

Abstract
Laboratory red blood cell (RBC) measurements are clinically important, heritable and differ among ethnic groups. To identify genetic variants that contribute to RBC phenotypes in African Americans (AAs), we conducted a genome-wide association study in up to ∼16 500 AAs. The alpha-globin locus on chromosome 16pter [lead SNP rs13335629 in ITFG3 gene; P < 1E−13 for hemoglobin (Hgb), RBC count, mean corpuscular volume (MCV), MCH and MCHC] and the G6PD locus on Xq28 [lead SNP rs1050828; P < 1E − 13 for Hgb, hematocrit (Hct), MCV, RBC count and red cell distribution width (RDW)] were each associated with multiple RBC traits. At the alpha-globin region, both the common African 3.7 kb deletion and common single nucleotide polymorphisms (SNPs) appear to contribute independently to RBC phenotypes among AAs. In the 2p21 region, we identified a novel variant of PRKCE distinctly associated with Hct in AAs. In a genome-wide admixture mapping scan, local European ancestry at the 6p22 region containing HFE and LRRC16A was associated with higher Hgb. LRRC16A has been previously associated with the platelet count and mean platelet volume in AAs, but not with Hgb. Finally, we extended to AAs the findings of association of erythrocyte traits with several loci previously reported in Europeans and/or Asians, including CD164 and HBS1L-MYB. In summary, this large-scale genome-wide analysis in AAs has extended the importance of several RBC-associated genetic loci to AAs and identified allelic heterogeneity and pleiotropy at several previously known genetic loci associated with blood cell traits in AAs.
INTRODUCTION
Laboratory red blood cell (RBC) measurements are important for the diagnosis and classification of various hematologic disorders. Some disorders of RBCs, such as sickle cell anemia and alpha thalassemia, are single-gene diseases with higher frequency among populations of African descent (1,2). Even among healthy individuals, African Americans (AAs) have lower hemoglobin (Hgb), hematocrit (Hct) and mean corpuscular volume (MCV) compared with other racial/ethnic groups across all ages (3–5).
Heritability studies suggest that RBC traits are under significant genetic influence. Genome-wide association studies (GWASs) of RBC indices have been reported among European and Japanese populations (6–8), but to our knowledge have not yet been reported for AA. In a gene-centric association study from the CARe consortium, the common African glucose-6-phosphate dehydrogenase (G6PD) A-variant on chromosome X and another variant of the α-globin (HBA2-HBA1) locus were associated with multiple RBC traits in AAs (9).
The genetic loci reported to date explain only a small fraction of heritability in RBC traits, highlighting the need for larger studies that include ethnic minorities and complementary analytic approaches (10). Thus, we performed a GWA meta-analysis of RBC traits among AA participants from cohorts of the Continental Origins and Genetic Epidemiology Network (COGENT). As AAs are an admixed population, the resulting genomic architecture can be leveraged to identify regions where either African or European ancestral alleles are associated with traits such as Hgb which differ significantly between European and African populations. Therefore, we performed admixture mapping for the association between available RBC traits (Hgb, Hct, MCHC) and local ancestry.
RESULTS
Descriptive analysis
Since not all RBC traits were available in every COGENT cohort, the numbers of individuals available for meta-analysis varied by each RBC trait (Supplementary Material, Table S1). Only Hgb (n = 16 485) and Hct (n = 16 496) were available in all cohorts. MCHC (n = 12 152), MCV (n = 6438), RBC count (4818), MCH (n = 4066) and RDW (n = 3811) were available in subsets of participating cohorts. There were varying degrees of pairwise correlation between RBC traits (Supplementary Material, Table S2). Pearson's correlation coefficients were highest (>0.95) between Hgb and Hct, and between MCV and MCH and were lowest between the RDW and RBC count (0.03).
GWAS of RBC traits in COGENT AAs
The GWA results for each RBC trait are summarized by Manhattan (Fig. 1) and quantile–quantile (Supplementary Material, Fig. S1) plots. The meta-analysis inflation factors were all near unity (0.998–1.005), suggesting that confounders and other technical artifacts were well-controlled. In total, seven independent genomic loci met the experiment-wide significance threshold (P < 1 × 10−8) for one or more RBC traits (Table 1 and Supplementary Material, Table S3). Three loci (1p31.1, 13q31.2, 16p13.3centromeric) have not been previously associated with RBC traits, whereas four loci (2p21, 6q21, 16p31.3telomeric, and Xq28) have been associated with at least one such trait in populations of European, Japanese or African descent.
Figure 1.

Manhattan plots of GWAS analysis for RBC traits (A) Hct; (B) Hgb; (C) MCHC; (D) MCH; (E) MCV; (F) RBC count and (G) RDW. The dashed horizontal red line indicates P = 1 × 10−8. The dashed horizontal blue line indicates P = 5 × 10−3.
Table 1.
