

Abstract
Purpose
Iododeoxyuridine (IdUrd), a halogenated nucleoside analog, produced clinical responses when administered as a radiosensitizer via continuous intravenous (c.i.v.) infusion over the course of radiation therapy. We conducted a Phase 0 trial of 5-iodo-2-pyrimidinone-2′-deoxyribose (IPdR), an oral prodrug of IdUrd, in patients with advanced malignances to assess whether the oral route was a feasible alternative to c.i.v. infusion prior to embarking on large-scale clinical trials. Plasma concentrations of IPdR, IdUrd, and other metabolites were measured after a single oral dose of IPdR.
Patients and Methods
Eligible patients had advanced refractory malignancies. A single oral dose of IPdR was administered per patient and patients were followed for 14 days for safety assessments; dose escalations were planned (150, 300, 600, 1200, and 2400 mg) with one patient per dose level (DL) and 6 patients at the highest DL. Blood sampling was performed over a 24-hour period for pharmacokinetic analysis.
Results
There were no drug-related adverse events. Plasma concentrations of IdUrd generally increased as the dose of IPdR escalated from 150 to 2400 mg. All patients at the 2400 mg dose achieved peak IdUrd levels of (mean ± SD) 4.0 μM ± 1.02 μM (25% CV) at 1.67 ± 1.21 hours after IPdR administration.
Conclusions
Adequate plasma levels of IdUrd were obtained to justify proceeding with a Phase I trial of IPdR in combination with radiation. This trial demonstrates the ability of a small, Phase 0 study to provide critical information for decision-making regarding future development of a drug.
Keywords: IPdR, IdUrd, phase 0, clinical trial, pharmacokinetics
INTRODUCTION
Development of radiosensitizing agents to improve the therapeutic outcome for cancer patients receiving definitive radiation therapy has been a longstanding area of research. Halogenated thymidine (TdR) analogs such as 5-iododeoxyuridine (IdUrd; NSC 39661) and 5-bromodeoxyuridine (BUdR) have been recognized as potential radiosensitizers since the early 1960s (1–3). Cellular uptake and metabolism of these analogs are dependent on the TdR salvage pathway (4). IdUrd produced responses in combination with radiation in Phase I/II clinical trials when given with radiation therapy to patients with high-grade anaplastic astrocytomas and sarcomas (5–7). However, due to the short half-life of IdUrd (in the order of minutes), continuous intravenous (c.i.v.) infusion over the course of radiation therapy was required to maintain adequate exposure (5).
5-iodo-2-pyrimidinone-2′-deoxyribose (IPdR; NSC 726188), an oral prodrug of IdUrd, has many advantages as compared to IdUrd, including ease of administration, a more favorable toxicity profile and a better therapeutic index in animals (8). Oral IPdR would present an attractive alternative to c.i.v. of IdUrd if adequate systemic exposures to IdUrd and its metabolites could be obtained. Here, we conducted a first in human, Phase 0, feasibility trial of IPdR in patients with advanced malignances prior to embarking on large-scale clinical trials of IPdR in combination with radiation therapy. Plasma concentrations of IPdR, IdUrd, and other metabolites were measured after a single oral dose of IPdR. Unlike micro-dosing studies and other variations of the Phase 0 concept, this study was designed to produce concentrations of IdUrd that are expected to be in the therapeutic range, based upon prior clinical studies with c.i.v. IdUrd. Since only a single dose of IPdR was administered, the exposure time to IdUrd is expected to be short, the probability of toxicity is minimized. Establishment of the toxicity profile and the maximum tolerated dose are in the realm of phase 1 studies.
