

Abstract
Huntington's disease (HD) is a fatal neurodegenerative disease characterized by abnormal motor coordination, cognitive decline and psychiatric disorders. This disease is caused by an expanded CAG trinucleotide repeat in the gene encoding the protein huntingtin. Reduced levels of brain-derived neurotrophic factor (BDNF) in the brain, which results from transcriptional inhibition and axonal transport deficits mediated by mutant huntingtin, have been suggested as critical factors underlying selective neurodegeneration in both HD patients and HD mouse models. BDNF activates its high-affinity receptor TrkB and promotes neuronal survival; restoring BDNF signaling is thus of particular therapeutic interest. In the present study, we evaluated the ability of a small-molecule TrkB agonist 7,8-dihydroxyflavone (7,8-DHF) and its synthetic derivative 4′-dimethylamino-7,8- dihydroxyflavone (4′-DMA-7,8-DHF) to protect neurons in the well-characterized N171-82Q HD mouse model. We found that chronic administration of 7, 8-DHF (5 mg/kg) or 4′-DMA-7,8-DHF (1 mg/kg) significantly improved motor deficits, ameliorated brain atrophy and extended survival in these N171-82Q HD mice. Moreover, 4′-DMA-7,8-DHF preserved DARPP32 levels in the striatum and rescued mutant huntingtin-induced impairment of neurogenesis in the N171-82Q HD mice. These data highlight consideration of TrkB as a therapeutic target in HD and suggest that small-molecule TrkB agonists that penetrate the brain have high potential to be further tested in clinical trials of HD.
INTRODUCTION
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by an expanded CAG repeat in the huntingtin gene. The huntingtin mutation induces selective massive degeneration of the medium spiny neurons (MSNs) in the striatum and, to a lesser extent, degeneration of the pyramidal neurons in the deep layer of the cortex that leads to characteristic motor dysfunction, cognitive decline and psychiatric disturbance (1). The onset of HD usually occurs at middle age, with death occurring 15–20 years later (2). Current treatment for HD is temporary symptomatic relief; thus far, no treatment has delayed the onset and/or slowed the progression of this disease (3–7).
Brain-derived neurotrophic factor (BDNF) has emerged as a candidate for treatment of HD, because reduced BDNF gene transcription and axonal transport of proteins have been implicated as key pathogenesis resulting in selective neurodegeneration and neuronal dysfunction in HD (8–10). Increasing BDNF levels by genetic manipulation or with agents that promote production of endogenous BDNF improves motor function, attenuates brain atrophy and/or extends survival in HD mice (11–16). Mature BDNF acts via two receptors, the TrkB tyrosine kinase receptor and p75 neurotrophin receptor (p75NTR) (17). Nevertheless, the poor pharmacokinetic profile limits the direct therapeutic potential of BDNF. Activation of TrkB receptor triggers cell survival signaling pathways, thus TrkB agonists that penetrate the brain might be valuable therapeutics for HD.
Our previous chemical screening found that 7,8-dihydroxyflavone (7,8-DHF), one of the flavone derivatives, is a TrkB agonist that can cross the blood–brain barrier (18). We further demonstrated that the synthetic derivative of 7,8-DHF, 4′-dimethylamino-7,8- dihydroxyflavone (4′-DMA-7,8-DHF) is a more potent TrkB agonist than 7,8-DHF and displays protective effects in neurological and psychiatric disorders such as Alzheimer's disease, Rett syndrome and immobilization stress (19–25). Our present study was undertaken to evaluate whether 7,8-DHF and 4′-DMA-7,8-DHF are protective in an HD mouse model. We found that activation of TrkB by these compounds improved motor deficits, attenuated brain atrophy and extended survival in HD mice. Moreover, we found that 4′-DMA-7,8-DHF preserved a protein marker of medium spiny neurons in the striatum and ameliorated impaired neurogenesis in HD mice.