Results of genome-wide significant SNPs for RBC traits in COGENT AA
| Trait | Chromo-some | Number of SNPs with P < 5 × 10−8 | Top SNP in the region | Position Hg18 | Candidate genes | Minor/major allele | MAF | Effect size (SE) | P-value |
|---|---|---|---|---|---|---|---|---|---|
| HCT | Xq28 | 6 | rs762516 | 153 417 857 | G6PD, TKTL1, MECP2, MPP1 | T/C | 0.148 | −0.452 (0.055) | 2.17E − 16 |
| HCT | 2p21 | 3 | rs13008603 | 46 209 352 | PRKCE | A/C | 0.163 | −0.277 (0.047) | 4.09E − 09 |
| HGB | 16p13.3 | 11 | rs13335629 | 250 381 | ITFG3, LUCL7, NPRL3, POLR3K, RPL2B, MPG | A/G | 0.120 | −0.190 (0.019) | 2.63E − 23 |
| HGB | Xq28 | 9 | rs762516 | 153 417 857 | G6PD, TKTL1, MECP2, MPP1 | T/C | 0.146 | −0.1614 (0.0186) | 3.73E − 18 |
| MCHC | 16p13.3 | 38 | rs13335629 | 250 381 | ITFG3, LUCL7, NPRL3, POLR3K, RPL2B, MPG, NME4, DECR | A/G | 0.117 | −0.3298 (0.0227) | 8.66E − 48 |
| MCH | 16p13.3 | 28 | rs13339636 | 238 589 | ITFG3, LUCL7, NPRL3, POLR3K, RPL2B, MPG, NME4, DECR | G/A | 0.132 | −0.6847 (0.0559) | 1.87E − 34 |
| MCH | 13q31.2 | 4 | rs9559892a | 88 166 665 | – | A/C | 0.253 | −0.2589 (0.0444) | 5.46E − 09 |
| MCH | 16p13.3 | 1 | rs7192051a | 4 482 118 | HMOX2 | G/T | 0.360 | −0.2396 (0.0411) | 5.70E − 09 |
| MCH | 6q21 | 10 | rs9386791 | 109 715 190 | CD164 | C/T | 0.416 | −0.2294 (0.0401) | 1.20E − 08 |
| MCV | 16p13.3 | 21 | rs13335629 | 250 381 | ITFG3, LUCL7, NPRL3, POLR3K, RPL2B, MPG, NME4, DECR, RHOT2, LMF1, WDR90 | A/G | 0.134 | −0.648 (0.0669) | 3.61E − 22 |
| MCV | Xq28 | 6 | rs762516 | 153 417 857 | G6PD, FAM3A, F8, MPP1 | T/C | 0.137 | 1.5768 (0.2083) | 3.76E − 14 |
| MCV | 16p13.3 | 1 | rs7192051a | 4 482 118 | HMOX2 | G/T | 0.363 | −0.259 (0.0475) | 4.83E − 08 |
| RBC | 16p13.3 | 12 | rs13335629 | 250 381 | ITFG3, LUCL7 | A/G | 0.120 | 0.1699 (0.0169) | 7.48E − 24 |
| RBC | Xq28 | 9 | rs1050828 | 153 417 411 | G6PD, F8, MPP1, MECP2, CTAG2 | T/C | 0.108 | −0.1424 (0.0159) | 4.00E − 19 |
| RDW | Xq28 | 1 | rs1050828 | 153 417 411 | G6PD | T/C | 0.116 | −0.0326 (0.0048) | 1.70E − 11 |
| RDW | 1p31.1 | 1 | rs10493739a | 83 698 745 | – | T/C | 0.334 | 0.0128 (0.0023) | 3.02E − 08 |
aNovel loci.
Previously reported RBC loci
The two top Xq28 single nucleotide polymorphisms (SNPs) (rs762516, rs1050828) for Hgb, Hct, MCV and RBC count are located in the G6PD gene. rs1050828 encodes the G6PD amino acid substitution Val68Met that results in the G6PD A− allele known to cause G6PD deficiency (MIM #305900). The G6PD A-variant has been previously associated with lower Hct, Hgb and RBC count, and with higher MCV in AAs (9). Here, we additionally report that the G6PD A− allele is associated with the lower RDW. Given the extent of the association signal at Xq28, we repeated the Hgb and Hct association analyses in women from Women Health Initiative (WHI), the largest AA cohort (n = 8304) for Hgb and conditioning the lead SNPs, rs1050828. After adjusting for rs1050828, the strength of association with Hgb for the remaining SNPs on Xq28 was greatly attenuated and no longer significant (data not shown).
The index SNP on 16p13, which encompasses the α-globin (HBA2-HBA1) locus, was rs13335629 within an intron of ITFG3. rs13335629 met the genome-wide significance threshold for association with lower Hgb, MCH, MCHC and MCV and also with a higher RBC count. The rs13335629 variant was also nominally associated with lower Hct (β = −0.215 ± 0.056; P = 1.33E−04) and higher RDW (β = 0.0053 ± 0.0021; P = 0.01). Lo et al. previously reported a common rs1211375 variant within the 16p13 region associated with lower Hgb, MCH and MCV in AAs, and that these associations were not present in Caucasians (9). Our index SNP rs13335629 is in moderate linkage disequilibrium (LD) with rs1211375 (r2 = 0.33 in HapMap YRI).
Three intronic variants of the protein kinase C (PKC)-epsilon gene PRKCE on 2p21 were associated with lower Hct. The index SNP rs13008603 was also nominally associated with a lower RBC count (β = −0.044 ± 0.013; P = 4.69E−04), but not with other RBC traits (P > 0.05 for Hgb, MCV, MCH, MCHC, RDW). The three Hct-associated PRKCE variants are in strong LD (pairwise r2 > 0.7). Another intronic variant of PRKCE (rs10495928) was previously associated with Hgb and Hct in Europeans (8) and with the RBC count in Japanese (6), but showed no evidence of association in AAs (P = 0.50 and 0.71 for Hct and Hgb, respectively). In European and African HapMap populations, there is no evidence of LD between rs10495928 and any of the three Hct-associated variants observed in COGENT AA. These results strongly suggest ethnicity-specific allelic heterogeneity for RBC traits at the PRKCE locus.
At 6q21, a haplotype comprised of 10 SNPs (lead SNP = rs9386791) was associated with a lower MCH, and nominally with a lower MCV (P = 1.09E−05), Hgb (P = 0.007), Hct (P = 0.03), MCHC (P = 0.02), RBC count (P = 0.01). These variants are located ∼50 kb upstream of CD164, which encodes a mucin-like molecule expressed by human CD34(+) hematopoietic progenitor cells that regulate erythropoiesis. Other variants of the CD164 5′ flanking region have been associated with RBC, MCH and MCV in Japanese (rs11966072) (6) and with MCV in Europeans (rs9374080) (8). In HapMap CEU, rs9374080 is in LD with our AA index SNP rs9386791 (r2 = 0.87).
Newly discovered RBC loci
Of the three novel loci associated with RBC traits, rs10493739 at 1p31.1 (associated with RDW) and rs9559892 at 13q31.2 (associated with MCH) are both located in regions devoid of known genes. TTLL7 is the closest gene to rs10493739 (400 kb away) and encodes a tubulin polyglutamylase, which modifies beta-tubulin (11). There are no known genes within 500 kb on either side of rs9559892. The lead SNP at the third locus, rs7192051 is located within the second intron of the heme oxygenase-2 gene (HMOX2) and was associated with lower MCH and MCV. Heme oxygenase 2, the protein product of HMOX2, degrades heme and is important in erythropoiesis (12). Although HMOX2 is located ∼4 Mb centromic to the alpha-globin locus, it is not in LD with the previously identified 16p13 association signals (maximum r2 = 0.004 with rs13335629).
We attempted to validate two of our three novel RBC loci discovered in COGENT in two independent population-based samples: ∼7700 AA youths ages 8–21 years from CHOP and 2010 AA adults from the Mount Sinai eMERGE study. There was no evidence of replication of rs9559892 with MCH, nor of rs7192051 with MCV or MCH (Supplementary Material, Table S4) in the validation sample. It was not possible to pursue replication of rs10493739 in CHOP and eMERGE because this SNP was not genotyped and it could not be imputed in the available replication samples. In over 20 000 Europeans from the CHARGE consortium and 14 000 Japanese from RIKEN, there was no evidence of association of rs9559892 with MCH. Similarly, there was no evidence of association of rs7192051 with MCV or MCH in CHARGE Europeans (Supplementary Material, Table S4).