MATERIALS AND METHODS
Eligibility Criteria
Adult patients with advanced malignancies refractory to at least one line of standard treatment were eligible. Patients were 18 years of age or older, had an Eastern Cooperative Oncology Group performance status of ≤ 2; and adequate liver, kidney, and marrow function defined as absolute neutrophil count ≥ 1,500/μL, platelets ≥ 100,000/μL, total bilirubin ≤ 1.5 X the upper limit of normal (ULN), aspartate aminotransferase and/or alanine aminotransferase < 3 X ULN, creatinine < 1.5 X ULN. Prior anti-neoplastic therapy must have been completed at least 2 weeks prior to enrollment. Patients unable to swallow pills or those with uncontrolled intercurrent illness or pregnant or lactating were not eligible.
Consent Process
Due to the non-therapeutic nature of the trial, the objectives and the consent form were discussed in detail with potential patients in advance. Patients were asked to verbalize their understanding of the nature of the trial prior to signing the consent form. This trial was conducted under an NCI-sponsored IND with approval from the NCI Institutional Review Board. Protocol design and conduct followed all applicable regulations, guidances, and local policies. ClinicalTrials.gov identifier: NCT01240577.
Study Design
IPdR was supplied by the Division of Cancer Treatment and Diagnosis, National Cancer Institute, Bethesda, MD, USA. A single oral dose of IPdR was administered on day 1, with serial blood and urine sampling performed before and during a 24-hour period after drug administration for pharmacokinetic (PK) analysis. Patients were followed for 14 days for safety assessments. Oral doses of 150, 300, 600, 1200, and 2400 mg, were explored with one patient enrolled successively to each dose level. At the highest dose (2400 mg), a total of 6 patients were enrolled. A new patient could only be enrolled once the prior patient had completed 2 weeks of participation with no drug related adverse events.
The starting dose of 150 mg was based on 10% of the tolerable dose from a repeat-dose study in the most sensitive animal species, the ferret. There was significant weight loss of 10 to 20% and gastrointestinal side effects in ferrets receiving 1500 mg/kg/day for 14 days.9 The tolerable dose for repeat dose studies in rats was higher, 2000 mg/kg/day for 28 days.8 In studies in athymic mice, no significant toxicities were reported after daily oral IPdR doses of ≤ 1500 mg/kg/day for 6 to 14 days.
Clinical toxicities were graded according to Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. Significant toxicities were defined as toxicities considered to be related to study medication occurring within 14 days of administering IPdR and met the following criteria: a) grade ≥ 2 non-hematologic toxicities other than easily correctable electrolyte abnormalities, b) grade ≥ 2 thrombocytopenia, c) grade ≥ 3 anemia, leucopenia, or neutropenia. If one patient developed significant toxicity, then no additional patients were to be enrolled, the study was to be put on hold, and all the safety and PK data analyzed.
History and physical examination, including performance status and vital signs, were performed at baseline and repeated at the end of 2 weeks. Complete blood counts with differential and serum chemistries were performed at baseline, on day 2, and on day 14 (at off-study).
Pharmacokinetic Evaluations
Peripheral blood samples (7 mL) were collected before and at multiple time points (5, 15, and 30 minutes, 1, 2, 4, 10, and 24 hours) post–IPdR administration. All samples were centrifuged and plasma was stored at −80°C for analysis. Urine (10 mL) was also collected just before drug administration, and then separately at each void; volume measured and recorded, and a sample (10–12 mL) was retained for analysis and stored at −80°C. A sensitive liquid chromatography coupled with tandem mass spectrometry detection (LC–MS/MS) method was developed to measure plasma and urinary concentrations of IPdR, IdUrd, and other metabolites.