Results
Peripheral administration of 7,8-DHF and 4′DMA-7,8-DHF activates TrkB receptors in the striatum of HD mice
In order to determine whether peripheral administration of 7,8-DHF or 4′-DMA-7,8-DHF can efficiently activate TrkB receptor in the striatum that is affected most in HD, 7,8-DHF(5 mg/kg) or 4′-DMA-7,8-DHF (1 mg/kg) was administered to HD mice once daily by oral gavage. Drugs were administered from 6 weeks of age, and mice were euthanized at 20 weeks; striatum samples were collected at 4 h after the last drug administration, and the activated TrkB levels were assessed by using Tyr 706 anti-pTrkB antibodies. We found that both compounds activated TrkB in the striatum of HD mice (Fig. 1A). We also observed that the TrkB downstream signaling pathway MAPK was activated in the samples treated with these compounds, and that the synthetic derivative 4′-DMA-7, 8-DHF was a more potent activator than 7,8-DHF, as we used five times higher doses of 7,8-DHF than were used for 4′-DMA-7,8-DHF. The results confirmed that these small molecule compounds activate TrkB receptor and its downstream MAPK signaling pathway in the striatum of HD mice (Fig. 1B).
Figure 1.

7,8-Dihydroxyflavone and its synthetic derivative 4′-dimethylamino-7,8- dihydroxyflavone activate TrkB receptor (A) and its downstream MAPK signaling pathway (B) in the striatum of HD mice. A cohort of mice were chronically administered 7, 8-DHF (5 mg/kg/day) or 4′-DMA-7,8-DHF (1 mg/kg/day) from 6 weeks of age. Mice were euthanized and striatum samples were collected 4 h after the last drug administration at 20 weeks of age, and western blotting (IB) was conducted with indicated antibodies. Values are expressed as mean ± SE from four mice in each group. *P< 0.05 compared with the vehicle value; one-way ANOVA with Scheffé post-hoc test.
7,8-DHF and 4′DMA-7,8-DHF improve motor function and prolong survival of N171-82Q mice
N171-82Q HD mice exhibit progressive motor deficits similar to those manifested in HD patients. In light of our finding that peripheral administration of 7,8-DHF and 4′-DMA-7,8-DHF activated the TrkB receptor in the striatum of HD mice, we next examined whether peripheral administration of these small molecules could improve the motor function that is impaired in the HD mice. We employed a balance beam to assess motor coordination. HD mice displayed extended transverse time on the balance beam compared with the age-matched wild-type control mice, indicating deficits in motor coordination in these mice. Administration of 7,8-DHF or 4′-DMA-7,8-DHF significantly improved the motor performance of HD mice, indicated by shorter transverse time on the beam (Fig. 2A and B).
Figure 2.

7,8-Dihydroxyflavone and 4′-dimethylamino-7,8- dihydroxyflavone rescue motor deficits and extend survival in N171-82Q HD mice. 7, 8-DHF (5 mg/kg/day) (A) or 4′-DMA-7,8-DHF (1 mg/kg/day) (B) was orally administered to mice daily from 6 weeks of age. Mice were tested on a 5 mm square-shaped beam at 26 weeks of age. n = 8–10 mice. The values are expressed as mean ± SE, *P< 0.01 compared with the values of the WT vehicle group; **P< 0.05 compared with the value of the HD vehicle group. One-way ANOVA with Scheffé post hoc tests. (C) Body weight was monitored weekly from indicated groups. n = 8–10. (D and E) 7,8-DHF (D) and 4′-DMA-7,8-DHF (E) extend life-span of N171-82Q mice. Survival was monitored daily. Mice were considered to be at the end of life when they were unable to back to their normal position after being placed on their backs and initiate movement after being gently prodded for 30 s n = 8–10.
In addition to neurological features, N171-82Q HD mice also display metabolic abnormalities, including body weight loss that is observed in other HD mouse models as well as in HD patients. We then determined whether these TrkB agonists affected body weight. We found that both TrkB agonists at the doses that we tested in the current study had no significant effect on body weight loss in HD mice throughout the treatment periods (Fig. 2C). We also did not see the effects of either TrkB agonist on mutant huntingtin aggregations in the brains of N171-82Q HD mice (data not shown).
