

Abstract
Transthyretin (TTR) is a homotetrameric transport protein, assembled from monomers that each contains two four-stranded β-sheets and a short α-helix and loop. In the tetramer, the ‘inner’ β-sheet forms a hydrophobic pocket while the helix and loop are solvent-exposed. Beta-amyloid (Aβ) aggregates bind to TTR, and the binding is significantly reduced in mutants L82A (on the loop) and L110A (on the inner β-sheet). Protection against Aβ toxicity was demonstrated for wild-type TTR but not L82A or L110A, providing a direct link between TTR-Aβ binding, and TTR-mediated cytoprotection. Protection is afforded at substoichiometric (1:100) TTR:Aβ molar ratios, and binding of Aβ to TTR is highest for partially aggregated materials and decreased for freshly-prepared or heavily aggregated Aβ, suggesting that TTR binds selectively to soluble toxic Aβ aggregates. A novel technique, nanoparticle tracking, is used to show that TTR arrests Aβ aggregation by both preventing formation of new aggregates and inhibiting growth of existing aggregates. TTR tetramers are normally quite stable; tetrameric structure is necessary for the protein’s transport functions, and mutations that decrease tetramer stability have been linked to TTR amyloid diseases. However, TTR monomers bind more Aβ than do tetramers, presumably because the hydrophobic ‘inner’ sheet is solvent-exposed upon tetramer disassembly. Wild-type and L110A tetramers, but not L82A, were destabilized when co-incubated with Aβ, suggesting that Aβ binding to L82 triggers tetramer dissociation. Taken together, these results suggest a novel mechanism of action for TTR: the EF helix/loop ‘senses’ the presence of soluble toxic Aβ oligomers, triggering destabilization of TTR tetramers and exposure of the hydrophobic inner sheet, which then ‘scavenges’ these toxic oligomers and prevents them from causing cell death
An important pathological feature of Alzheimer’s disease (AD) is deposition of aggregates of beta-amyloid (Aβ) in extracellular plaques, primarily in the hippocampus and cerebral cortex. These amyloid deposits figure prominently in the dominant current hypothesis regarding the cause of AD: specifically, that aggregates of Aβ are toxic to neurons, by a still-undefined mechanism. Aβ is a proteolytic cleavage product of the transmembrane amyloid precursor protein (APP). Most cases of AD arise sporadically in late life; however, there are genetically linked cases of early-onset AD that are linked to mutations in APP, where there is aggressive Aβ deposition. Early efforts to establish a transgenic mouse as an AD model, by engineering expression of the Swedish mutation of APP (APPSw), were not entirely successful. Although amyloid deposits were abundant, the mice did not progress to develop other characteristics of the disorder such as neurofibrillary tangles or widespread neuronal loss. As a possible explanation for the lack of AD-like pathology, Stein and Johnson observed a spontaneous 8-fold increase in the expression of the gene for transthyretin (TTR) in APPSw mice, and showed that infusion of anti-TTR antibodies led to increased tau phosphorylation and neuronal loss. These results strongly suggest that increased TTR expression protects APPSw mice from AD-like pathologies (1, 2). Increased TTR expression in mouse models of AD has been confirmed by other groups (3–5). Furthermore, neurons from human AD patients, but not age-matched controls, secrete TTR (5). The protective effect of TTR against Aβ toxicity has been observed in vitro (5–8), and supported by other animal studies. For example, progeny from APPSw mice crossed with mice engineered to express human TTR performed as well as wt and better than APPSw mice in cognitive tests (9), and AD mice raised in an enriched environment expressed more TTR and performed better on cognitive tests than those raised in a control environment (10).
TTR is a 55 kDa homotetrameric transport protein that is synthesized in the liver and choroid plexus and is present in both blood (3–7 μM) and cerebrospinal fluid (CSF, 0.1–0.4 μM) (11, 12). Each monomer contains two four-stranded β-sheets, an inner sheet of strands D, A, G and H, and an outer sheet of strands C, B, E and F, as well as a short α-helix and loop between E and F strands. Monomer assembly into dimers is stabilized by extensive hydrogen bonding. The assembly of dimers into tetramers is via a small contact region between the AB loop and H strands. Tetramer assembly creates a hydrophobic pocket in which thyroxine binds. TTR is the primary carrier for thyroxine in CSF and a secondary carrier in blood, with about 15–20% of TTR containing thyroxine. TTR serves also as carrier for retinol-binding protein (RBP), which binds near residues in the EF loop. RBP and thyroxine do not compete for binding to TTR, and both ligands reportedly stabilize TTR tetramers and reduce TTR misfolding and aggregation.
We and others have shown that TTR binds to Aβ (13–15). L82 and L110 were identified as two critical residues on TTR that mediate the interaction between Aβ and TTR (13). L82 is on the EF helix/loop, while L110 is in strand G, part of the inner sheet that lines the hydrophobic thyroxine-binding pocket (Figure 1). Aβ binding to alanine mutants L82A and L110A was significantly reduced compared to binding to wt TTR (13). In this study we used these mutants to further elucidate the mode of interaction between Aβ and TTR, and the means by which TTR mediates protection against Aβ-induced neuronal toxicity.
Figure 1.
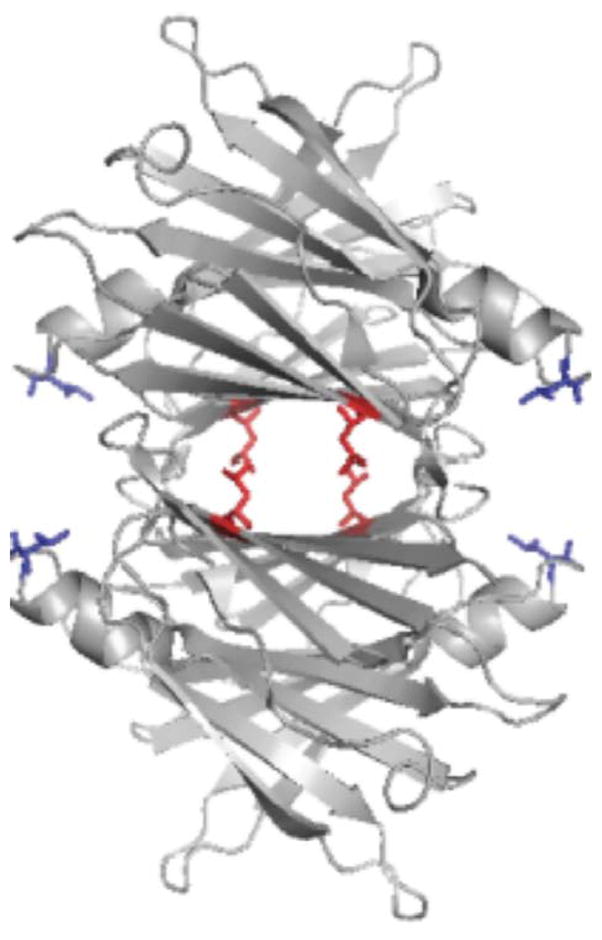
Ribbon structure of TTR tetramers, showing Leu82 (blue, on end of EF helix) and Leu110 (red, on strand G). Mutation of either of these leucines to alanine resulted in significantly less binding of Aβ (13).
EXPERIMENTAL PROCEDURES
TTR production and purification
Recombinant human transthyretin was produced and purified as previously described in detail (13). Wild-type (wt) and three mutants were prepared: F87M/L110M (mTTR), L82A, and L110A. F87M/L110M is an engineered TTR mutant that is stable as a monomer (16). L82A and L110A are mutants with native-like secondary, tertiary and quaternary structure, but both show substantially decreased binding of Aβ relative to wt (13).