Admixture mapping analysis of Hgb, Hct and MCHC traits in WHI AA
As a complementary approach to identifying variants associated with RBC traits in AAs that occur at disparate frequencies in ancestral African versus European populations, we performed admixture mapping for Hgb, Hct and MCHC in WHI, the largest cohort comprising COGENT. For MCHC, there was one genome-wide significant association signal at the p-term of 16 containing the alpha-globin locus (Supplementary Material, Fig. S2). Local African ancestry in this region was associated with lower MCHC. The admixture association peak is at rs7203694 (P = 2.78e−06) located within RAB40C, and the genome-wide significant region spans 0–0.78 mb (build 36). There were no genome-wide significant admixture associations for Hct (data not shown). For Hgb, a 2 mb region on chromosome 6p22.2–6p22.1 (25.2–27.1 mb, build 36) reached genome-wide significance, with increased European ancestry associated with higher Hgb levels (Fig. 2A). The Hgb admixture signal appears to be comprised of two peaks (Fig. 2B). Underlying the centromeric peak is HFE, the hemochromatosis protein-coding gene, which regulates iron absorption by modulating the interaction of the transferrin receptor with transferrin. Two known HFE mutations C282Y (rs1800562) and H63D (rs1799945) cause hereditary hemochromatosis, an autosomal recessive iron storage disorder (13). Among individuals of European descent, the frequencies of C282Y and H63D are 3.23 and 16.6 %, respectively. Both the mutations are essentially absent in the HapMap YRI populations, and therefore the frequency in AAs is low and is the result of European admixture. C282Y was directly typed in WHI SHARe and other COGENT cohorts (total N = 15 584); the genotype association test yielded an Hgb association P = 0.0003 in WHI alone and 4.3 × 10−6 in COGENT overall (minor allele frequency = 0.015; β = 0.239 ± 0.052). H63D was not directly typed; however, it is tagged by rs129128 (r2 = 1.0 in CEU), which was associated with Hgb levels in WHI (P = 0.008) but not when all COGENT cohorts were analyzed together (P = 0.07).
Figure 2.
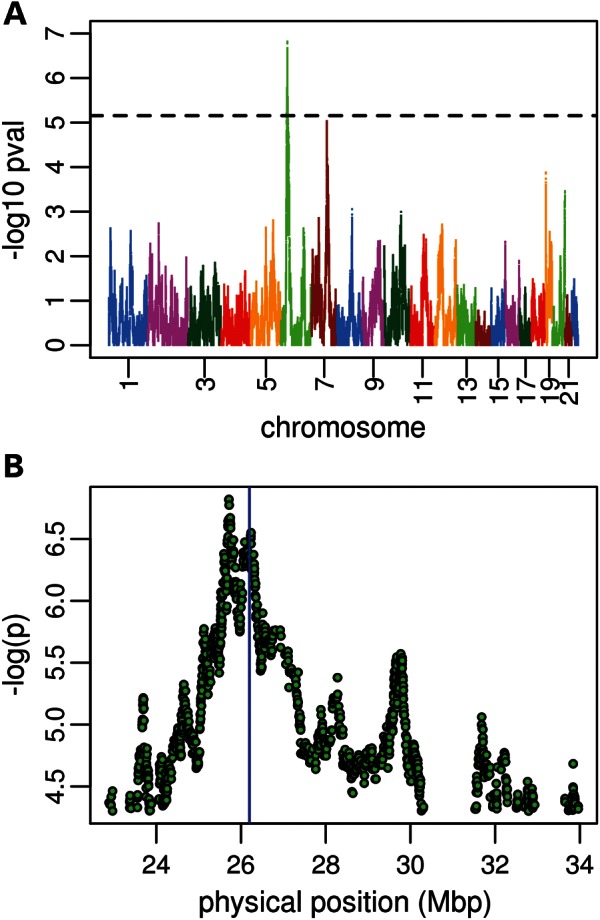
Admixture scan of Hgb concentration. (A) shows a genome-wide plot of −log(P-values) for local-ancestry association with Hgb. The dashed horizontal line indicates the experiment-wide admixture scan significance threshold of P < 7 × 10−6. (B) indicates a zoom-in of the genome-wide significant region on chromosome 6, where there appears to be a broad, bimodal admixture peak. The region corresponding to the HFE gene is shown in blue.
After adjusting for genotypes at C282Y and the H63D proxy rs129128 in WHI, the admixture P value for chromosome 6p22 was attenuated, but remained significant (from 3.28 × 10−7 to 1.12 × 10−4), suggesting the existence of additional variants in this region that contribute to inter-population differences in Hgb levels. Located within the telomeric peak of the admixture signal (Fig. 2B) are a number of additional variants that have Fst > 0.3, including several near LRRC16A, which has been associated with both serum transferrin levels in whites and the platelet count in AAs from COGENT (14). The most strongly associated LRRC16A variant rs9356970 is located ∼25 kb upstream of the 5′ flanking region (MAF = 0.09; β = 0.118 ± 0.028; P = 2.7 × 10−5). According to the HapMap, the minor allele is present in 30% of European chromosomes, but only 2.5% of YRI chromosomes. In a regression model simultaneously adjusting for LRRC16A rs9356970 in addition to HFE C282Y and rs129128, the association signal for local African ancestry at 6p22 was further attenuated, but remained nominally associated with lower Hgb (β = −0.058 ± 0.029; P = 0.045). Together, these results suggest that several European-derived alleles in the 6p22 region, including those of HFE and LRRC16A, may contribute to higher Hgb levels observed in populations of European descent compared with AAs.