Plasma samples were processed by solvent deproteinization, while the urine samples were processed by liquid-liquid extraction. Separation of IPdR and its metabolites was performed on an Agilent 1200LC system (Agilent Technologies, Palo Alto, CA) using a 4.6 x 250 mm Synergi Hydro-RP C18 column. The reference compounds 5-iodo-2-pyrimidinone-2′-deoxyribose (IPdR, NSC726188), 5-iodo-2′-deoxyuridine (IdUrd, NSC39661), 5-iodo-2-pyrimidinone (IP, NSC754229), and 5-iodouracil (IUra, NSC57848) were supplied by the NCI Developmental Therapeutics Program (Bethesda, MD). 5-Bromo-2′-deoxyuridine (BrdUrd) and 5-bromouracil (BrUra) used as internal standards for quantitations were obtained from the Sigma-Aldrich Company (St. Louis, MO). All solvents were HPLC grade. All other reagents were also obtained from the Sigma-Aldrich Company (St. Louis, MO). Calibration curves were constructed by adding known amounts of IPdR, IP, IdUrd, and IUra to control human plasma or to patient pre-dose urine in order to give samples containing concentrations ranging 0.1 to 50 μM of each compound. The response factor was linear over the range of 0.1 to 50 μM. Samples were diluted 1:10 in control matrix and reanalyzed when calculated concentrations exceeded 50 μM. Absolute recoveries of analytes from plasma were 76% for IPdR, 75% for IdUrd, 92% for IUra and 87% for IP. For urine, absolute recoveries of analytes were 92% for IPdR, 93% for IdUrd, and 89% for IUra. The lower limit of quantitation was 0.1uM for IdUrd and IPdR, and 0.25uM for IP and IUra. Accuracy was found to be greater than 98% for these compounds, with precision of 95%.
RESULTS
A total of 10 patients participated in the study and all patients tolerated study drug well with no drug related adverse events. Patient demographics are presented in Table 1.
Table 1.
Patient Demographics
| Parameter | Value |
|---|---|
| No. of patients enrolled | 10 |
| No. enrolled by sex | |
| Female | 2 |
| Male | 8 |
| Age range, years | 37–73 |
| Diagnosis, No. of patients | |
| Adenoid cystic carcinoma | 1 |
| Bladder cancer | 1 |
| Breast cancer | 1 |
| Colorectal cancer | 2 |
| Hepatocellular carcinoma | 1 |
| Lymphoma | 1 |
| Neuroendocrine neoplasm | 1 |
| Thyroid | 2 |
Pharmacokinetics
Plasma concentrations of IdUrd, the active metabolite, generally increased as oral doses of IPdR escalated from 150 to 2400 mg (Table 2 and Figure 1). At the highest IPdR dose of 2400 mg, the 6 patients achieved peak IdUrd plasma levels of 4.0 ± 1.02 μM (25% CV) after 1.67 ± 1.21 hours. Plasma concentrations remained above 1 μM for 3–4 hours, and declined with a half-life of 1.5 hours. For plasma AUC of IdUrd, the CV was 32%.
Table 2.
Plasma Cmax and AUC values. Cmax units are μM; AUC units are μM-hr. One patient per dose from 150–1200 mg, and 6 patients at 2400mg. The patient at 300 mg did not have quantifiable concentrations of IPdR.
| DOSE mg | IPdR AUC | IPdR Cmax | IdUrd AUC | IdUrd Cmax | IUra AUC | IUra Cmax |
|---|---|---|---|---|---|---|
| 150 | 3.4 | 0.6 | 1.7 | 0.8 | 1.0 | 0.2 |
| 300 | n.d. | n.d. | 0.3 | 0.3 | 4.5 | 0.8 |
| 600 | 8.3 | 0.93 | 3.2 | 1.8 | 17.2 | 4.5 |
| 1200 | 0.7 | 0.95 | 4.7 | 2.1 | 102.7 | 34.2 |
|
| ||||||
| 2400 | 15.6 | 4.3 | 12.2 | 4.0 | 1057.8 | 133 |
| SD | +/−18.8 | +/−4.3 | +/−3.9 | +/−1.0 | +/− 483.5 | +/−42.2 |
Figure 1.

Plasma exposures of the active metabolite IdUrd following a single dose of IPdR over the range of 150 to 2400 mg. Plasma concentrations remained above 1 μM for 3 to 4 hours and declined with a half-life of 1.5 hours.