In vitro cellular toxicity
Primary cortical neuronal cultures were generated from individual E15.5 embryos derived from C57/Bl6 female mice. Briefly, individual cortices were dissected and dissociated with trypsin, and plated on individual poly-D-lysine coated 96-well plates or on coverslips in 24-well plates at a density of 100,000 cells/well or 400,000 cells/well, respectively. Cells were initially plated in modified eagle media containing fetal bovine serum, horse serum, L-glutamine and penicillin/streptomycin (PS). After 45 min, the media was replaced with neurobasal media (NBM) supplemented with B27, PS, and L-glutamine for the duration of the experiment in the humidified tri-gas incubator under normoxic condition. Half NBM was changed with fresh NBM every 2–3 days. 1 mg/ml Aβ(1–42) (American Peptide, Sunnyvale, CA) was prepared in phosphate buffered saline (PBS) and then diluted to a final concentration of 10 μM with wt or mutant TTR (mTTR, L82A and L110A) for one hour in NBM supplemented with B27 minus antioxidant, L-glutamine and PS. The TTR+Aβ mixture was then added to cells at 6 days in vitro (DIV), and viability was assessed at 8 DIV.
The MTS [3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)-2H-tetrazolium salt] assay (Promega) was used according to the manufacturer’s instructions to assess cell viability. Briefly, phenazine methosulfate (PMS) and MTS were mixed together at 1:20 ratio immediately before use. 20μl of MTS/PMS mixture was added per well of 96-well plate and incubated with cells for 2 hrs in humidified tri-gas incubator. The quantity of resulting formazan product was measured by 490 nm absorbance on a UV-Vis plate reader. Triplicate measurements were taken at each condition; data are reported relative to medium-treated cells.
To assess apoptotic cell death, terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL; in situ cell death detection kit, TMR red; Roche) staining was used per manufacturer’s instruction. Apoptotic cells were identified by fluorescein labeling of DNA strand breaks with the TdT enzyme. Briefly, cells on coverslips were washed and fixed in 4% paraformaldehyde for 60 min. Following three washes the cells were permeabilized with freshly prepared 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice. 50 μl enzyme solution and 450 μl label solution from the kit were mixed together and incubated with the cells in ahumidified chamber for one hour . Positive and negative controls were run along with the samples. The cells were washed post-incubation and mounted on the glass slides with aqueous mounting media containing DAPI to counterstain the nuclei. The samples were visualized using a Zeiss fluorescence microscope at 595 nm emission for TUNEL and 350 nm emission for DAPI.
Peptide arrays
A series of peptides with overlapping sequences derived from Aβ, each 10 amino acids long, was synthesized onto a cellulose membrane (Sigma-Genosys, St. Louis, MO). The membrane was wetted with a few drops of methanol, rinsed with water, and then washed with 10 mL Tris-buffered saline (TBS: 20 mM Tris, 150 mM NaCl, pH 7.6) three times for 10 min each. After overnight incubation in casein blocking buffer (Thermo Scientific), the membrane was washed with 10 mL TBS and incubated with 10 mL TTR or mTTR (5 μg/mL in blocking buffer) at room temperature overnight. The membrane was washed three times with 10 mL T-TBS (TBS with 0.05% (v/v) Tween-20) for 10 min each, and bound protein was transferred onto 0.2 μm poly(vinylidene difluoride) (PVDF) membrane (Millipore Corp, Billerica, MA) at 70 mA three times: for 30 min, 30 min and 60 min, sequentially. The three PVDF membranes were blocked with 10 mL blocking buffer overnight and then reacted with anti-human TTR antibody (DAKO, Glostrup, Denmark) at 1:1500 dilution in the blocking buffer for 2 h. PVDF membranes were subsequently treated with anti-rabbit immunoglobulins/HRP antibody (DAKO) at 1:1500 dilution in blocking buffer for 2 hr. Bound TTR was visualized by means of ECL™ Western Blotting Analysis System (GE Healthcare, Buckinghamshire, UK).
ELISA measurement of Aβ-TTR binding
ELISA plates (Corning Inc, Corning, NY) were coated with 5 μg/mL of wt TTR (100 μL/well) in coating buffer (10 mM sodium carbonate, 30 mM sodium bicarbonate, 0.05% NaN3, pH 9.6) overnight at room temperature. The plate was washed three times with phosphate-buffered saline (PBS: 10 mM Na2HPO4/NaH2PO4, 150 mM NaCl, pH 7.4, with 0.05% Tween 20) and incubated with blocking buffer (5% non-fat dry milk in PBS-T) for 2 hr at room temperature. For a negative control, TTR was not coated but wells were incubated with blocking buffer. Four replicate wells were prepared at each condition. Aβ stock solution was prepared by dissolving lyophilized Aβ (Aβ(1–40), Anaspec, San Jose, CA) to 2.8 mM in 8 M urea pH 10 as described (17). Aβ samples were prepared by dilution of Aβ stock into PBS with 0.02% sodium azide to a final concentration of 0.8 mg/mL. Freshly-prepared Aβ or Aβ aggregated for 1–7 days was diluted to 5 μg/mL in PBS and then immediately added to TTR-coated or control wells (50 μL/well). The plate was incubated at 37°C for 1 hr. After washing, anti-Aβ antibody 6E10 (1:3000 in PBS-T, Covance, Princeton, New Jersey) was added to each well (100 μL/well), and the plate was incubated at room temperature for 1 hr with gentle shaking. After washing, anti-mouse HRP antibody (1:3000 dilution; Pierce, Rockford, IL) was added to each well (100 μL/well) and the plate was incubated for 1 hr at room temperature with gentle shaking. The plate was washed three times, then 100 μL 3,3′,5,5′ – tetramethylbenzidine (TMB) substrate solution (Pierce, Rockford, IL) was added to each well. The plate was incubated at room temperature for 15–30 min; color development was stopped by adding 100 μL 2 M sulfuric acid. Absorbance was measured at 450 nm with EL800 Universal Microplate Reader (Bio-tek Instruments Inc, Winooski, VT). Aβ binding was calculated as the mean of 4 replicate wells by subtracting the absorbance of the negative control from the sample absorbance.
Immunoblot assays
28 μM Aβ in PBS was mixed with 0.5 μM TTR and incubated up to 24 h at room temperature. 1.5 μL was dotted onto 0.45 μm nitrocellulose membrane (Pierce) after 0, 5 and 24 h of incubation. Dried membrane was blocked with 5% non-fat dry milk in T-TBS overnight at 4°C and reacted with OC antibody (Millipore Corp) diluted to 1:2000 in 5% non-fat dry milk in T-TBS for 1 hr at room temperature. After washing three times with T-TBS, the membrane was incubated with anti-rabbit immunoglobulins/HRP antibody diluted to 1:1000 in 5% non-fat dry milk in T-TBS for 30 min at room temperature. After washing, the membrane was visualized by means of ECL™ Western Blotting Analysis System.
Photo-induced cross-linking of Aβ
Photo-induced cross-linking of unmodified proteins (PICUP) experiment was performed according to the protocol of Fancy and Kodadek (18) with some modifications. Aβ samples at different aggregation states were prepared by diluting to 0.8 mg/mL in PBS from Aβ stock and incubating at room temperature for 0 to 7 days. Aβ samples were then diluted to 30 μM in PBS prior to PICUP analysis. 1 μL of 1 mM Tris(2,2′-bipyridyl)dichlororuthenium(II) (Sigma) and 1 μL of 20 mM ammonium persulfate (Sigma) in PBS were added to 18 μL sample. The cross-linking reaction was carried out by irradiation with visible light for 10 s, and the reaction was quenched immediately by addition of 1 μL of 1 MDTT. A flashlight (Mag-lite) with krypton lamp was used as a light source and the reaction mixture in the tube was placed parallel to the beam of light at a distance of 5 cm. Samples were separated by SDS-PAGE on a Precise 4–20% polyacrylamide gradient gel (Pierce), and transferred onto 0.45 μm nitrocellulose membrane (Pierce) at 60 V for 1 hr. The membrane was blocked with 5% non-fat dry milk in T-TBS overnight at 4°C, washed three times with T-TBS, and reacted with 6E10 antibody diluted to 1:7500 in T-TBS for 1 hr at room temperature. After washing, the membrane was incubated with anti-mouse HRP antibody diluted to 1:7500 in T-TBS for 30 min at room temperature. After washing for 30 min, the membrane was visualized by means of ELC™ Western Blotting Analysis System.