CNV analysis and assessment of allelic heterogeneity at 16p13 alpha-globin region
Given the genetic complexity of the alpha-globin locus on chromosome 16p13, including the presence of a common 3.7 kb alpha-thalassemia deletion in AAs (15), and the extent and magnitude of the observed GWAS signal for RBC traits at 16p13, we assessed structural variation at the 16p13 alpha-globin locus using data from 1000 genomes. First, we confirmed the presence of a common deletion (−α3.7) among African Americans (AAs) and West Africans that removes one alpha-globin gene copy (HBA2) (Supplementary Material, Fig. S3). Using pooled sequence data from 16 samples (9 YRI, 5 LWK, 1 ASW, 1 CLM) that appear to be homozygous for −α3.7 deletion, we further localized the breakpoints, which appear to be bounded by ∼300 bp of nearly identical sequence located within the 5′ flanking regions of HBA1 and HBA2 (Supplementary Material, Fig. S4). Second, we identified a rare deletion spanning HBM through HBQ1, in three Han Chinese individuals, and several other possible (but uncertain) rare copy number variations (CNVs) including one duplication (Supplementary Material, Fig. S5) and a very rare deletion that deletes a known regulatory element MCS-R1 (16) (Supplementary Material, Fig. S6).
Among all typed and imputed SNPs in the WHI dataset, the strongest correlation with alpha37 in 63 YRI samples from 1000 Genomes was observed in the region of ITFG3. This includes rs13335629 (r-squared = 0.6), which is also the top Hgb- and MCHC-associated SNP in COGENT AA. We repeated the association analyses in WHI (n = 8304) for Hgb and MCHC conditioning on rs13335629. The top SNP for Hgb in the conditional analysis was POLR3K rs798693 (P = 8.7E−06). For MCHC, rs2541612 in NPRL3 remained genome-wide significantly associated with lower MCHC (P = 1.14E−09). When both ITFG3 rs13335629 and NPRL3 rs2541612 were included as covariates in the conditional analysis, the SNP most strongly associated with lower MCHC was LUC7L rs1211375 (P = 1.50E−06). Taken together, the 1000 Genomes CNV analyses and the results of conditional regression analyses for Hgb and MCHC suggest that while some of the red cell GWAS association signal may be due to the common African alpha37 deletion, there appears to be independent signals coming from other structural variants and/or SNP(s) in the region.
Cross-ethnic transferability of previously reported RBC to COGENT AA
We assessed whether 72 SNPs previously associated with RBC traits in European or Japanese populations are associated with RBC traits in COGENT AA (Supplementary Material, Table S5). Using the conservative Bonferroni multiple comparison corrected significance threshold (P < 0.0001), we validated four associations. In addition to the association of HFE rs1800562 with Hgb, these include ITFG3 rs1122794 (previously associated with MCH in Europeans) with higher MCHC (P = 1.5 × 10−8), MCH (P = 7.2 × 10−6) and MCV (P = 7.1 × 10−5) in AAs; ITFG3 rs7189020 (previously associated with MCV in Europeans) with higher MCH (P = 1.0 × 10−5) and MCV (P = 1.5 × 10−5) in AAs; and HBS1L-MYB rs7775698 (previously associated with HCT, MCH, MCHC, MCV and RBC count in Japanese) with a lower RBC count (P = 3.3 × 10−5) in AAs.
RBC-associated genetic variants and anemia in AA women
To identify genetic variants associated with anemia, defined dichotomously as Hgb < 12 g/dl, we performed a GWA scan in 8304 AA women 50–79 years old from WHI. Two loci, Xq28 and 16p13, met the threshold of genome-wide significance. The G6PD rs1050828 A-variant was associated with a 1.49-fold (95% CI: 1.33–1.67) increased risk of anemia (P = 3.3 × 10−12). The index SNP at 16p13 (rs1088638) is located ∼20 kb 3′ to POLR3K, and was associated with a 1.42-fold (95% CI: 1.26–1.60) increased risk of anemia (P = 1.2 × 10−8). We also constructed a composite RBC genetic risk score (GRS) by summing genotyped or imputed allele dosage at the 15 SNPs associated with at least one RBC trait in AA through the GWA scan, admixture mapping scan, conditional analyses or cross-ethnic transferability analyses described above. The GRS ranged from 5 to 19, with a median of 12. When modeled as a quantitative trait, the GRS was strongly associated with anemia (P = 3.5 × 10−18), explaining 1.4% of the anemia phenotypic variance, or 2.2% of the variance in Hgb concentration. When WHI participants were grouped into four GRS categories, those in the highest GRS category had a 1.95-fold increased risk of anemia (95% CI: 1.56–2.42) compared with those in the lowest GRS category (P = 3.6 × 10−9).
DISCUSSION
In this first reported GWAS meta-analysis of RBC traits in AAs, we report genome-wide associations for four loci (G6PD on Xq28, alpha-globin locus on 16pter, PRKCE on 2p21 and CD164 on 6q21). We also validated the association in AAs of variants in genes such as HFE and HBS1L-MYB, which have previously been associated with RBC traits in other ethnicities. At the alpha-globin locus, there appears to be allelic heterogeneity (particularly for MCHC), with both copy number variants and SNPs having apparent independent effects. At PRKCE, the variants associated with lower Hct in our AA sample appear to be distinct from another set of PRKCE variants that have been associated with Hgb, Hct and RBC count in Europeans and Japanese.
Hemizygous males and in some instances female carriers of the X-linked G6PD A- allele are predisposed to acute episodes of drug- or infection-induced hemolytic anemia. Under basal conditions, however, the G6PD A-allele is not generally thought to be associated with RBC abnormalities, and hemizygous G6PD A-individuals have been reported to have normal baseline red cell survival in the absence of oxidant stress (17). Nonetheless, the association of low RDW with G6PD deficiency may be due to low grade hemolysis resulting in an increase in the MCV with rightward shift of the overall distribution of RBC volume without change in the shape of the distribution (18). The G6PD A-variant is in LD with other nearby genetic variants that plausibly could influence Hgb or RBC morphology. TKTL1 encodes a transketolase enzyme that links the pentose phosphate pathway with anaerobic glycolysis, which constitutes the two major metabolic pathways for glucose utilization in human erythrocytes. MPP1 encodes the red cell membrane protein p55, a scaffolding protein that anchors the actin cytoskeleton to the plasma membrane by forming a ternary complex with protein 4.1R and glycophorin C (19).
Aside from genes involved in Hgb synthesis or metabolism, other genetic loci such as CD164 and PRKCE may be associated with RBC traits through effects on erythropoiesis. CD164 (endolyn) is an adhesive receptor present on early hematopoietic progenitors and maturing erythroid cells that regulates the adhesion of CD34+ cells to bone marrow stroma and affects migration and proliferation of hematopoietic stem cells and progenitor cells (20,21). The upstream region harboring the RBC trait-associated variants contains an erythroleukemia cell line (K562)-specific cluster of histone modifications and ENCODE transcription factor ChIP-seq binding sites including those for GATA-2 and c-Jun. PRKCE encodes an isoform of PKC, PKC epsilon, which is expressed in hematopoietic progenitor cells in a lineage- and stage-specific manner and appears to influence erythroid and megakaryocytic progenitor proliferation and differentiation by modulating the response of hematopoietic precursors to a tumor necrosis factor-related apoptosis-inducing ligand (22–24).