For the prodrug, IPdR, both Cmax and AUC for plasma were variable at the lower doses that enrolled single patient cohorts (Table 2), including one patient with undetectable levels. At the highest dose, the coefficients of variation for IPdR were 100% for both Cmax and AUC. Both Cmax and AUC for plasma of IUra, the major metabolite, increased more than proportionally as doses were doubled (Table 2 and Figure 2). At the 2400mg dose of IPdR, Cmax values for IUra were 133 ± 42 μM after 3.3 ± 1.0 hours post IPdR administration (Table 2 and Figure 2). IUra concentrations remained near 100 μM for 10 hours in 4 of 6 patients receiving the 2400 mg dose. The pathways of formation for metabolites of IPdR are presented in Figure 3.
Figure 2.

Plasma exposures of the secondary metabolite, IUra, by dose level. Peak levels of IUra were reached 3.3 (± 1.0) hours post IPdR administration.
Figure 3.
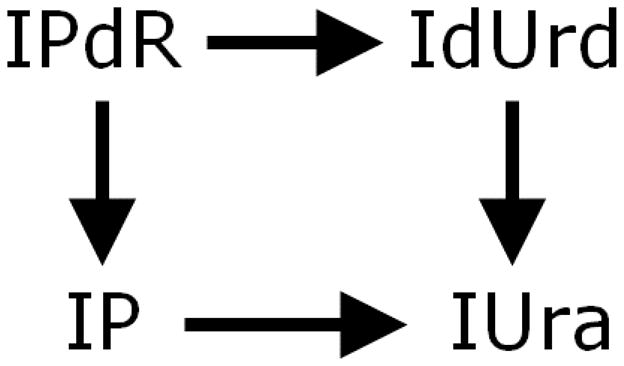
Formation of metabolites of IPdR.
Patient 10 had a history of gastric bypass surgery as part of a bariatric surgery procedure 20 years prior to enrollment on study. Plasma levels of IPdR and its metabolites in this patient were comparable to other patients on the 2400 mg dose.
IPdR and its metabolites, IdUrd, IP, and IUra, were detectable in urine samples from patients receiving 2400 mg of IPdR. Twelve percent (± 8%) of the dose was recovered over 24 hours; of the metabolites measured in urine, 90 ± 13%, was IUra.
DISCUSSION
Radiosensitization to improve curative rates in cancer has been an area of ongoing research. Chemotherapies such as fluorouracil (5FU) and gemcitabine have been co-administered to increase the effectiveness of radiation therapy; however, this comes at the cost of increasing toxicities, both systemic and local. Halogenated thymidine analogs have been studied as radiosensitizers and incorporation of their phosphorylated forms into DNA, by DNA polymerase during the process of replication, is necessary for radiosensitization (4, 10). When halogenated thymidine analogs are incorporated into DNA, there is increased sensitivity to damage by the highly reactive uracil free radicals generated by radiation (11). However, due to the short half-life of halogenated analogs, and the need for a high labeling index to derive clinical benefit, c.i.v. administration of such analogs throughout the course of radiation has been evaluated (2, 12–14). IdUrd has shown radiopotentiation in sarcomas and brain tumors with clinical benefit (5, 15–17). A long term study of patients with anaplastic astrocytoma treated with a combination of radiation and IdUrd reported a median survival of 3.2 years, with 33% of patients surviving at 5 years (5). Even though clinical benefit was observed in these trials, the need to administer an agent by c.i.v. infusion over weeks presents practical challenges.
The ability to administer IdUrd orally would circumvent the practical limitations of c.i.v. administration and allow development of this class of agents as potential radiosensitizers. IPdR was formulated as an oral prodrug of IdUrd and, in preclinical models; adequate exposures to IdUrd were obtained following oral administration of IPdR (7, 18). The initial goal of early clinical development of IPdR is to determine safe and effective doses capable of being administered along with radiation. However, prior to proceeding with the definitive dose finding safety study of IPdR with radiation, we conducted a small phase 0 trial of IPdR in patients with advanced malignancies to determine the pharmacokinetics of IPdR and its metabolites in humans.