Dynamic light scattering (DLS)
All buffers and TTR samples were filtered through 0.02 μm filters before use. Lyophilized Aβ(1–40) (Anaspec, Inc. San Jose, CA) was dissolved in 8 M urea/0.01 M glycine-NaOH, pH 10 (17) to 2.8 mM, then snap-frozen and stored at −80°C. Immediately before each experiment, frozen stocks were thawed and sonicated for 2 minutes before dilution into PBSA. Aβ alone (23 μM) or mixed with TTR (7 μM) was filtered through a 0.45 μm filter directly into a light-scattering cuvette and then placed into a bath of the index-matching solvent decahydronaphthalene with temperature controlled to 25°C. Light scattering data were collected using a Brookhaven BI-200SM system (Brookhaven Instruments Corp., Holtsville, NY) and an Innova 90C-5 argon laser (Coherent, Santa Clara, CA) operating at 488 nm and 150 mW. The z-averaged hydrodynamic diameter was determined from the autocorrelation function using the method of cumulants.
Nanoparticle tracking (NTA)
NTA measurements were collected with a Nanosight LM10 (Nanosight, Amesbury, United Kingdom) equipped with a 405 nm laser. All buffers were filtered through 0.02 μm filters prior to use as were TTR samples. Aβ solutions were prepared as described for DLS. Aβ alone (28 μM in PBSA) or mixed with TTR (0.7 μM) was injected into the sample chamber using a syringe. All measurements were collected at room temperature with the camera level set to the maximum value to allow for the detection of smaller particles. One 90-second video was taken every 30 min for 6 hours, and then hourly for another 18 hours. Data were captured and analyzed using NTA 2.3 software.
Native gel electrophoresis
TTR alone (3.6 μM) or with Aβ (82 μM) were incubated for 0 or 2 days at 25°C or 37°C, then diluted into SDS to a final concentration of 2% (w/v). Samples were loaded on a Precise 4–20% polyacrylamide gradient gel (Pierce, Rockford, IL) along with EZ-Run Protein Ladder (Fisher BioReagents, Fair Lawn, NJ) and electrophoresed using SDS buffer for 45 min at 125V. Gels were stained with Coomassie blue. TTR tetramers are normally stable under these electrophoresis conditions.
Circular dichroism (CD) analysis
TTR stock solutions were dialyzed against phosphate-NaF buffer (10 mM Na2HPO4/NaH2PO4 and 150 mM NaF, pH 7.4). TTR alone (3.6 μM) or with Aβ (82 μM) were incubated for 0 to 2 days at 37°C. Samples were then diluted six-fold and transferred into a 1-mm cell. CD spectra were collected on an Aviv 202SF CD spectrophotometer from Aviv Biomedical (Lakewood, NJ) at 22 °C. Blank solvent spectra, and spectra for Aβ alone, were collected and subtracted.
Crosslinking and analysis by gel electrophoresis and Western blotting
50 μl of TTR alone (3.6 μM) or with Aβ (82 μM) in PBS were incubated for 2 h at 37°C, and cross-linked with glutaraldehyde by adding 2 μL 25% glutaraldehyde solution. After 2 min, 2 μL of 7% (w/v) NaBH4 in 0.5 M NaOH was added to quench the reaction. The samples were heated at 95°C for 5 min and analyzed on a Precise 4–20% polyacrylamide gradient gel (Pierce, Rockford, IL). One gel was stained with Coomassie blue. A second gel was transferred onto 0.45 μm nitrocellulose membranes (Pierce) at 60 V for 1 h. Membranes were blocked with 5% non-fat dry milk in T-TBS overnight at 4°C. After washing three times with T-TBS, the membrane was incubated with anti-human TTR antibody at 1:1500 dilution in T-TBS and then with anti-rabbit immunoglobulins/HRP antibody at 1:1500 dilution in T-TBS. After washing, membranes were visualized by means of ECL™ Western Blotting Analysis System (GE Healthcare).
TTR tetramer dissociation kinetics
The S-Trap method was used to measure the rate of tetramer dissociation (19). TTR (0.4 mg/mL in PBSA) was diluted with SDS buffer (25% glycerol, 2% SDS, 0.01% bromophenol blue and 62.5 mM Tris-HCl, pH 6.8) so that the final concentration of protein and SDS were 0.2 mg/mL (3.6 μM) and 1%, respectively. SDS buffer was pre-heated at 90°C to quickly achieve the desired temperature (80°C) after mixing. After incubation for different time periods, the samples were quickly placed in an ice water bath for 5–10 seconds and then centrifuged for 5 seconds at 3000 rpm. Samples were loaded on a Precise 4–20% polyacrylamide gradient gel (Pierce, Rockford, IL) along with EZ-Run Protein Ladder (Fisher Bioreagents, Fair Lawn, NJ) and electrophoresed for 45 min at 125 V. Gels were stained with Coomassie blue. Destained gels were photocopied and the images were analyzed using ImageJ. The fraction of unfolded protein was determined by the ratio between the amount of unfolded and total protein. The tetramer dissociation rate (kd) was obtained by fitting the fraction of unfolded protein versus incubation time to a single exponential equation.
RESULTS
Loss of binding sites reduces TTR protection against Aβ toxicity
In cortical cultures exposed to 10 μM Aβ, we observed a reduction to 50% cell viability as measured by the MTS assay (Figure 2a). Wt TTR protected Aβ toxicity in a dose-dependent manner, achieving complete protection at ~6 μg/mL (~0.1 μM, or 1:100 TTR:Aβ molar ratio (Figure 2), consistent with previously reported data from our own and other laboratories (7, 20, 21). A stable monomeric mutant, F87M/L110M (mTTR), also afforded complete protection against Aβ toxicity at ~3 μg/mL (~0.2 μM). Both wt and mTTR bind Aβ, with greater binding of Aβ to mTTR than wt (14). In previous work, leucine residues L82 and L110 on TTR (Figure 1) were identified as important contributors to TTR-Aβ association, and binding of Aβ to TTR alanine mutants L82A and L110A was significantly diminished compared to wt, despite the fact that these mutants retained native secondary, tertiary and quaternary structure (13). L82A and L110A were much less effective at protecting neurons against Aβ toxicity (Figure 2a). In control experiments, TTR alone (wt, mTTR, L82A or L110A) had no effect on cell viability (data not shown).
Figure 2.

Effect of TTR on Aβ toxicity. (a) Neuron-enriched cortical cultures were established from mouse embryos and then exposed to Aβ alone (10 μM) mixed with varying concentrations of wt TTR (➂), mTTR (▲), L82A (○), and L110A (□). Viability was assessed relative to vehicle-treated cells using MTS. (b) Cells were exposed to Aβ alone (top panel) or Aβ plus wt, L82A, or L110A, then assessed for apoptosis using TUNEL staining.
TUNEL staining was used to corroborate the MTS results. Apoptotic cells are clearly present in the Aβ-treated cortical cultures, as evidenced by red staining of DNA strand breaks (Figure 2b). The addition of wt TTR to Aβ virtually eliminated red staining. However, neither L82A nor L110A was effective at preventing Aβ-mediated apoptosis. These results connect the loss of binding sites on TTR to a reduction in TTR’s ability to inhibit Aβ toxicity, demonstrate that Aβ binding to TTR is causally linked to TTR-mediated protection, and support the role of leucine residues L82 and L110 in mediating biologically-relevant Aβ-TTR association.
Aβ soluble aggregates bind preferentially to TTR
In previous work, we synthesized peptide arrays containing linear sequences of TTR and used these arrays to identify specific sequences, corresponding to strand G and the EF helix/loop, to which soluble Aβ bound (13). In an attempt to identify residues on Aβ that contribute to TTR-Aβ binding, we reversed the experiment and constructed peptide arrays containing linear sequences of Aβ 10 residues long. Despite repeated attempts, we observed no binding of either TTR or mTTR to any spots on the peptide arrays (data not shown). Using anti-Aβ antibodies directly, we were able to easily detect spots, indicating that the peptides were correctly synthesized and were available for binding. Possible (non-exclusive) explanations for this result include: (a) that the TTR binding site on Aβ is non-contiguous, (b) that a specific conformation of Aβ is required for TTR binding, and /or (c) that Aβ must be aggregated in order to bind to TTR.