Both Trk B agonists also extended the mean lifespan of N171-82Q HD mice by 18∼23% (Fig. 2D and E). It is not precisely known why HD mice die. Notably, patients with HD most often die from complications of pneumonia. It can, therefore, be argued that survival as an outcome measure in HD-like mice, although very useful, does not always directly inform us about human disease. We believe that metabolic abnormalities also contribute to the death of N171-82Q HD mice, which might explain why both TrkB agonists dramatically improved motor function, but the extended the survival is relatively modest.
7,8-DHF and 4′DMA-7,8-DHF attenuate brain atrophy in N171-82Q HD mice
It is widely accepted that changes in brain volumes observed in HD-like mice strongly resemble those in human disease. We next determined whether the improved motor function and survival with our TrkB agonists were associated with preventing brain atrophy, by obtaining in vivo structural MRI scans and measuring brain volumes after 20 weeks of drug administration. As we reported previously, N171-82Q mice show significant regional and whole brain regional atrophy (26). Both 7,8-DHF and 4′-DMA-7,8-DHF administration significantly attenuated the magnitude of atrophy in the whole brain (Fig. 3A), neocortex (Fig. 3B) and periform cortex (Fig. 3D). 4′-DMA-7,8-DHF also ameliorated the atrophy in the striatum (Fig. 3C) and hippocampus (Fig. 3E) of HD mice. These results are consistent with the stronger effects of 4′-DMA-7,8-DHF on HD mice compared with effects of 7,8-DHF. Brain atrophy is region-specific in N171-82Q mice, as we did not detect atrophy in the cerebellum of HD mice (Fig. 3F).
Figure 3.

Effects of 7,8-dihydroxyflavone and 4′-dimethylamino-7,8- dihydroxyflavone on brain atrophy in N171-82Q HD mice. 7, 8-DHF (5 mg/kg/day) or 4′-DMA-7,8-DHF (1 mg/kg/day), was orally administered to mice daily from 6 weeks of age, and in vivo structural MRI were obtained at 26 weeks of age. Brain volumes were calculated by Large Deformation Diffeomorphic Metric Mapping (LDDMM). The transformations encode morphological differences between subjects and template images were analyzed with DBM to detect regional changes in brain volumes. Values are mean ± SE from five mice in each group. * P< 0.05 compared with the WT vehicle value; **P< 0.05 compared with the HD vehicle value, One-way ANOVA with Scheffé or Holm-Sidak post hoc test.
4′-DMA-7,8-DHF preserves DARPP32 levels in the striatum of N171-82Q HD mice
Dopamine- and cyclic AMP-regulated phosphoprotein of molecular weight 32 kDa (DARPP32) is a fundamental component of the dopamine-signaling cascade, and HD pathology is marked by an extensive loss of MSNs in the striatum that express high levels of DARPP32; therefore, DARPP32 can serve as a marker of neuronal loss as well as neuronal dysfunction in HD (27,28). We observed significantly reduced levels of DARPP-32 in the striatum of N171-82Q HD mice, as we reported previously (16), and these were restored by administration of 4′-DMA-7,8-DHF (Fig. 4), suggesting that 4′-DMA-7,8-DHF preserved medium spiny neuronal function and protected these neurons from degeneration.
Figure 4.

4′-Dimethylamino-7,8-dihydroxyflavone preserves DARPP32 levels in the striatum of N171-82Q HD mice. 4′-DMA-7,8-DHF (1 mg/kg) was administered to mice daily from 6 weeks of age; at 20 weeks of age the mice were euthanized and the striatum was dissected for measurement of DARPP32 levels by western blotting. Top, representative blots and bottom, densitometry results from three mice in each group. All values are expressed as mean ± SE. *P< 0.05 compared with the value of WT vehicle group; **P< 0.05 compared with the value of HD vehicle group. #P< 0.05 compared with the value of WT-vehicle group. One-way ANOVA with Scheffé post-hoc test.
It is noteworthy that most HD mouse models, including N171-82Q, show brain atrophy, but not striking neuronal death. The phenotypes possibly result from neuronal dysfunction instead of neuronal death, such as loss of DARPP32. Therefore, 4′-DMA-7,8-DHF prevented loss of DARPP32, and preserved the function of MSNs in the striatum, although it did not completely prevent striatal atrophy. In addition, 4′-DMA-7,8-DHF also increased DARPP32 levels in the striatum of wild-type mice (Fig. 4).