Though the finding was not validated in independent AA samples, one of our novel genome-wide significant associations in our discovery cohorts was the association of MCH and MCV with HMOX2, which encodes heme oxygenase-2, a constitutively expressed enzyme with a major role in heme catabolism. Heme induces expression of globin genes in erythrocyte progenitor cells and thus plays an important role in erythropoiesis (12,25,26). The lead SNP in this region, rs7192051, is within 5 kb of predicted HMOX2 regulatory elements such as transcription factor binding sites, DNase sites and histone modification sites (27). Therefore, further study of this variant in larger, independent samples of AA may be warranted.
Our findings have potential clinical implications. Although previous studies have explored the role of common genetic variation in the regulation of these RBC phenotypes in populations of European and Asian descent (6,7,10), no systematic genetic association studies of these traits have been reported in African-ancestry populations. This is particularly important, as there are marked differences in these RBC indices among ethnic groups, and anemia is more prevalent in populations of African descent (28). While it appears that some of the phenotypic variations for RBC and other hematologic traits are controlled by genetic variation shared across ethnic groups (29), other RBC loci are relatively unique to Africans. Rare variants, which are not well captured by GWASs, and undetected common variants of more modest effect may account for additional genetic variance. Discovery and validation of these and additional genetic variants associated with RBC traits in other ethnic populations are likely to uncover new mechanisms and pathways that affect hematopoiesis and RBC turnover, offering insights that may inform further research into red cell biology. Indeed, recent reports have shown that genetic loci uncovered through an unbiased genome-wide study in human populations, together with follow-up functional studies incorporating gene expression, bioinformatic analyses and insights from mouse models and gene knockdown experiments, can greatly contribute to our understanding of the biological mechanisms underlying RBC production (30,31).
MATERIALS AND METHODS
Primary subjects and data collection
We performed GWA analysis of RBC traits in over 16 000 AAs from seven population-based cohorts that comprise the Continental Origins and Genetic Epidemiology Network (COGENT). The characteristics of each cohort were described in previous publications (14,29). Fasting blood samples were drawn and analyzed for RBC traits at designated clinical laboratories using an automated electronic cell counter. These counters directly measure Hgb concentration (in grams per deciliter), RBC count (in millions per microliter) and MCV, the average size of the RBC in femtoliters. Electronic cell counters calculate MCH, MCHC, Hct and RDW. Hct is the percentage of blood by volume that is occupied by RBC and is calculated by multiplying the RBC count in millions/microliter by the MCV in femtoliters. MCH is the average amount of Hgb inside an RBC expressed in picograms and is calculated by dividing the Hgb concentration by the RBC count in millions per microliter, then multiplying by 10. The MCHC is the average concentration of Hgb in RBCs and is calculated by dividing Hgb in grams per deciliter by Hct. The RDW is a measure of the variance in RBC size and is calculated by dividing the standard deviation of RBC volume by the MCV and multiplying by 100.
All participants self-reported their race/ethnicity. Additional clinical information was collected by self-report and clinical examination. Participants provided written informed consent as approved by local Human Subjects Committees. Study participants who were pregnant or had a diagnosis of cancer or AIDS at the time of blood count were excluded.
Replication subjects and data collection
For validation of novel, genome-wide significant associations identified in the COGENT discovery sample, we performed association analyses in two independent population-based samples: ∼7700 AA youths ages 8–21 years from Children's Hospital of Philadelphia (CHOP) and 2010 AA adults from the Mount Sinai electronic Medical Records and Genomics (eMERGE) study. We also attempted to replicate novel loci in two other ethnic populations: 14 088 Japanese from RIKEN and up to 30 000 European Americans from CHARGE. Details of each validation cohort are provided under Supplementary Material.
Genotyping and quality-control
Genomic DNA was extracted from peripheral blood leukocytes and genotyping was performed on the Affymetrix 6.0 array or Illumina Omni or 1 M platforms within each cohort using methods described previously (14,29). DNA samples with a genome-wide genotyping success rate of <90% or sex discordance were excluded, as were genetic ancestry outliers (identified by cluster analysis using principal components analysis or multi-dimensional scaling). SNPs with a genotyping success rate of <95% or MAF <1%, monomorphic SNPs and SNPs that map to several genomic locations were removed from the analyses. Participants and SNPs passing basic quality control thresholds were imputed to >2.2 million autosomal SNPs based on HapMap2 haplotype data using a 1:1 mixture of Europeans (CEU) and Africans (YRI) as the reference panel. Details of the genotype imputation procedure have been described previously (14,29). Prior to discovery meta-analyses, SNPs were excluded if imputation quality metrics (equivalent to the squared correlation between proximal imputed and genotyped SNPs) were <0.30.
Data analyses
For all cohorts, GWA analysis was performed on the raw, untransformed RBC trait using linear regression adjusted for covariates, implemented in either PLINK v1.07 or MACH2QTL v1.08. In GeneSTAR, the family structure was accounted for in the association tests using linear mixed-effects models implemented in R (32). For the 22 autosomes, analysis was performed using genotyped and imputed SNPs. For the X chromosome, only genotyped SNPs were analyzed due to the technical limitations of imputing X-linked SNPs. All analyses were performed under an additive genetic model using allelic dosage (genotyped or imputed) at each SNP, adjusted for age, age-squared, sex and clinic site (if applicable), 4–10 principal components.
For each phenotype, meta-analysis was conducted using inverse-variance weighted fixed-effects models to combine β coefficients and standard errors from study-level regression results for each SNP, to derive pooled estimates. Study-level results were corrected for genomic inflation factors (λ) by multiplying the standard errors (SEs) of the regression coefficients by the square-root of the study-specific λ. Meta-analyses were implemented in the METAL software. Between-study heterogeneity of results was assessed by using Cochran’s Q statistic and the I2 inconsistency metric. A threshold of α = 1 × 10−8 was used to declare genome-wide statistical significance. This statistical threshold accounts for the greater nucleotide diversity and lower LD in African descent populations combined with testing of multiple, correlated RBC traits (31,33). We carried out replication testing of ‘suggestive’ SNPs selected on the basis of a more liberal significance threshold in our primary AA discovery GWAS (P < 5 × 10−8).