To test whether Aβ must be in a specific conformation or state of aggregation in order to bind TTR, we incubated Aβ for 0 to 7 days and then characterized its binding to TTR by ELISA and its aggregation state by cross-linking and gel electrophoresis. Freshly prepared Aβ bound poorly to TTR-coated wells (Figure 3a), consistent with previous observations (14). Binding increased for samples aggregated 1–5 days prior to contact with TTR, but then partially decreased if Aβ was aggregated for 7 days (Figure 3a). Crosslinking studies revealed that freshly prepared Aβ contained only low molecular weight oligomers (primarily apparent dimers and trimers with some monomers and tetramers, Figure 3b). On day 1, some high molecular-weight aggregates (>100 kDa) were in evidence, along with low molecular weight oligomers. The distribution between low molecular weight oligomers and high molecular weight aggregates remained roughly constant from day 1 through day 5. On day 7, the Aβ sample was very viscous and much of the material did not enter through the gel, indicative of a highly aggregated population. The low molecular weight oligomers were substantially diminished. All samples stained positive with the OC antibody (not shown), a conformationally specific antibody that detects soluble and insoluble fibrillar aggregates (22). From the combined data, we conclude that soluble fibrillar Aβ aggregates (~100 kDa and above) bind much more to TTR than low-molecular weight oligomers, but that very mature, highly aggregated Aβ binds poorly to TTR.
Figure 3.

Effect of Aβ aggregation state on binding to TTR. (a) Relative amount of Aβ bound to immobilized TTR, as determined by ELISA. Aβ was freshly prepared (day 0) or pre-aggregated for the indicated number of days prior to contact with TTR. (b) Size distribution of Aβ as function of number of days aggregated, ascertained by PICUP crosslinking and gel electrophoresis.
TTR slows down aggregation of Aβ
We tested the effect of TTR (wt, L82A and L110A) on the growth of Aβ aggregates, using dynamic light scattering. For these experiments, we used snap-frozen and thawed Aβ samples. Aβ prepared in this manner contains some aggregates initially. Experiments were run at moderate (~3:1) molar Aβ excess, to accentuate the effect of TTR. Growth rates were monitored for 45 minutes. The fastest growth rate, as measured by the increase in the mean hydrodynamic diameter of aggregates, was observed with Aβ alone. Wt TTR significantly slowed the rate of growth, as did L110A. L82A was less effective at slowing down the initial growth rate than L110A (Figure 4).
Figure 4.

Effect of TTR (wt and mutants) on Aβ aggregate growth kinetics. The mean hydrodynamic diameter of aggregates was measured by dynamic light scattering. Samples contained 23 μM Aβ alone (▢) or with 7 μM wt (■), with L82A (➂), with L110A (▲).
Nanoparticle tracking measures Aβ aggregate size distribution and number concentration
We here introduce the use of nanoparticle tracking analysis (NTA) to detect Aβ aggregates and measure the growth of aggregates over time. To our knowledge this is the first reported use of NTA to monitor Aβ aggregation. Briefly, in NTA, scattering from individual particles is detected and the trajectory of those particles is tracked over a short time, from which a diffusion coefficient, and an equivalent hydrodynamic diameter, is calculated (23). Nanoparticle tracking has advantages over conventional DLS for several reasons: a particle number concentration can be measured, smaller and weaker scatterers are detected even in the presence of larger stronger scatterers, and a size distribution is directly obtained because single particles are tracked. NTA has some advantages over AFM: particles remain in solution, so surface effects are not a significant concern. There are some limitations as well; in particular, smaller (<50 nm) particles cannot be reliably tracked, so Aβ monomers or smaller Aβ oligomers are not measured.
We first checked the buffer only. No aggregates were detected in these samples: the video was completely dark (not shown). We next collected data for a solution of Aβ (28 μM) at several time points, up to 24 hours. Particle counts at each particle size (1 nm resolution) are summed and converted to number of particles per mL. Representative size distributions at 0.5 h and 12 h are shown in Figure 5a. With time, the number of particles increased, and the size distribution broadened and shifted towards larger aggregates. The absence of particles at sizes <50 nm is due to limitations in detecting and tracking smaller aggregates; because of this the mean size of aggregates detected by NTA is larger than that detected by DLS. By modeling the aggregates as wormlike chains with 100 nm persistence lengths and 10 nm diameter (17), we estimated that about 3% of the total mass of Aβ is incorporated into these large aggregates (>50 nm hydrodynamic diameter, or ~200 nm or longer contour length) at early times, increasing to ~8% after 24 h. To simplify presentation of the data, we binned particles (50 nm bin size for diameters < 400 nm, 100 nm bin size for diameters >400 nm). Figure 5b illustrates binned size distributions for Aβ aggregates taken at 4 time points. The growth in both number and size of aggregates over time is evident.
Figure 5.

Nanoparticle tracking analysis of Aβ aggregate growth kinetics. Aβ (28 μM) was prepared in PBSA from stock solutions and incubated at room temperature. (a) Size distribution of Aβ aggregates after 0.5 hours (○) and after 12 hours (▢). Distributions are shown at 1 nm resolution. (b) Size distributions of Aβ aggregates were binned and compared after 0.5 h (solid), 3.5 h (open), 7 h (cross) and 12 h (stripe) incubation.
TTR affects both Aβ aggregate size distribution and number concentration
We first confirmed that TTR did not contribute any signal to the NTA measurement. Aβ was mixed with TTR (wt, L82A, or L110A) in PBSA and the effect of TTR on Aβ aggregation was evaluated using NTA. The addition of wt TTR completely arrested the growth of aggregates: the size distribution did not change (Figure 6), and the particle number concentration did not increase (Figures 6 and 8), for at least 12 h. These data show that wt TTR effectively suppresses both the growth in size of pre-existing aggregates and the appearance of new aggregates.
Figure 6.

Size distributions of Aβ alone (open bar) or incubated with wt TTR (solid bar) as a function of incubation time, as measured by NTA. (a) 0.5 hours. (b) 3.5 hours. (c) 7 hours. (d) 12 hours.
Figure 8.

Total aggregate number concentration as a function of time, as measured by NTA. Aβ was incubated alone (▢) or with wt TTR (■), with L82A (➂), with L110A (▲) Insert. Immunoblots of Aβ mixed with and without wt TTR and stained with OC antibody.
We next compared the effect of L82A and L110A on Aβ aggregation, to that of wt TTR. Although both mutants had some effect on slowing the growth of Aβ aggregates, neither was as effective as wt (Figure 7). Unlike with wt, the size distribution shifted towards larger aggregates, albeit less than with Aβ alone (Figure 7a–d). Over time, L110A was less effective than L82A at suppressing Aβ growth. This is the opposite of the behavior observed in DLS, but that data were taken over only 45 minutes.
Figure 7.

Size distributions of Aβ with wt TTR (solid), Aβ with L82A (cross) and Aβ with L110A (stripe) as a function of incubation time, as measured by NTA. (a) 0.5 hours. (b) 3.5 hours. (c) 7 hours. (d) 12 hours.
The total particle number concentration versus time is shown in Figure 8. This analysis again illustrates that Aβ aggregates grow in number over time, that wt completely prevents new aggregates from forming (aggregates of the size that can be detected by NTA), and that both L82A and L110A partially suppress growth in particle number, but not as effectively as wt.
Aβ preparations used in these experiments were characterized by immunoblot. Two conformationally specific polyclonal antibodies were used. OC binds to soluble fibrillar oligomers and fibrils, including both thioflavin S–positive and -negative deposits (24). OC-positive deposits are significantly higher in AD than age-matched controls, and increase with severity of dementia, and OC-positive staining increases dramatically in Tg2576 but not wt mice after 12 months (25). A11 reportedly binds to ‘prefibrillar oligomers’ but not to fibrillar oligomers or mature fibrils (26). Our Aβ samples were OC-positive and A11-negative. The addition of TTR did not affect the OC-positive status, despite the slowdown in aggregation rate (Figure 8). This result suggests that TTR binding does not cause a significant change or re-arrangement in the conformation of Aβ aggregates.