4′-DMA-7,8-DHF ameliorates impaired neurogenesis in N171-82Q HD mice
In the hippocampus of both rodents and primates, adult-generated neuronal cells arise from progenitor cells in the subgranular zone (SGZ) and migrate into the granule cell layer, where they differentiate into granular neurons. These neurons have been shown to be capable of functional integration into the hippocampal circuitry. There are some controversial reports on the status of neurogenesis between HD human brains and mouse models (29–35). Altered neurogenesis has been reported in HD mouse models such as R6/2 mice (29,30,32) and R6/1 mice (34). We previously demonstrated impaired survival of newly generated neurons in the hippocampus of N171-82Q HD mice (14). We therefore examined whether 4′-DMA-7,8-DHF improved the survival of newly generated neurons in the hippocampus of HD mice by administering 4′-DMA-7,8-DHF(1 mg/kg) or vehicle by oral gavage daily for 14 weeks. BrdU was then given by i.p. injection for five consecutive days, and mice were perfused at 3 weeks after the last BrdU injection. We found that survival of the newly generated neurons in the dentate gyrus of HD mice was significantly improved by chronic administration of 4′-DMA-7,8-DHF (Fig. 5).
Figure 5.
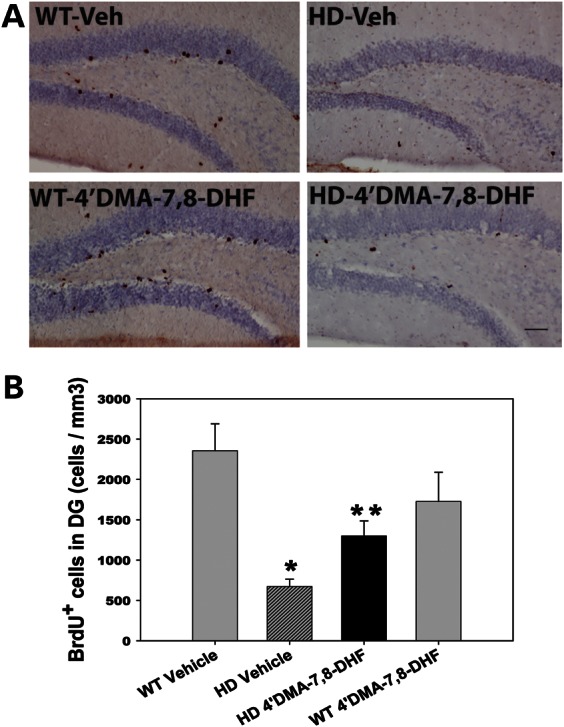
4′-Dimethylamino-7,8- dihydroxyflavone increases neurogenesis in N171-82Q HD mice. 4′-DMA-7,8-DHF (1 mg/kg/day) was administered to mice from 6 weeks of age for 14 weeks. Mice then received 5-bromo-2-deoxyuridine (BrdU, 100 mg/kg/day) injection for five consecutive days and were perfused at 3 weeks after the last BrdU injections for quantifying survival of newly generated cells. (A) Representative microscopic images showing BrdU-positive staining. Scale bar, n = 100 μm. (B) Quantification of newly generated cells in the dentate gyrus of the hippocampus. All values are expressed as mean and SE. n = 3–4 mice per group. * P< 0.05 compared with the WT vehicle value; **P< 0.05 compared with the HD vehicle value (One-way ANOVA with Scheffé post-hoc test).
DISCUSSION
BDNF deficiency, including down-regulation of gene transcription and disturbed BDNF transport from the cortex to the striatum, has been implicated as key HD pathogenesis. Decreased levels of BDNF in the HD brains also suggest reduced ability of neurons to cope with stress in HD. Moreover, alteration of BDNF-TrkB receptor signaling also contributes to disease progression in HD (36,37). Our present finding that pharmacological activation of TrkB in HD mice improves motor function, attenuates brain atrophy and extends survival supports the role of BDNF/TrkB signaling deficits in motor dysfunction and brain atrophy in HD, and provides proof of concept for the therapeutic potential of small-molecule TrkB agonists to improve HD motor symptoms and brain pathology, as well as to slow down disease progression.