To assess the potential existence of multiple, independent variants influencing a trait at the same locus (allelic heterogeneity), regression analyses were repeated in the largest sample (WHI, n = 8095), conditional on the most strongly associated (index) SNP in that region.
We also assessed the transferability to AAs of SNPs previously associated with RBC traits in populations of European or Japanese ancestry by assessing association with RBC traits in the COGENT discovery meta-analyses. For validation, we considered consistency of direction of effect, and assessed statistical significance using a simple Bonferroni adjustment for the total number of SNPs assessed, using a two-sided hypothesis test.
Local ancestry estimation and admixture mapping in WHI
For each AA individual in the WHI sample, locus-specific ancestry was estimated using an extension of the model described by Tang et al. (34). We used phased haplotype data from HapMap3 CEU and YRI individuals as reference panels. An admixture mapping analysis was performed in WHI to test for association between Hgb levels and ancestry at each genomic location (local ancestry), while adjusting for the first 10 principal components, regions of recruitment, clinical trial, age and age-squared. The critical value for genome-wide significance level of admixture mapping is substantially lower than the genotype test due to the extensive correlation in local ancestry between adjacent markers that result from the recent admixture in AAs. We therefore adopted an empirically determined genome-wide significance threshold of P < 7.1 × 10−6, which corresponds to a Bonferroni correction of ∼7000 independent tests (35).
Copy number variation (CNV) analysis using 1000 genomes data
We used the 1000 Genomes sequencing data to investigate CNVs at the chromosome 16 p31 alpha-globin locus, studying 946 African-ancestry samples at roughly 4× sequencing coverage. As a result of noise in depth-based genotyping at this locus (due to low-pass sequencing, high %GC and potential overlapping variants), some of our analyses were confined to the 76 YRI samples, which have higher sequence coverage in 1000 Genomes data and more complete genotyping (call rate 84 % at 95 % CI).
SUPPLEMENTARY MATERIAL
FUNDING
Additional support for this work was provided by NIH (R01 HL71862-06 and ARRA N000949304 to A.P.R.). Some of the results of this paper were obtained by using the program package S.A.G.E., which is supported by a US Public Health Service Resource Grant (RR03655) from the National Center for Research Resources. Additional support came from the National Cancer Institute (grant R25CA094880 to U.M.S.).
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to acknowledge the support of the National Heart, Lung and Blood Institute and the contributions of the involved research institutions, study investigators, field staff and study participants of Atherosclerosis Risk in Communities (ARIC), Coronary Artery Risk in Young Adults (CARDIA), Jackson Heart Study (JHS) and Broad Institute in creating the Candidate-gene Association Resource for biomedical research (CARe; http://public.nhlbi.nih.gov/GeneticsGenomics/home/care.aspx).
The authors also wish to thank the investigators, staff and participants of GeneSTAR, Health ABC, Healthy Aging in Neighborhoods of Diversity across the Life Span Study (HANDLS) and Women Health Initiative (WHI) for their important contributions. A listing of WHI investigators can be found at http://www.whiscience.org/publications/WHI_investigators_shortlist.pdf.
We thank all the children who donated blood samples for genetic research purpose. The CHOP study was funded by the Institute Development Funds to the Center for Applied Genomics at the Children's Hospital of Philadelphia and an Adele S. and Daniel S. Kubert Estate gift to the Center for Applied Genomics.
The Mount Sinai IPM Biobank Program is supported by The Andrea and Charles Bronfman Philanthropies.
The authors acknowledge the essential role of the Cohorts for Heart and Aging Research in Genome Epidemiology (CHARGE) Consortium in development and support of this manuscript. CHARGE members include the Rotterdam Study (RS), Framingham Heart Study (FHS), Cardiovascular Health Study (CHS), the NHLBI's Atherosclerosis Risk in Communities (ARIC) Study and the NIA's Iceland Age, Gene/Environment Susceptibility (AGES) Study. The collaboration of studies such as the Health Aging and Body Composition Study (Health ABC), the Baltimore Longitudinal Study of Aging (BLSA), the Invecchiare in Chianti Study (InChianti), and the Heart and Vascular Health Study (HVH) also played a vital role.
The following parent studies contributed study data, ancillary study data and DNA samples through the Broad Institute (N01-HC-65226) to create this genotype/phenotype data base for wide dissemination to the biomedical research community:
Atherosclerosis Risk in Communities (ARIC): University of North Carolina at Chapel Hill (N01-HC-55015), Baylor Medical College (N01-HC-55016), University of Mississippi Medical Center (N01-HC-55021), University of Minnesota (N01-HC-55019), Johns Hopkins University (N01-HC-55020), University of Texas, Houston (N01-HC-55017), University of North Carolina (N01-HC-55018). Other NIH support contributing to the GWAS in ARIC are: R01HL087641, R01HL59367, R01HL86694, U01HG004402 and HHSN268200625226C.
Coronary Artery Risk in Young Adults (CARDIA): University of Alabama at Birmingham (N01-HC-48047), University of Minnesota (N01-HC-48048), Northwestern University (N01-HC-48049), Kaiser Foundation Research Institute (N01-HC-48050), University of Alabama at Birmingham (N01-HC-95095), Tufts-New England Medical Center (N01-HC-45204), Wake Forest University (N01-HC-45205), Harbor-UCLA Research and Education Institute (N01-HC-05187), University of California, Irvine (N01-HC-45134, N01-HC-95100).
Jackson Heart Study (JHS): Jackson State University (N01-HC-95170), University of Mississippi (N01-HC-95171), Tougaloo College (N01-HC-95172).
Healthy Aging in Neighborhoods of Diversity across the Life Span Study (HANDLS): this research was supported by the Intramural Research Program of the NIH, National Institute on Aging and the National Center on Minority Health and Health Disparities (intramural project Z01-AG000513 and human subjects protocol 2009-149). Data analyses for the HANDLS study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD, USA (http://biowulf.nih.gov).
Health ABC: this research was supported by NIA contracts N01AG62101, N01AG62103 and N01AG62106. The GWAS was funded by NIA grant 1R01AG032098-01A1 to Wake Forest University Health Sciences and genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C. This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging.
GeneSTAR: this research was supported by the National Heart, Lung and Blood Institute (NHLBI) through the PROGENI (U01 HL72518) and STAMPEED (R01 HL087698-01) consortia. Additional support was provided by grants from the NIH/National Institute of Nursing Research (R01 NR08153), and the NIH/National Center for Research Resources (M01-RR000052) to the Johns Hopkins General Clinical Research Center.