Binding of Aβ triggers conformational change in TTR
We incubated TTR (wt, L82A, and L110A) with Aβ for up to 2 days, and collected CD spectra for these mixtures as well as for Aβ or TTR alone. We subtracted the spectra for Aβ alone from that for TTR+Aβ, and then compared the CD spectra for TTR alone to that of TTR in the presence of Aβ. Spectra for wt, L82A, and L110A were very similar, as reported previously (13), indicating no changes in secondary structure are caused by the alanine mutations. At day 0, we saw no change in the TTR spectra in the presence of Aβ (data not shown). After two days incubation, however, we observed a measurable shift in the spectra for wt TTR in the presence of Aβ (Figure 9). In contrast, CD spectra of L82A or L110A were minimally affected by Aβ (Figure 9). For comparison, we produced two TTR mutants: F87M/L110M, an engineered TTR mutant that is stably monomeric, and S112I, a dimeric mutant. We compared CD spectra for these two mutants (without Aβ) to wt TTR. The CD spectra for the monomeric and dimeric TTR variants are shifted upward (less negative ellipticity) relative to the wt, similar to the shift observed for wt TTR in the presence of Aβ (data not shown). A similar shift upward in CD spectra was previously reported for the TTR mutant S85A, which assembles into unstable tetramers (13).
Figure 9.

CD spectra of TTR alone (open symbols) or after 2 days incubation with Aβ (closed symbols). Buffer spectra was subtracted from TTR alone, while CD spectra of Aβ in buffer was subtracted from TTR with Aβ. Wt (□), L82A (○), L110A (△), wt+Aβ (■), L82A+Aβ (➂), L110A+Aβ (▲).
We considered two possible explanations for these results. First, binding of Aβ could induce a change in wt TTR structure, leading to destabilization of the tetramer. Weaker binding of Aβ to L82A and L110A could be insufficient to trigger this destabilization, and/or binding to both sites could be required for destabilization. Alternatively, Aβ secondary structure could be changed due to TTR binding, so subtraction of the spectra for Aβ alone from the (wt+Aβ) spectra does not correctly account for the contribution of Aβ to the spectra. We tested the first hypothesis by looking for other evidence of tetramer destabilization.
Aβ binding facilitates TTR tetramer destabilization/dissociation
TTR is natively a very stable tetramer, but it undergoes slow monomer and dimer subunit exchange, with half-times on the order of 18–24 h (27). To explore whether TTR tetramer stability is affected by Aβ, we incubated TTR (wt, L82A, and L110A) alone or with Aβ at room temperature for 0, 1 or 2 days, and then analyzed samples by native gel electrophoresis, under conditions where TTR normally runs as a tetramer and Aβ as a monomer. As expected, TTR alone (wt or mutant) was tetrameric, and remained tetrameric for 2 days (Figure 10). When Aβ was added to TTR and the sample was analyzed shortly after mixing, bands corresponding to TTR tetramer and Aβ monomer were observed, as expected (Figure 10a). After 1 day (not shown) or 2 days (Figure 10b) co-incubation, however, partial dissociation of TTR tetramers to monomers was observed for wt and L110A, but not for L82A. Similar results were obtained with incubation at 37°C (not shown).
Figure 10.

Native gel electrophoretic analysis of TTR tetramer stability w/o and w/Aβ over time. (a) 0 days, 25°C. (b) day 2, 25°C. Lanes 1,3 and 5 correspond to wt, L110A and L82A without Aβ, respectively. Lanes 2, 4 and 6 correspond to wt, L110A and L82A with Aβ, respectively. Lane 7 corresponds to Aβ alone.
We next explored why Aβ destabilized wt and L110A, but not L82A. We previously showed that L110A and L82A mutations did not cause a loss of secondary, tertiary, or quaternary structure in TTR (13). We tested whether these mutations caused a more subtle effect on tetramer stability by measuring dissociation at high temperature using the S-Trap method (19). This method prevents re-association or aggregation of dissociated monomer. Dissociation rate constants kd for wt and L82A were indistinguishable (Table 1), and the rate constant for wt is consistent with published data (19). Thus, the lack of tetramer dissociation for L82A incubated with Aβ is not due to an increase in L82A stability relative to wt. With L110A, kd increased by roughly 30%. Thus there is a modest reduction in tetramer stability of L110A that may account, in part, for the enhanced dissociation of L110A in the presence of Aβ.
Table 1.
Comparison of wt and mutant TTR tetramer dissociation rate at 80°C, measured using the S-trap method. Both the first-order rate constant (kd) and the half-time (t1/2) are reported.
| kd (min−1) [t1/2 (min)] | |
|---|---|
| wt | 0.31±0.04 [2.2±0.2] |
| L82A | 0.30±0.03 [2.3±0.2] |
| L110A | 0.42±0.05 [1.6±0.2] |
To see if Aβ is simply acting nonspecifically, like a detergent, and trapping TTR monomers during subunit exchange, we incubated TTR (wt and mutants) with SDS (either 0.5% or 2% SDS) for 0 or 2 days. At 0 day incubation the protein was fully tetramer, as expected, but after 2 days a significant fraction was dissociated into monomers. The monomer:tetramer ratio was about the same for all 3 proteins (wt, L110A and L82A, data not shown). This result indicates that Aβ is acting specifically, because Aβ affects each TTR mutant differently whereas SDS has similar effects on all.
To see if the lag in formation of TTR monomers in the presence of Aβ was attributable to the time it takes to form Aβ aggregates, we pre-aggregated Aβ for one to two days prior to adding TTR. After one hour of co-incubation of TTR with pre-aggregated Aβ, we observed no TTR monomer bands (data not shown). These data show that the slow appearance of the TTR monomer bands is not due to the time scale for Aβ aggregation but rather to slow TTR tetramer dissociation.
We conducted a similar experiment where Aβ and TTR were incubated and then analyzed by gel electrophoresis, except we crosslinked with glutaraldehyde before running gels, and boiled the samples prior to application to the gel. Under these conditions, TTR runs as a monomer in the absence of crosslinker. Gels were analyzed by Coomassie staining as well as Western blots with anti-TTR antibodies (Figure 11). With wt TTR in the absence of Aβ, we observed tetramers and dimers. (The dimer-dimer contacts are relatively small in area, while monomer-monomer contact is extensive. Thus dimers are crosslinked readily while not all tetramers are efficiently crosslinked; based on preliminary experiments we know that some protein that is tetrameric in solution will not be crosslinked under our conditions.) When Aβ was added to wt, the tetramer band became weaker, a stronger dimer band emerged, and a monomer band was apparent. This pattern is clear with both Coomassie and anti-TTR staining (Figure 11a and 11b). With L110A, there was also an increase in dimer and monomer bands in the presence of Aβ, although the change may be somewhat less than that observed for wt. In contrast to wt or L110A, there was no discernable destabilization for L82A caused by addition of Aβ. These data are consistent with the native gel data obtained in the absence of crosslinking.
Figure 11.
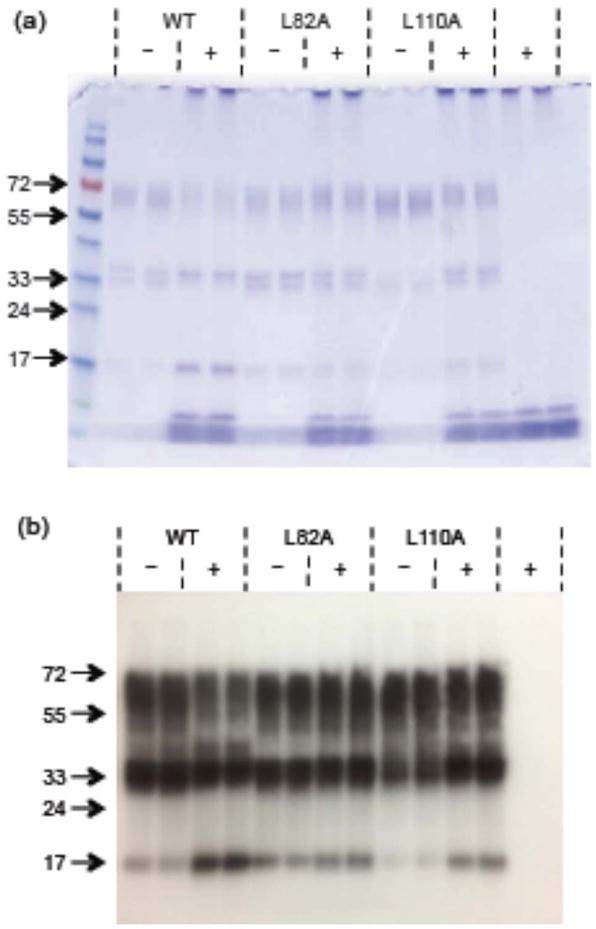
Cross-linking of TTR (wt and mutants) incubated alone (−) and with (+) Aβ. Molecular weight markers are shown in the first lane. (a) Stained with Coomassie Blue. (b) Western blot with anti-TTR antibody.