The data in Table 2, as well as Figures 1 and 2, exhibit substantial differences for IPdR and its metabolites in the patterns for their changes in systemic exposure with dose. Due to the nature of this study design, which intended to minimize the number of patients, it must be recognized that definitive conclusions are not possible. Nonetheless, sufficient information was generated to guide further efforts. In particular, the disproportional increases for IUra exposure in this study are very consistent with prior work that demonstrated disproportional accumulation due to saturation of its metabolic elimination (11). The lack of a consistent pattern for exposure to IPdR is a signal that absorption and/or first-pass metabolism should continue to be monitored in future studies. For the most important compound, IdUrd, the modest variation in both AUC and Cmax among the 6 patients at the highest dose are positive findings for further pursuit of this oral approach. The apparent absence major trends toward disproportional changes in exposure for IdUrd are tempered by the small numbers of patients, but are consistent with larger studies for direct administration of IdUrd (11).
First-in-human ‘Phase 0’ clinical trials offer an opportunity to evaluate the pharmacology of novel agents in humans well in advance of dose-finding Phase 1 trials. Such trials present a platform for clinical testing of novel compounds with less pharmacology and toxicology data than traditionally required, as drug exposure is limited in Phase 0 trials providing the safety margin (19). Depending on the circumstances, Phase 0 trials can be conducted either under a standard IND or the FDA’s exploratory IND guidance (20). Subsequent clinical development decisions can be based on data generated from patient samples rather than solely from preclinical models. Such data can inform drug development decisions including systematic deprioritization of compounds that do not satisfy pre-specified PK or pharmacodynamic criteria.
Previous clinical studies (11,16) have defined a minimum target concentration of 1 μM for IdUrd in plasma. If this phase zero study had found that oral dosing with IPdR could not reach this target concentration of IdUrd, then it would be futile to continue further trials. Because the mean Cmax of IdUrd in plasma was 4.0 μM for the 6 patients at the 2400 mg dose, we can move ahead to consideration of the second drug delivery question: What is the optimal pattern for exposure to IdUrd? Prior work showed that prolonged exposure to IdUrd is essential to derive therapeutic benefit, and continuous infusions have been for 2–4 weeks have been utilized in most clinical studies of IdUrd. However, the length of exposure or time sequence with once-a-day radiation treatment remains largely unexplored. Given the ease of administration of oral IPdR, clinical trials of IPdR in combination with radiation could easily evaluate various sequences and durations of exposure to optimize the therapeutic index of this radiosensitizer. Initiating the clinical evaluation of IPdR with dosing once per day would be a reasonable strategy to generate initial safety data in combination with prolonged periods of radiation. This approach could be followed with dosing twice per day, which would reduce the length of time below the nominal target concentration. Pharmacodynamic studies or diagnostic approaches, as described below, could be combined with clinical assessments to determine the preferred schedule.
When IdUrd was first assessed as a chemotherapeutic, Calabresi et al reported tumor shrinkage in 6 out of 11 patients treated with c.i.v. (2). In 4 of 9 patients treated with intrahepatic arterial infusions of IdUrd, Cheng et al reported tumor shrinkage to be between 40 to 65% (13). Morgan et al reported disappearance of ascites in patients with ovarian cancer treated with intraperitoneal IdUrd (12). The pharmacologic activity of parent IPdR has not been firmly established, and it is primarily viewed as a prodrug to provide oral delivery of its active metabolite, IdUrd. IPdR was originally synthesized as an antiviral agent. Based upon its pyrimidinone structure and our unpublished data, IPdR might also serve as an inhibitor of cytidine deaminase. Previous reports indicated that plasma concentrations of IPdR in excess of 100 μM could be sustained for at least 6 hours following oral administration of IPdR in mice or rats (7, 8). In contrast, this clinical study found lower and more transient exposure for IPdR, which diminishes the potential for any direct pharmacologic activity from the parent compound.