WHI: the WHI program is funded by the National Heart, Lung and Blood Institute, National Institutes of Health, US Department of Health and Human Services through contracts N01WH22110, 24152, 32100–2, 32105–6, 32108–9, 32111–13, 32115, 32118–32119, 32122, 42107–26, 42129–32 and 44221.
AGES: the Age, Gene/Environment Susceptibility Reykjavik Study is funded by NIH contract N01-AG-12100, the NIA Intramural Research Program, Hjartavernd (the Icelandic Heart Association) and the Althingi (the Icelandic Parliament).
Framingham: the National Heart, Lung and Blood Institute's Framingham Heart Study is a joint project of the National Institutes of Health and Boston University School of Medicine and was supported bythe National Heart, Lung, and Blood Institute's Framingham Heart Study (contract No. N01-HC-25195) and its contract with Affymetrix, Inc. for genotyping services (contract No. N02-HL-6-4278). Analyses reflect the efforts and resource development from the Framingham Heart Study investigators participating in the SNP Health Association Resource (SHARe) project. A portion of this research was conducted using the Linux Cluster for Genetic Analysis (LinGA-II) funded by the Robert Dawson Evans Endowment of the Department of Medicine at Boston University School of Medicine and Boston Medical Center.
InChianti: the InChianti Study was supported as a “targeted project” (ICS 110.1RS97.71) by the Italian Ministry of Health, by the US National Institute on Aging (Contracts N01-AG-916413, N01-AG-821336, 263 MD 9164 13 and 263 MD 821336) and in part by the Intramural Research Program, National Institute on Aging, National Institutes of Health, USA.
Rotterdam: Rotterdam Study GWAS database of the Rotterdam Study was funded through the Netherlands Organization of Scientific Research NWO (no. 175.010.2005.011, 911.03.012) and the Research Institute for Diseases in the Elderly (RIDE). This study was supported by the Netherlands Genomics Initiative (NGI)/NWO project number 050 060 810 (Netherlands Consortium for Healthy Ageing). We thank Dr Michael Moorhouse, Pascal Arp, Mila Jhamai, Marijn Verkerk and Sander Bervoets for their help in creating the genetic database. We thank the laboratory technicians Jeannette M Vergeer—Drop, Bernadette H M van Ast—Copier, Andy A L J van Oosterhout, Sue Ellen Mauricia, Andrea J M Vermeij—Verdoold, Els Halbmeijer—van der Plas, Debby M S Lont and Hasna Kariouh for their help in phenotype assessment. The Rotterdam Study is supported by the Erasmus Medical Center and Erasmus University, Rotterdam; the Netherlands organization for scientific research (NWO), the Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Netherlands Heart Foundation, the Ministry of Education, Culture and Science, the Ministry of Health, Welfare and Sports, the European Commission (DG XII) and the Municipality of Rotterdam.
RIKEN: we would like to thank all the staff of the Laboratory for Statistical Analysis at RIKEN for their technical assistance. The BioBank Japan Project was supported by Ministry of Education, Culture, Sports, Science and Technology, Japan.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106:3710–3717. doi: 10.1182/blood-2005-05-1857. [DOI] [PubMed] [Google Scholar]
- 2.Melis M.A., Cau M., Congiu R., Sole G., Barella S., Cao A., Westerman M., Cazzola M., Galanello R. A mutation in the TMPRSS6 gene, encoding a transmembrane serine protease that suppresses hepcidin production, in familial iron deficiency anemia refractory to oral iron. Haematologica. 2008;93:1473–1479. doi: 10.3324/haematol.13342. [DOI] [PubMed] [Google Scholar]
- 3.Patel K.V., Longo D.L., Ershler W.B., Yu B., Semba R.D., Ferrucci L., Guralnik J.M. Haemoglobin concentration and the risk of death in older adults: differences by race/ethnicity in the NHANES III follow-up. Br. J. Haematol. 2009;145:514–523. doi: 10.1111/j.1365-2141.2009.07659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schechter G.P. Hemoglobin levels in African-Americans. Blood. 2006;107:2208. doi: 10.1182/blood-2005-07-3025. author reply 2208–2209. [DOI] [PubMed] [Google Scholar]
- 5.Beutler E., Duparc S. Glucose-6-phosphate dehydrogenase deficiency and antimalarial drug development. Am. J. Trop. Med. Hyg. 2007;77:779–789. [PubMed] [Google Scholar]
- 6.Kamatani Y., Matsuda K., Okada Y., Kubo M., Hosono N., Daigo Y., Nakamura Y., Kamatani N. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat. Genet. 2010;42:210–215. doi: 10.1038/ng.531. [DOI] [PubMed] [Google Scholar]
- 7.Soranzo N., Spector T.D., Mangino M., Kühnel B., Rendon A., Teumer A., Willenborg C., Wright B., Chen L., Li M., et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41:1182–1190. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganesh S.K., Zakai N.A., van Rooij F.J., Soranzo N., Smith A.V., Nalls M.A., Chen M.H., Kottgen A., Glazer N.L., Dehghan A., et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat. Genet. 2009;41:1191–1198. doi: 10.1038/ng.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lo K.S., Wilson J.G., Lange L.A., Folsom A.R., Galarneau G., Ganesh S.K., Grant S.F., Keating B.J., McCarroll S.A., Mohler E.R., III, et al. Genetic association analysis highlights new loci that modulate hematological trait variation in Caucasians and African Americans. Hum. Genet. 2011;129:307–317. doi: 10.1007/s00439-010-0925-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kullo I.J., Ding K., Jouni H., Smith C.Y., Chute C.G. A genome-wide association study of red blood cell traits using the electronic medical record. PLoS One. 2010;5:e13011. doi: 10.1371/journal.pone.0013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukai M., Ikegami K., Sugiura Y., Takeshita K., Nakagawa A., Setou M. Recombinant mammalian tubulin polyglutamylase TTLL7 performs both initiation and elongation of polyglutamylation on beta-tubulin through a random sequential pathway. Biochemistry. 2009;48:1084–1093. doi: 10.1021/bi802047y. [DOI] [PubMed] [Google Scholar]