DISCUSSION
TTR inhibits toxicity of Aβ as a direct result of binding to Aβ oligomers
TTR inhibits Aβ toxicity in in vitro cell culture, as shown in this study as well as in several other reports (5) (7, 20, 21). Brouillette et al. demonstrated that TTR inhibits Aβ toxicity in vivo using an animal model of repeated intrahippocampal injections of Aβ oligomers (28). Here, we observed that TTR mutants L82A and L110A, which bind less Aβ than wt, were ineffective at inhibiting toxicity. These results provide strong evidence of a direct cause-and-effect connection between TTR-Aβ binding, and TTR-mediated protection against Aβ neurotoxicity.
With both wt and mTTR, complete recovery of cell viability was achieved at substoichiometric TTR:Aβ ratios; in fact, mTTR was somewhat more protective at equivalent mass concentration than wt. The fact that complete protection is afforded at ~ 1:100 TTR:Aβ molar ratio suggests strongly that TTR is preferentially binding to toxic Aβ oligomers. To further test this hypothesis, we characterized Aβ aggregates over 7 days by both crosslinking (size) and immunoblot (conformation). OC-positive aggregates, which can be either soluble or insoluble and are believed to have a fibrillar β-sheet structure (22), were observed in all samples. Only low molecular weight oligomers were present on day 0, and this sample bound poorly to TTR. With 1–5 days incubation some high-molecular weight (~100 kDa or above) soluble Aβ aggregates appeared, and binding of Aβ to TTR increased significantly. Longer (7 day) incubation led to the appearance of very large, insoluble aggregates, and binding to TTR was again reduced. Thus, binding of Aβ to TTR correlates best with the presence of soluble Aβ aggregates containing roughly twenty-five to a few hundred monomers. These data strongly support our hypothesis that TTR binds selectively to soluble fibrillar Aβ oligomers, which are widely believed to be the most toxic form of Aβ. This hypothesis explains how TTR can fully protect against Aβ toxicity even when present at substoichiometric ratios (relative to Aβ monomers). We estimate an apparent equilibrium dissociation constant for TTR-Aβ binding of 10–100 nM from the toxicity data. It is not possible to define a true single equilibrium constant: the molar concentration of Aβ oligomers and the stoichiometry of TTR-Aβ binding are not known, there are two binding sites on TTR (EF helix and strand G), Aβ binding to TTR to one binding site triggers exposure of the second binding site, and it is possible that different Aβ oligomeric species differ in their binding strength.
TTR suppresses Aβ aggregate growth, as a direct result of binding
We introduce the use of nanoparticle tracking (NTA) to monitor Aβ aggregate growth. A particular advantage of this technique over DLS is that both aggregate size distribution and aggregate number concentration are quantified simultaneously. (DLS has an advantage in its sensitivity to smaller aggregates less than 50 nm in diameter.) Our results demonstrate clearly that Aβ aggregates grow in both number and size over time. The NTA data provide the insight that wt TTR suppresses Aβ aggregation in two ways: by preventing formation of new aggregates and by inhibiting growth of pre-existing aggregates. However, pre-formed aggregates are not dissociated. Nor does TTR cause a major re-structuring of aggregates, since they are still OC-positive. We propose that TTR-mediated protection of cells from Aβ-induced neurotoxicity is accomplished by sequestration of high-molecular weight soluble oligomers, coating them and preventing interaction with cells.
Both L110A and L82A slow but do not stop Aβ aggregate growth, and neither mutant is as effective as wt TTR in suppressing aggregation. With these data, the reduced binding of the mutants to Aβ is connected to their reduced efficacy at inhibiting aggregation. DLS experiments (collected for 45 minutes) indicated L110A was somewhat more effective than L82A, while the longer (12 h) NTA experiments suggested L110A is somewhat less effective than L82A. Aβ binds to L110A at the EF helix/loop site (where L82 is), and to L82A at the interior site (strand G, where L110 is). The DLS and NTA data combined could be explained as follows: Aβ binding to the exposed L82 site (present in L110A) is faster, but ultimately thermodynamically weaker and less effective, while Aβ binding to the interior L110 site (present in L82A) is slower due to greater steric restriction, but stronger and more effective for suppression of growth.
Aβ binding induces destabilization of TTR quaternary structure
We observed, by circular dichroism, a change in wt TTR secondary structure when incubated with excess Aβ for 2 days; this change was absent with short incubation or if Aβ was incubated with L82A and L110A. TTR tetramers are remarkably stable, but it is well established that tetramers undergo slow monomer subunit exchange with a time constant of approximately 18–24 h (27). The change in wt TTR secondary structure caused by Aβ was very similar to the difference in secondary structure between wt TTR and TTR mutants that are unstable tetramers (e.g., S85A), dimers (S112I), or monomers (F87M/L110M). We therefore hypothesize that Aβ induces destabilization of TTR tetramers. We obtained direct support for this hypothesis from both native gel electrophoresis and crosslinking experiments, demonstrating that wt TTR tetramers partially dissociate after incubation with Aβ for 1–2 days.
L82A tetramers were not destabilized when incubated with Aβ. From high temperature experiments we determined that the monomer exchange rates for L82A and wt are equal, so the lack of tetramer dissociation of L82A cannot be explained by differences in tetramer stability. Further experiments established that tetramer destabilization is not simply due to a detergent-like action of Aβ, since incubation of TTR with a detergent for 1–2 days caused partial dissociation of all 3 TTR variants equally. Finally, our data show that the slow kinetics of Aβ-induced TTR dissociation is not due to time-dependent changes in Aβ aggregates. Based on these combined results, we propose that binding of Aβ to the L82 site on TTR triggers tetramer destabilization and dissociation to monomers and/or dimers.
We have previously shown that monomeric and dimeric TTR mutants bind more Aβ than tetramers (14). Furthermore, TTR that is chemically crosslinked to stabilized tetramers will bind less Aβ than uncrosslinked TTR (unpublished results). Thus, by destabilizing tetramers, Aβ binding to L82 has the fortuitous effect of exposing the interior hydrophobic β-sheet (including L110), a potent binding site for Aβ.
Previously we showed that Aβ can compete to some extent with re-association of acid-dissociated TTR (14). Thus, it is possible that the observed tetramer destabilization is due to Aβ binding to TTR monomers during subunit exchange (in particular to the interior hydrophobic β-sheet) that competes with monomer re-association to tetramers. L110A contains the L82 binding site and is destabilized by Aβ; this dissociation is likely further facilitated by its modestly higher tetramer instability (as measured by kd). Aβ does not bind to the exposed interior β-sheet in L110A, but this mutant still is destabilized. Together with the lack of dissociation of L82A, this suggests that TTR destabilization is not strictly attributable to Aβ binding to the hydrophobic interior β-sheet of dissociated TTR subunits and subsequent competition with TTR re-association. However, this competitive binding could be an additional mechanism that maintains wt TTR in dissociated form.