Oral IPdR, as a convenient method of delivery of IdUrd, also offers the opportunity to reexamine previous diagnostic applications for IdUrd. As a probe for assessing the tumor proliferation rate, IPdR could be an alternative to intravenous bolus of bromodeoxyuridine (BUdR). BUdR is currently the most widely used measure of cellular proliferation in vitro (21), and the same techniques are applicable to IdUrd in vivo. Utilization of radioiodine isotopes, that have a long half-life (e.g., 4 days for 124I), could provide a major logistical advantage as compared to the current use of 3′-deoxy-18F-fluorothymidine (18F-FLT) for imaging tumor proliferation, since 18F-FLT has a half-life of only 110 minutes (22). Iodine isotopes of IdUrd have been shown to assess proliferation of tumors in in vivo models (23). However, release of free radioiodine creates background signal that obscures the imaging of DNA synthetic pathways. The secondary metabolite of IPdR, IUra, does not have established pharmacologic activity, but inhibits the enzyme dihydropyrimidine dehydrogenase. Inhibition of this enzyme could prevent the release of radioiodine from labeled IdUrd, overcoming the long-standing limitation to noninvasive measurement of tumor proliferation using radioiodine labeled IdUrd. In view of prior response heterogeneity and DNA incorporation, either an oral test dose of IPdR or PET imaging with 124I could provide an enrichment strategy for patient selection.
This trial demonstrates the ability of a small, Phase 0 study to provide critical information for decision-making regarding future development of a drug. Adequate plasma levels of IdUrd were obtained to justify proceeding with a Phase I trial of oral IPdR in combination with radiation, and assessment of other diagnostic and therapeutic applications.
Statement of Translational Relevance.
Iododeoxyuridine (IdUrd), a halogenated nucleoside analog, produced clinical responses when administered as a radiosensitizer via continuous intravenous (c.i.v.) infusion over the course of radiation therapy. We conducted a Phase 0 trial of IPdR, an oral prodrug of IdUrd, in patients with advanced malignances to assess whether the oral route was a feasible alternative to c.i.v. infusion prior to embarking on large-scale clinical trials. Plasma concentrations of IPdR, IdUrd, and other metabolites were measured after a single oral dose of IPdR. Adequate plasma levels of IdUrd were obtained to justify proceeding with a Phase I trial of IPdR in combination with radiation. This trial demonstrates the ability of a small, Phase 0 study to provide critical information for decision-making regarding future development of a drug.
Acknowledgments
Research Support: This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. There are no other directly related manuscripts, published or unpublished, by any authors of this paper.
We are grateful to Timothy Kinsella, M.D., for his many efforts to bring IPdR to the clinic, including his helpful comments and encouragement of this study. We thank Hana Biosciences, Inc., and Dr. Kinsella for facilitating the transfer of the regulatory documentation to enable this study. We also thank Heather Gorby, SAIC-Frederick, Inc., for editorial assistance in the preparation of this manuscript.
Footnotes
Disclosure of Potential Conflicts of Interest: None.