- 12.Alves L.R., Costa E.S., Sorgine M.H., Nascimento-Silva M.C., Teodosio C., Barcena P., Castro-Faria-Neto H.C., Bozza P.T., Orfao A., Oliveira P.L., et al. Heme-oxygenases during erythropoiesis in K562 and human bone marrow cells. PLoS One. 2011;6:e21358. doi: 10.1371/journal.pone.0021358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feder J.N., Gnirke A., Thomas W., Tsuchihashi Z., Ruddy D.A., Basava A., Dormishian F., Domingo R., Jr., Ellis M.C., Fullan A., et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 14.Qayyum R., Snively B.M., Ziv E., Nalls M.A., Liu Y., Tang W., Yanek L.R., Lange L., Evans M.K., Ganesh S., et al. A meta-analysis and genome-wide association study of platelet count and mean platelet volume in african americans. PLoS Genet. 2012;8:e1002491. doi: 10.1371/journal.pgen.1002491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beutler E., West C. Hematologic differences between African-Americans and whites: the roles of iron deficiency and alpha-thalassemia on hemoglobin levels and mean corpuscular volume. Blood. 2005;106:740–745. doi: 10.1182/blood-2005-02-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viprakasit V., Harteveld C.L., Ayyub H., Stanley J.S., Giordano P.C., Wood W.G., Higgs D.R. A novel deletion causing alpha thalassemia clarifies the importance of the major human alpha globin regulatory element. Blood. 2006;107:3811–3812. doi: 10.1182/blood-2005-12-4834. [DOI] [PubMed] [Google Scholar]
- 17.Beutler E. G6PD deficiency. Blood. 1994;84:3613–3636. [PubMed] [Google Scholar]
- 18.Nakhaee A., Dabiri S., Noora M. Survey of the prevalence of glucose-6-phosphate dehydrogenase (G6PD) deficiency in admitted men for premarriage tests in Zahedan-Iran Reference Laboratory. Zahedan J. Res. Med. Sci. 2009;11:0–0. [Google Scholar]
- 19.Chishti A.H. Function of p55 and its nonerythroid homologues. Curr. Opin. Hematol. 1998;5:116–121. doi: 10.1097/00062752-199803000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Watt S.M., Buhring H.J., Rappold I., Chan J.Y., Lee-Prudhoe J., Jones T., Zannettino A.C., Simmons P.J., Doyonnas R., Sheer D., et al. CD164, a novel sialomucin on CD34(+) and erythroid subsets, is located on human chromosome 6q21. Blood. 1998;92:849–866. [PubMed] [Google Scholar]
- 21.Forde S., Tye B.J., Newey S.E., Roubelakis M., Smythe J., McGuckin C.P., Pettengell R., Watt S.M. Endolyn (CD164) modulates the CXCL12-mediated migration of umbilical cord blood CD133+ cells. Blood. 2007;109:1825–1833. doi: 10.1182/blood-2006-05-023028. [DOI] [PubMed] [Google Scholar]
- 22.Klingmuller U., Wu H., Hsiao J.G., Toker A., Duckworth B.C., Cantley L.C., Lodish H.F. Identification of a novel pathway important for proliferation and differentiation of primary erythroid progenitors. Proc. Natl Acad. Sci. USA. 1997;94:3016–3021. doi: 10.1073/pnas.94.7.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gobbi G., Mirandola P., Sponzilli I., Micheloni C., Malinverno C., Cocco L., Vitale M. Timing and expression level of protein kinase C epsilon regulate the megakaryocytic differentiation of human CD34 cells. Stem Cells. 2007;25:2322–2329. doi: 10.1634/stemcells.2006-0839. [DOI] [PubMed] [Google Scholar]
- 24.Mirandola P., Gobbi G., Ponti C., Sponzilli I., Cocco L., Vitale M. PKC epsilon controls protection against TRAIL in erythroid progenitors. Blood. 2006;107:508–513. doi: 10.1182/blood-2005-07-2676. [DOI] [PubMed] [Google Scholar]
- 25.Kollia P., Noguchi C.T., Fibach E., Loukopoulos D., Schechter A.N. Modulation of globin gene expression in cultured erythroid precursors derived from normal individuals: transcriptional and posttranscriptional regulation by hemin. Proc. Assoc. Am. Phys. 1997;109:420–428. [PubMed] [Google Scholar]
- 26.Melefors O., Goossen B., Johansson H.E., Stripecke R., Gray N.K., Hentze M.W. Translational control of 5-aminolevulinate synthase mRNA by iron-responsive elements in erythroid cells. J. Biol. Chem. 1993;268:5974–5978. [PubMed] [Google Scholar]
- 27.Rosenbloom K.R., Dreszer T.R., Long J.C., Malladi V.S., Sloan C.A., Raney B.J., Cline M.S., Karolchik D., Barber G.P., Clawson H., et al. Nucleic Acids Res. 2012;Vol. 40:D912–D917. doi: 10.1093/nar/gkr1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zakai N.A., McClure L.A., Prineas R., Howard G., McClellan W., Holmes C.E., Newsome B.B., Warnock D.G., Audhya P., Cushman M. Correlates of anemia in American blacks and whites: the REGARDS Renal Ancillary Study. Am. J. Epidemiol. 2009;169:355–364. doi: 10.1093/aje/kwn355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reiner A.P., Lettre G., Nalls M.A., Ganesh S.K., Mathias R., Austin M.A., Dean E., Arepalli S., Britton A., Chen Z., et al. Genome-wide association study of white blood cell count in 16,388 African Americans: the continental origins and genetic epidemiology network (COGENT) PLoS Genet. 2011;7:e1002108. doi: 10.1371/journal.pgen.1002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sankaran V.G., Ludwig L.S., Sicinska E., Xu J., Bauer D.E., Eng J.C., Patterson H.C., Metcalf R.A., Natkunam Y., Orkin S.H., et al. Cyclin D3 coordinates the cell cycle during differentiation to regulate erythrocyte size and number. Genes Dev. 2012;26:2075–2087. doi: 10.1101/gad.197020.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Harst P., Zhang W., Mateo Leach I., Rendon A., Verweij N., Sehmi J., Paul D.S., Elling U., Allayee H., Li X., et al. Seventy-five genetic loci influencing the human red blood cell. Nature. 2012;492:369–375. doi: 10.1038/nature11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen M.H., Yang Q. GWAF: an R package for genome-wide association analyses with family data. Bioinformatics. 2010;26:580–581. doi: 10.1093/bioinformatics/btp710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pe'er I., Yelensky R., Altshuler D., Daly M.J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol. 2008;32:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 34.Tang H., Coram M., Wang P., Zhu X., Risch N. Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet. 2006;79:1–12. doi: 10.1086/504302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang H., Siegmund D.O., Johnson N.A., Romieu I., London S.J. Joint testing of genotype and ancestry association in admixed families. Genet. Epidemiol. 2010;34:783–791. doi: 10.1002/gepi.20520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.