TTR is both sensor and scavenger of toxic Aβ oligomers
Our hypothesis can be stated briefly: the EF loop of TTR is the sensor, and the inner β-sheet is the scavenger, for Aβ oligomers. TTR must be tetrameric to carry out its normal functions as a transporter of thyroxine and retinol-binding protein. Furthermore, TTR monomers are prone to self-aggregation and amyloid formation (29). There are several known natural TTR mutants that destabilize TTR tetramers; these mutations lead to aggregation and amyloid deposition, and are associated with diseases such as familial amyloid polyneuropathy (30, 31). Thus, normally stable TTR tetramers are preferable. But, TTR monomers bind more Aβ (14), presumably because of greater solvent exposure of the hydrophobic ‘inner’ β-sheet. In our proposed scenario, then, TTR remains safely tetrameric unless and until it detects unwanted Aβ oligomers. Exposure of the interior binding site is triggered when, and only when, the exterior loop of TTR senses the presence of Aβ oligomers.
This hypothesis is supported indirectly by our discovery of S85A, a TTR alanine mutant where the mutation is in the EF loop. This mutant folds into less-stable tetramers, binds more Aβ than wt (13), and protects against Aβ toxicity in vitro (unpublished data). Similarly, the mutation F87M on the EF loop produced a mixture of tetramers and monomers (16). Others have shown that low pH (3.5–4) causes conformational changes in the EF helix and loop that destabilize the tetramer; this is believed to be the first step in the TTR amyloidogenic pathway (32). Together, these data support the hypothesis that changes in the EF helix/loop can destabilize tetramers. Initial binding between Aβ and TTR could be mediated through the EF helix/loop, triggering a conformational change that makes TTR more amyloidogenic, and thus more able to interact with Aβ Furthermore, we previously showed that a monomeric TTR mutant (F87M/L110M) is more effective than TTR tetramers at arresting Aβ aggregate growth (14). Thus, Aβ-mediated destabilization of tetramers leads to ever greater TTR-Aβ association. While normally destabilized TTR is undesirable, in this case this is a favorable outcome, since the interaction between TTR and Aβ prevents Aβ toxicity.
Comparison of TTR to synthetic Aβ-sequence-derived inhibitors
We and others have reported that short sequences derived from full-length Aβ can act as ‘recognition elements’, binding to and disrupting normal Aβ aggregation pathways (33–37). In our most effective compounds, we linked the recognition element KLVFF (residues 16–20 of Aβ) to a hexamer of charged polar amino acids or derivatives. Full protection against Aβ toxicity was obtained at 10-fold Aβ excess, and partial protection at 100-fold Aβ excess (250 nM inhibitor to 25 μM Aβ) (38). In work reported here, we observed that TTR afforded complete protection at 100-fold Aβ excess, and partial protection at 1000-fold excess (about 20 nM or 1 μg/mL, Figure 2). Thus TTR is 10-fold more protective than our best synthesized compounds. (However, it must be recognized that different cell lines were used to assess toxicity.)
For the Aβ-sequence-derived compounds, our previous data were consistent with a mechanism by which the compounds inhibited toxicity by accelerating growth of large aggregates. We hypothesized that the compounds reversibly associated with beta-amyloid aggregates and, through effects on local solvent properties, enhanced hydrophobic interactions to drive association of smaller Aβ aggregates into larger, less toxic aggregates (39, 40). Such a mechanism would reduce exposure of cells to smaller soluble oligomers that have greater hydrophobicity and are presumably more toxic. TTR appears to act differently, because it arrests Aβ aggregation. We hypothesize that hydrophobic sites of TTR (particularly the inner β-sheet) bind tightly to exposed hydrophobic patches on Aβ oligomers, preventing both further Aβ-Aβ association as well as unwanted Aβ-cell association.
Relationship between TTR and AD
As described earlier, in transgenic mice engineered to express APP and develop amyloid deposits, TTR is upregulated; increased synthesis of TTR may protect these mice from the neuronal death of full-blown AD (1, 2). Although normally CSF TTR is synthesized in the choroid plexus, a recent investigation demonstrated TTR synthesis in choroid-plexus-free cortical cultures established from these transgenic mice, whereas minimal TTR synthesis was observed in wt controls (5). Another study showed that APPsα mediates TTR expression (41). These data suggest that APP and/or its proteolytic products trigger TTR synthesis, at least in transgenic mice.
In humans, plasma TTR levels fall in middle age or under conditions of malnutrition or chronic inflammation (11). A statistically significant decrease in TTR plasma levels was observed in AD patients relative to age-matched controls, and among a cohort of AD sufferers, TTR levels were lower in those with more rapid cognitive decline (42). These data, although very limited, hint that the drop in TTR levels with age or disease could reduce TTR’s normally protective role in sensing and scavenging toxic Aβ soluble oligomers. Perhaps restoring or replacing TTR’s natural protective role would provide a safe therapeutic approach to treating AD.
Acknowledgments
Funding Source: National Institutes of Health Grant 2R01AG033493
ABBREVIATIONS
- Aβ
beta-amyloid
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- CD
circular dichroism
- CSF
cerebrospinal fluid
- DLS
dynamic light scattering
- DIV
days in vitro
- MTS
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)-2H-tetrazolium salt
- NBM
neurobasal medium
- NTA
nanoparticle tracking analysis
- PMS
phenazine methosulfate
- PBS
phosphate-buffered saline
- PICUP
photo-induced cross-linking of unmodified proteins
- PS
penicillin/streptomycin
- RBP
retinol-binding protein
- TBS
Tris-buffered saline
- TTR
transthyretin
- TUNEL
terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
- wt
wild-type
References
- 1.Johnson JA, Stein TD. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22:7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson JA, Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APP(Sw) mice resulting in tau phosphorylation and loss of hippocampal neurons: Support for the amyloid hypothesis. J Neurosci. 2004;24:7707–7717. doi: 10.1523/JNEUROSCI.2211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen CKJ, Tsai KJ, Yang CH, Lee PC, Wang WT, Chiu MJ. Asymmetric expression patterns of brain transthyretin in normal mice and a transgenic mouse model of Alzheimer’s Disease. Neuroscience. 2009;159:638–646. doi: 10.1016/j.neuroscience.2008.12.045. [DOI] [PubMed] [Google Scholar]
- 4.Wu ZL, Ciallella JR, Flood DG, O’Kane TM, Bozyczko-Coyne D, Savage MJ. Comparative analysis of cortical gene expression in mouse models of Alzheimer’s disease. Neurobiol Aging. 2006;27:377–386. doi: 10.1016/j.neurobiolaging.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Li XY, Masliah E, Reixach N, Buxbaum JN. Neuronal Production of Transthyretin in Human and Murine Alzheimer’s Disease: Is It Protective? J Neurosci. 2011;31:12483–12490. doi: 10.1523/JNEUROSCI.2417-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu L, Hou J, Du JL, Chumanov RS, Xu QG, Ge Y, Johnson JA, Murphy RM. Differential modification of Cys10 alters transthyretin’s effect on beta-amyloid aggregation and toxicity. Protein Eng Des Sel. 2009;22:479–488. doi: 10.1093/protein/gzp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giunta S, Valli MB, Galeazzi R, Fattoretti P, Corder EH, Galeazzi L. Transthyretin inhibition of amylold beta aggregation and toxicity. Clin Biochem. 2005;38:1112–1119. doi: 10.1016/j.clinbiochem.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Costa R, Ferreira-da-Silva F, Saraiva MJ, Cardoso I. Transthyretin protects against A-beta peptide toxicity by proteolytic cleavage of the peptide: A mechanism sensitive to the Kunitz Protease Inhibitor. PLOS One. 2008;3 doi: 10.1371/journal.pone.0002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci USA. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Potter H, Costa DA, Craechiolo JR, Bachstetter AD, Hughes TF, Bales KR, Paul SM, Mervis RF, Arendash GW. Enrichment improves cognition in AD mice by amyloid-related and unrelated mechanisms. Neurobiol Aging. 2007;28:831–844. doi: 10.1016/j.neurobiolaging.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 11.Hamilton JA, Benson MD. Transthyretin: a review from a structural perspective. Cell Mol Life Sci. 2001;58:1491–1521. doi: 10.1007/PL00000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richardson SJ. Cell and molecular biology of transthyretin and thyroid hormones. Int Rev Cytol. 2007;258:137–193. doi: 10.1016/S0074-7696(07)58003-4. [DOI] [PubMed] [Google Scholar]
