Ford AA, Cleaver KM, Yassaee M, Cameron HA. Short-term and long-term survival of new neurons in the rat dentate gyrus. J Comp Neurol. 2003;460:563–572. doi: 10.1002/cne.10675. [DOI] [PubMed] [Google Scholar]
- Dieni CV, Chancey JH, Overstreet-Wadiche LS. Dynamic functions of GABA signaling during granule cell maturation. Front Neural Circuits. 2012;6:113. doi: 10.3389/fncir.2012.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- Duveau V, Laustela S, Barth L, Gianolini F, Vogt KE, Keist R, Chandra D, Homanics GE, Rudolph U, Fritschy JM. Spatiotemporal specificity of GABAA receptor-mediated regulation of adult hippocampal neurogenesis. Eur J Neurosci. 2011;34:362–373. doi: 10.1111/j.1460-9568.2011.07782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espósito MS, Piatti VC, Laplagne DA, Morgenstern NA, Ferrari CC, Pitossi FJ, Schinder AF. Neuronal differentiation in the adult hippocampus recapitulates embryonic development. J Neurosci. 2005;25:10074–10086. doi: 10.1523/JNEUROSCI.3114-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Goh EL, Sailor KA, Kitabatake Y, Ming GL, Song H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;439:589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Pradhan DA, Ming GL, Song H. GABA sets the tempo for activity-dependent adult neurogenesis. Trends Neurosci. 2007a;30:1–8. doi: 10.1016/j.tins.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Ge S, Yang CH, Hsu KS, Ming GL, Song H. A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron. 2007b;54:559–566. doi: 10.1016/j.neuron.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Sulser A, Wang J, Motamedi GK, Avoli M, Vicini S, Dzakpasu R. The 4-aminopyridine in vitro epilepsy model analyzed with a perforated multi-electrode array. Neuropharmacology. 2011;60:1142–1153. doi: 10.1016/j.neuropharm.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FL, Huang KP, Wu J, Boucheron C. Environmental enrichment enhances neurogranin expression and hippocampal learning and memory but fails to rescue the impairments of neurogranin null mutant mice. J Neurosci. 2006;26:6230–6237. doi: 10.1523/JNEUROSCI.1182-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/S0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- Jagasia R, Steib K, Englberger E, Herold S, Faus-Kessler T, Saxe M, Gage FH, Song H, Lie DC. GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J Neurosci. 2009;29:7966–7977. doi: 10.1523/JNEUROSCI.1054-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee N, Teixeira CM, Wang AH, Frankland PW. Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat Neurosci. 2007;10:355–362. doi: 10.1038/nn1847. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Saitoh Y, Murayama A, Sugiyama H, Inokuchi K. LTP induction within a narrow critical period of immature stages enhances the survival of newly generated neurons in the adult rat dentate gyrus. Mol Brain. 2010;3:13. doi: 10.1186/1756-6606-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel X, Medina I, Khalilov I, Ben-Ari Y, Khazipov R. Ca2+ oscillations mediated by the synergistic excitatory actions of GABA(A) and NMDA receptors in the neonatal hippocampus. Neuron. 1997;18:243–255. doi: 10.1016/S0896-6273(00)80265-2. [DOI] [PubMed] [Google Scholar]
- Leveille F, Papadia S, Fricker M, Bell KF, Soriano FX, Martel MA, Puddifoot C, Habel M, Wyllie DJ, Ikonomidou C, Tolkovsky AM, Hardingham GE. Suppression of the intrinsic apoptosis pathway by synaptic activity. J Neurosci. 2010;30:2623–2635. doi: 10.1523/JNEUROSCI.5115-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Malinow R. Deficiency in induction but not expression of LTP in hippocampal slices from young rats. Learn Mem. 1996;3:138–149. doi: 10.1101/lm.3.2-3.138. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Bensen AL, Washburn EK, Perederiy JV, Su KG, Li Y, Kernie SG, Parada LF, Westbrook GL. miR-132 mediates the integration of newborn neurons into the adult dentate gyrus. PLoS One. 2011;6:e19077. doi: 10.1371/journal.pone.0019077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwardt SJ, Wadiche JI, Overstreet-Wadiche LS. Input-specific GABAergic signaling to newborn neurons in adult dentate gyrus. J Neurosci. 2009;29:15063–15072. doi: 10.1523/JNEUROSCI.2727-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwardt SJ, Dieni CV, Wadiche JI, Overstreet-Wadiche L. Ivy/neurogliaform interneurons coordinate activity in the neurogenic niche. Nat Neurosci. 2011;14:1407–1409. doi: 10.1038/nn.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz K, Herold S, Lie DC. CREB in adult neurogenesis: master and partner in the development of adult-born neurons? Eur J Neurosci. 2011;33:1078–1086. doi: 10.1111/j.1460-9568.2011.07606.x. [DOI] [PubMed] [Google Scholar]