DARPP-32 is a fundamental component of the dopamine-signaling cascade (38,39), and HD pathology is marked by extensive loss of medium spiny striatal neurons that express high levels of DARPP32; therefore, DARPP32 serves as a marker of neuronal loss as well as neuronal dysfunction in HD (40,41). BDNF has been shown to up-regulate DARPP32 (42,43) by activating the MAPK (ERK) and/or AKT pathways (44). Our results show that TrkB agonists activate MAPK in the striatum, suggesting that activation of the TrkB receptor downstream signaling MAPK pathway might contribute to its regulation of DARPP32 levels. It has been suggested that BDNF regulates morphologic maturation of MSNs and controls levels of DARPP32 in these neurons. A further mechanistic study indicated that PI3K is required for BDNF-induced DARPP32 increase. (42). A recent study in which TrkB receptors were ablated specifically in the striatum provided solid evidence that TrkB depletion in the striatum led to reduction of DARPP32 and severe deficits in striatal dopamine signaling through DARPP32 (45). Taken together, it is possible that 4′-DMA-7,8-DHF activates TrkB receptor, and provokes its downstream signaling pathway, and therefore increases DARPP32 levels in the WT mice. In addition, it is also possible that the activity of other TrkB signaling partners not examined here, such as AKT and PLCγ, is also affected by mutant HTT and plays a role in the response to these TrkB agonists in HD mice.
Adult neurogenesis might be involved in the physiological regenerative response and thereby alter or alleviate symptoms; it becomes impaired by the disease mechanism and thereby contributes to the symptoms of HD. Impaired neurogenesis has been reported in multiple HD mouse models, including N171-82Q HD mice (14,32,34,46–48). At the earlier stage of disease, when neurons self-regenerate, neurodegeneration can be overcome, cells are still functional and no severe symptoms appear. As the disease progresses, however, the regeneration ability may not be sufficient to cope with the neurodegenerative process and symptoms will become evident. Previous studies reported that 4′-DMA-7,8-DHF increased neurogenesis in normal mice (19). BDNF/TrkB signaling has been shown to up-regulate hippocampal neurogenesis (49,50); indeed, in the present study, the TrkB agonist 4′-DMA-7,8-DHF rescued compromised neurogenesis in N171-82Q HD mice. Notably, our previous study indicated that a higher dose of 4′-DMA-7,8-DHF (5 mg/kg) increased cell proliferation in normal mice (19), and our current study shows that although 4′-DMA-7,8-DHF at 1 mg/kg did not affect neurogenesis in normal mice, it did improve survival of newly generated neurons in HD mice.
In terms of the clinical applicability of the TrkB agonists, we have recently examined in vivo pharmacokinetic profiles and metabolism of 7,8-DHF (Liu et al., Pharmacology In press) and 4′-DMA-7,8-DHF (51). There is a significant discrepancy between their remarkable therapeutic efficacy and in vivo oral bioavailability. This might be due to the extreme potency of these compounds and that a low concentration in the brain is sufficient to promote its pharmacological effect, and it is also possible that metabolites of these compounds are active as well. On the other hand, it remains unclear how 4′-DMA-7,8-DHF, which has a short half-life, affects its activities in animals. Its O-methylated- or B-ring-hydroxylated metabolites in mouse brain might contribute to its actions. Clearly, further structure–activity studies are needed for continuing development of compounds that are metabolically stable and/or reduce plasma protein-binding affinity, thereby improving their bioavailability in the brain.