- 13.Du JL, Cho PY, Yang DT, Murphy RM. Identification of beta-amyloid-binding sites on transthyretin. Protein Eng Des Sel. 2012;25:337–345. doi: 10.1093/protein/gzs026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du JL, Murphy RM. Characterization of the interaction of beta-amyloid with transthyretin monomers and tetramers. Biochemistry. 2010;49:8276–8289. doi: 10.1021/bi101280t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwarzman AL, Gregori L, Vitek MP, Lyubski S, Strittmatter WJ, Enghilde JJ, Bhasin R, Silverman J, Weisgraber KH, Coyle PK, Zagorski MG, Talafous J, Eisenberg M, Saunders AM, Roses AD, Goldgaber D. Transthyretin sequesters amyloid-beta protein and prevents amyloid formation. Proc Natl Acad Sci USA. 1994;91:8368–8372. doi: 10.1073/pnas.91.18.8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC, Kelly JW. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- 17.Pallitto MM, Murphy RM. A mathematical model of the kinetics of beta-amyloid fibril growth from the denatured state. Biophys J. 2001;81:1805–1822. doi: 10.1016/S0006-3495(01)75831-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fancy DA, Kodadek T. Chemistry for the analysis of protein-protein interactions: Rapid and efficient cross-linking triggered by long wavelength light. Proc Natl Acad Sci USA. 1999;96:6020–6024. doi: 10.1073/pnas.96.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia K, Zhang SJ, Bathrick B, Liu SQ, Garcia Y, Colon W. Quantifying the kinetic stability of hyperstable proteins via time-dependent SDS trapping. Biochemistry-Us. 2012;51:100–107. doi: 10.1021/bi201362z. [DOI] [PubMed] [Google Scholar]
- 20.Murphy RM, Liu L, Hou J, Du JL, Chumanov RS, Xu QG, Ge Y, Johnson JA. Differential modification of Cys10 alters transthyretin’s effect on beta-amyloid aggregation and toxicity. Protein Eng Des Sel. 2009;22:479–488. doi: 10.1093/protein/gzp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costa R, Goncalves A, Saralva MJ, Cardoso I. Transthyretin binding to A-Beta peptide - Impact on A-Beta fibrillogenesis and toxicity. Febs Lett. 2008;582:936–942. doi: 10.1016/j.febslet.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 22.Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filipe V, Hawe A, Jiskoot W. Critical Evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the Measurement of Nanoparticles and Protein Aggregates. Pharm Res-Dordr. 2010;27:796–810. doi: 10.1007/s11095-010-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glabe CG, Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2 doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarsoza F, Saing T, Kayed R, Dahlin R, Dick M, Broadwater-Hollifield C, Mobley S, Lott I, Doran E, Gillen D, Anderson-Bergman C, Cribbs DH, Glabe C, Head E. A fibril-specific, conformation-dependent antibody recognizes a subset of A beta plaques in Alzheimer disease, Down syndrome and Tg2576 transgenic mouse brain. Acta Neuropathol. 2009;118:505–517. doi: 10.1007/s00401-009-0530-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glabe CG, Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 27.Keetch CA, Bromley EHC, McCammon MG, Wang N, Christodoulou J, Robinson CV. L55P transthyretin accelerates subunit exchange and leads to rapid formation of hybrid tetramers. J Biol Chem. 2005;280:41667–41674. doi: 10.1074/jbc.M508753200. [DOI] [PubMed] [Google Scholar]
- 28.Brouillette J, Caillierez R, Zommer N, Alves-Pires C, Benilova I, Blum D, De Strooper B, Buee L. Neurotoxicity and Memory Deficits Induced by Soluble Low-Molecular-Weight Amyloid-beta(1–42) Oligomers Are Revealed In Vivo by Using a Novel Animal Model. J Neurosci. 2012;32:7852–7861. doi: 10.1523/JNEUROSCI.5901-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammarstrom P, Sekijima Y, White JT, Wiseman RL, Lim A, Costello CE, Altland K, Garzuly F, Budka H, Kelly JW. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: A prescription for central nervous system amyloidosis? Biochemistry-Us. 2003;42:6656–6663. doi: 10.1021/bi027319b. [DOI] [PubMed] [Google Scholar]
- 30.McCutchen SL, Colon W, Kelly JW. Transthyretin mutation Leu-55-Pro significantly alters tetramer stability and increases amyloidogenicity. Biochemistry. 1993;32:12119–12127. doi: 10.1021/bi00096a024. [DOI] [PubMed] [Google Scholar]
- 31.Babbes ARH, Powers ET, Kelly JW. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: The relationship between stability and amyloidosis. Biochemistry. 2008;47:6969–6984. doi: 10.1021/bi800636q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palaninathan SK, Mohamedmohaideen NN, Snee WC, Kelly JW, Sacchettini JC. Structural Insight into pH-induced Conformational Changes within the Native Human Transthyretin Tetramer. J Mol Biol. 2008;382:1157–1167. doi: 10.1016/j.jmb.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 33.Ghanta J, Shen CL, Kiessling LL, Murphy RM. A strategy for designing inhibitors of beta-amyloid toxicity. J Biol Chem. 1996;271:29525–29528. doi: 10.1074/jbc.271.47.29525. [DOI] [PubMed] [Google Scholar]
- 34.Findeis MA, Musso GM, Arico-Muendel CC, Benjamin HW, Hundal AM, Lee JJ, Chin J, Kelley M, Wakefield J, Hayward NJ, Molineaux SM. Modified-peptide inhibitors of amyloid beta-peptide polymerization. Biochemistry. 1999;38:6791–6800. doi: 10.1021/bi982824n. [DOI] [PubMed] [Google Scholar]
- 35.Tjernberg LO, Naslund J, Lindqvist F, Johansson J, Karlstrom AR, Thyberg J, Terenius L, Nordstedt C. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J Biol Chem. 1996;271:8545–8548. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- 36.Gordon DJ, Sciarretta KL, Meredith SC. Inhibition of beta-amyloid(40) fibrillogenesis and disassembly of beta-amyloid(40) fibrils by short beta-amyloid congeners containing N-methyl amino acids at alternate residues. Biochemistry. 2001;40:8237–8245. doi: 10.1021/bi002416v. [DOI] [PubMed] [Google Scholar]
- 37.Rangachari V, Davey ZS, Healy B, Moore BD, Sonoda LK, Cusack B, Maharvi GM, Fauq AH, Rosenberry TL. Rationally designed dehydroalanine (delta Ala)-containing peptides inhibit Amyloid-beta (Abeta) peptide aggregation. Biopolymers. 2009;91:456–465. doi: 10.1002/bip.21151. [DOI] [PubMed] [Google Scholar]
- 38.Lowe TL, Strzelec A, Kiessling LL, Murphy RM. Structure-function relationships for inhibitors of beta-amyloid toxicity containing the recognition sequence KLVFF. Biochemistry. 2001;40:7882–7889. doi: 10.1021/bi002734u. [DOI] [PubMed] [Google Scholar]
- 39.Kim JR, Gibson TJ, Murphy RM. Targeted control of kinetics of beta-amyloid self-association by surface tension-modifying peptides. J Biol Chem. 2003;278:40730–40735. doi: 10.1074/jbc.M305466200. [DOI] [PubMed] [Google Scholar]
- 40.Kim JR, Murphy RM. Mechanism of accelerated assembly of beta-amyloid filaments into fibrils by KLVFFK6. Biophys J. 2004;86:3194–3203. doi: 10.1016/S0006-3495(04)74367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li HM, Wang BP, Wang ZL, Guo QX, Tabuchi K, Hammer RE, Sudhof TC, Zheng H. Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. P Natl Acad Sci USA. 2010;107:17362–17367. doi: 10.1073/pnas.1012568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Velayudhan L, Killick R, Hye A, Kinsey A, Guentert A, Lynham S, Ward M, Leung R, Lourdusamy A, To AWM, Powell J, Lovestone S. Plasma transthyretin as a candidate marker for Alzheimer’s Disease. J Alzheimers Dis. 2012;28:369–375. doi: 10.3233/JAD-2011-110611. [DOI] [PubMed] [Google Scholar]