References
- 1.Prusoff WH. Synthesis and biological activities of iododeoxyuridine, an analog of thymidine. Biochim Biophys Acta. 1959;32:295–6. doi: 10.1016/0006-3002(59)90597-9. [DOI] [PubMed] [Google Scholar]
- 2.Calabresi P, Cardoso SS, Finch SC, Kligerman MM, Von Essen CF, Chu MY, et al. Initial clinical studies with 5-iodo-2′-deoxyuridine. Cancer Res. 1961;21:550–9. [PubMed] [Google Scholar]
- 3.Erikson RL, Szybalski W. Molecular Radiobiology of Human Cell Lines. V. Comparative Radiosensitizing Properties of 5-Halodeoxycytidines and 5-Halodeoxyuridines. Radiat Res. 1963;20:252–62. [PubMed] [Google Scholar]
- 4.Kinsella TJ, Dobson PP, Mitchell JB, Fornace AJ., Jr Enhancement of X ray induced DNA damage by pre-treatment with halogenated pyrimidine analogs. Int J Radiat Oncol Biol Phys. 1987;13:733–9. doi: 10.1016/0360-3016(87)90292-6. [DOI] [PubMed] [Google Scholar]
- 5.Urtasun RC, Kinsella TJ, Farnan N, DelRowe JD, Lester SG, Fulton DS. Survival improvement in anaplastic astrocytoma, combining external radiation with halogenated pyrimidines: final report of RTOG 86-12, Phase I–II study. Int J Radiat Oncol Biol Phys. 1996;36:1163–7. doi: 10.1016/s0360-3016(96)00429-4. [DOI] [PubMed] [Google Scholar]
- 6.Prados MD, Scott CB, Rotman M, Rubin P, Murray K, Sause W, et al. Influence of bromodeoxyuridine radiosensitization on malignant glioma patient survival: a retrospective comparison of survival data from the Northern California Oncology Group (NCOG) and Radiation Therapy Oncology Group trials (RTOG) for glioblastoma multiforme and anaplastic astrocytoma. Int J Radiat Oncol Biol Phys. 1998;40:653–9. doi: 10.1016/s0360-3016(97)00770-0. [DOI] [PubMed] [Google Scholar]
- 7.Kinsella TJ, Kunugi KA, Vielhuber KA, Potter DM, Fitzsimmons ME, Collins JM. Preclinical evaluation of 5-iodo-2-pyrimidinone-2′-deoxyribose as a prodrug for 5-iodo-2′-deoxyuridine-mediated radiosensitization in mouse and human tissues. Clin Cancer Res. 1998;4:99–109. [PubMed] [Google Scholar]
- 8.Kinsella TJ, Kinsella MT, Hong S, Johnson JP, Burback B, Tosca PJ. Toxicology and pharmacokinetic study of orally administered 5-iodo-2-pyrimidinone-2′deoxyribose (IPdR) x 28 days in Fischer-344 rats: impact on the initial clinical phase I trial design of IPdR-mediated radiosensitization. Cancer Chemother Pharmacol. 2008;61:323–34. doi: 10.1007/s00280-007-0518-4. [DOI] [PubMed] [Google Scholar]
- 9.Kinsella TJ, Vielhuber KA, Kunugi KA, Schupp J, Davis TW, Sands H. Preclinical toxicity and efficacy study of a 14-day schedule of oral 5-iodo-2-pyrimidinone-2′-deoxyribose as a prodrug for 5-iodo-2′-deoxyuridine radiosensitization in U251 human glioblastoma xenografts. Clin Cancer Res. 2000;6:1468–75. [PubMed] [Google Scholar]
- 10.Lawrence TS, Davis MA, Maybaum J, Stetson PL, Ensminger WD. The effect of single versus double-strand substitution on halogenated pyrimidine-induced radiosensitization and DNA strand breakage in human tumor cells. Radiat Res. 1990;123:192–8. [PubMed] [Google Scholar]
- 11.Klecker RW, Jr, Jenkins JF, Kinsella TJ, Fine RL, Strong JM, Collins JM. Clinical pharmacology of 5-iodo-2′-deoxyuridine and 5-iodouracil and endogenous pyrimidine modulation. Clin Pharmacol Ther. 1985;38:45–51. doi: 10.1038/clpt.1985.132. [DOI] [PubMed] [Google Scholar]