- Michelson HB, Wong RK. Synchronization of inhibitory neurones in the guinea-pig hippocampus in vitro. J Physiol. 1994;477:35–45. doi: 10.1113/jphysiol.1994.sp020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongiat LA, Esposito MS, Lombardi G, Schinder AF. Reliable activation of immature neurons in the adult hippocampus. PLoS One. 2009;4:e5320. doi: 10.1371/journal.pone.0005320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller W, Misgeld U. Picrotoxin- and 4-aminopyridine-induced activity in hilar neurons in the guinea pig hippocampal slice. J Neurophysiol. 1991;65:141–147. doi: 10.1152/jn.1991.65.1.141. [DOI] [PubMed] [Google Scholar]
- Nitz D, McNaughton B. Differential modulation of CA1 and dentate gyrus interneurons during exploration of novel environments. J Neurophysiol. 2004;91:863–872. doi: 10.1152/jn.00614.2003. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Hentges ST, Bumaschny VF, de Souza FS, Smart JL, Santangelo AM, Low MJ, Westbrook GL, Rubinstein M. A transgenic marker for newly born granule cells in dentate gyrus. J Neurosci. 2004;24:3251–3259. doi: 10.1523/JNEUROSCI.5173-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet Wadiche L, Bromberg DA, Bensen AL, Westbrook GL. GABAergic signaling to newborn neurons in dentate gyrus. J Neurophysiol. 2005;94:4528–4532. doi: 10.1152/jn.00633.2005. [DOI] [PubMed] [Google Scholar]
- Overstreet-Wadiche LS, Bensen AL, Westbrook GL. Delayed development of adult-generated granule cells in dentate gyrus. J Neurosci. 2006;26:2326–2334. doi: 10.1523/JNEUROSCI.4111-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatti VC, Davies-Sala MG, Espósito MS, Mongiat LA, Trinchero MF, Schinder AF. The timing for neuronal maturation in the adult hippocampus is modulated by local network activity. J Neurosci. 2011;31:7715–7728. doi: 10.1523/JNEUROSCI.1380-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh P, Adlaf E, Zhao CS, Markwardt S, Gavin C, Wadiche J, Overstreet-Wadiche L. Enhanced integration of newborn neurons after neonatal insults. Front Neurosci. 2011;5:45. doi: 10.3389/fnins.2011.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Represa A, Ben-Ari Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 2005;28:278–283. doi: 10.1016/j.tins.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, Meletis K, Huang ZJ, Ge S, Enikolopov G, Deisseroth K, Luscher B, Christian KM, Ming GL, Song H. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature. 2012;489:150–154. doi: 10.1038/nature11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocca G, Schmidt-Hieber C, Bischofberger J. Differential dendritic Ca2+ signalling in young and mature hippocampal granule cells. J Physiol. 2008;586:3795–3811. doi: 10.1113/jphysiol.2008.155739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Sandler VM, Toni N, Zhao C, Gage FH. NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature. 2006;442:929–933. doi: 10.1038/nature05028. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Makino H, Gage FH. Experience-specific functional modification of the dentate gyrus through adult neurogenesis: a critical period during an immature stage. J Neurosci. 2007;27:3252–3259. doi: 10.1523/JNEUROSCI.4941-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoeringer CK, Erhardt A, Sillaber I, Mueller MB, Ohl F, Holsboer F, Keck ME. Long-term anxiolytic and antidepressant-like behavioural effects of tiagabine, a selective GABA transporter-1 (GAT-1) inhibitor, coincide with a decrease in HPA system activity in C57BL/6 mice. J Psychopharmacol. 2010;24:733–743. doi: 10.1177/0269881109103091. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Kriegstein AR. GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J Neurosci. 2008;28:5547–5558. doi: 10.1523/JNEUROSCI.5599-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]