- 12.Morgan RJ, Jr, Newman EM, Doroshow JH, McGonigle K, Margolin K, Raschko J, et al. Phase I trial of intraperitoneal iododeoxyuridine with and without intravenous high-dose folinic acid in the treatment of advanced malignancies primarily confined to the peritoneal cavity: flow cytometric and pharmacokinetic analysis. Cancer Res. 1998;58:2793–800. [PubMed] [Google Scholar]
- 13.Chang AE, Collins JM, Speth PA, Smith R, Rowland JB, Walton L, et al. A phase I study of intraarterial iododeoxyuridine in patients with colorectal liver metastases. J Clin Oncol. 1989;7:662–8. doi: 10.1200/JCO.1989.7.5.662. [DOI] [PubMed] [Google Scholar]
- 14.Sondak VK, Robertson JM, Sussman JJ, Saran PA, Chang AE, Lawrence TS. Preoperative idoxuridine and radiation for large soft tissue sarcomas: clinical results with five-year follow-up. Ann Surg Oncol. 1998;5:106–12. doi: 10.1007/BF02303842. [DOI] [PubMed] [Google Scholar]
- 15.Kinsella TJ, Russo A, Mitchell JB, Collins JM, Rowland J, Wright D, et al. A phase I study of intravenous iododeoxyuridine as a clinical radiosensitizer. Int J Radiat Oncol Biol Phys. 1985;11:1941–6. doi: 10.1016/0360-3016(85)90275-5. [DOI] [PubMed] [Google Scholar]
- 16.Kinsella TJ, Collins J, Rowland J, Klecker R, Jr, Wright D, Katz D, et al. Pharmacology and phase I/II study of continuous intravenous infusions of iododeoxyuridine and hyperfractionated radiotherapy in patients with glioblastoma multiforme. J Clin Oncol. 1988;6:871–9. doi: 10.1200/JCO.1988.6.5.871. [DOI] [PubMed] [Google Scholar]
- 17.Sullivan FJ, Herscher LL, Cook JA, Smith J, Steinberg SM, Epstein AH, et al. National Cancer Institute (phase II) study of high-grade glioma treated with accelerated hyperfractionated radiation and iododeoxyuridine: results in anaplastic astrocytoma. Int J Radiat Oncol Biol Phys. 1994;30:583–90. doi: 10.1016/0360-3016(92)90944-d. [DOI] [PubMed] [Google Scholar]
- 18.Kinsella TJ, Kunugi KA, Vielhuber KA, McCulloch W, Liu SH, Cheng YC. An in vivo comparison of oral 5-iodo-2′-deoxyuridine and 5-iodo-2-pyrimidinone-2′-deoxyribose toxicity, pharmacokinetics, and DNA incorporation in athymic mouse tissues and the human colon cancer xenograft, HCT-116. Cancer Res. 1994;54:2695–700. [PubMed] [Google Scholar]
- 19.Kummar S, Kinders R, Gutierrez ME, Rubinstein L, Parchment RE, Phillips LR, et al. Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009;27:2705–11. doi: 10.1200/JCO.2008.19.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.United States Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research. Guidance for Industry, Investigators, and Reviewers. Exploratory IND Studies [Internet] Rockville: Food and Drug Administration; 2006. [cited 2012 Jul 27]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM078933.pdf. [Google Scholar]
- 21.Neef AB, Luedtke NW. Dynamic metabolic labeling of DNA in vivo with arabinosyl nucleosides. Proc Natl Acad Sci USA. 2011;108:20404–9. doi: 10.1073/pnas.1101126108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibson RE, Burns HD, Hamill TG, Eng WS, Francis BE, Ryan C. Non-invasive radiotracer imaging as a tool for drug development. Curr Pharm Des. 2000;6:973–89. doi: 10.2174/1381612003399987. [DOI] [PubMed] [Google Scholar]
- 23.Dupertuis YM, Vazquez M, Mach JP, De Tribolet N, Pichard C, Slosman DO, Buchegger F. Fluorodeoxyuridine improves imaging of human glioblastoma xenografts with radiolabeled iododeoxyuridine. Cancer Res. 2001;61:7971–7. [PubMed] [Google Scholar]