To our knowledge, this is the first report that small-molecular TrkB activators provide beneficial effects in an HD mouse model. Since the discovery of the brain-penetrable TrkB agonist 7,8-DHF, multiple studies have described the beneficial effects of 7,8-DHF in a variety of models of neurological disorders, including Alzheimer's disease (19), Parkinson's disease (18), stroke (23), Rett Syndrome (21), depression and stress or aging-related cognitive dysfunction (22,25). Other groups are also studying TrkB agonists in brain disease models. For example, small-molecule BDNF mimetics identified by in silico screening with a BDNF loop-domain pharmacophore, followed by low-throughput in vitro screening in mouse fetal hippocampal neurons, have been shown to prevent neuronal degeneration in rodents (52) and improved respiratory function in a mouse model of Rett Syndrome (53). Nevertheless, the chemical structures of the compounds identified by in silico screening are quite different from the structure of 7,8-DHF. 7,8-DHF has been widely tested in a variety of cellular and animal models of neurological disorders. The consensus conclusion is that 7,8-DHF and 4′-DMA-7,8-DHF mimic the biological and physiological functions of BDNF, specifically activate TrkB receptors and provide potent therapeutic efficacy toward numerous neurological and mental diseases, in which BDNF activity has been previously demonstrated. Most importantly, these compounds are safe for chronic treatment without any noticeable side effects or toxicity.
In summary, our data establish that activation of TrkB receptors restores the molecular defects induced by mutant huntingtin including depleted DARPP32 levels and impaired neurogenesis, improves motor function, attenuates brain atrophy and increases survival of HD mice. These findings provide validation for the use of TrkB as a therapeutic target in mouse models of HD, and indicate that TrkB agonists can effectively overcome functional deficits associated with reduced BDNF levels in HD.
MATERIALS AND METHODS
Mice and drug administration
Male N171-82Q HD mice were mated to female hybrid mice (B6C3F1/J, Jackson Laboratory, ME, USA). Male mice were used in our studies because we found gender-dependent phenotypic differences in N171-82Q HD mice (13). Seventy-nine male mice were used in the current study. Mice were randomly divided into different groups. All mice were used in these analyses. 7,8-DHF and 4′-DMA-7,8-DHF were synthesized in our laboratory and freshly prepared by dissolving in water and adjusting the pH value to 7.4. Drugs were orally administered to the mice by gavage from 6 weeks old to the end of study. All animal experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee of the Johns Hopkins University.
Behavioral test and survival study
All mice were randomly divided into different groups. Each group contained 10 mice at the beginning of experiments for survival and behavioral tests. Motor function was assessed on an 80-cm long and 5-mm wide square-shaped balance beam that was mounted on supports 50 cm in height. A bright light illuminated the start platform, and a darkened enclosed 1728 cm3 escape box (12 × 12 × 12 cm) was situated at the end of the beam. Disposable pads placed under the beam provided cushioning if an animal fell off the beam. Mice were trained to walk across the beam twice at least 1 h prior to testing. If a mouse stopped during training, the tail was gently pressed to encourage movement. After the training trial, mice were left undisturbed for at least an hour before testing. The time for each mouse to traverse the balance beam was recorded with a 60 s maximum cut-off, if the traverse time is more than 60 s, and falls were scored as 60 s.
Survival was monitored daily by experienced investigators (M.J. and Q.P.). The mice were considered at the end of life when they were unable to back to their normal position after being placed on their backs and initiate movement after being gently prodded for 30 s.
In vivo structural MRI acquisition and quantification of brain volume
In vivo structural MRI studies were performed on a horizontal 9.4 T magnetic resonance imager (Bruker Biospin, Billerica) with a triple-axis gradient and an animal imaging probe. The detailed image capture and analysis were described in our previous study (54). Briefly, mice were anesthetized with isoflurane (1%), respiration was monitored and the temperature was maintained during the entire scan. Images were acquired by a three-dimensional T2-weighted fast spin echo sequence with the following parameters: echo time (TE)/repetition time (TR) = 40/700 ms, resolution = 0.1 × 0.1 × 0.1 mm, echo train length = 4, number of average = 2 and flip angle = 40°. The imaging resolution and contrast were sufficient for automatic volumetric characterization of the mouse brains and substructures. The intensity-normalized images were submitted by the Diffeomap software to a linux cluster, which runs Large Deformation Diffeomorphic Metric Mapping (LDDMM). The transformations encode morphological differences between subject and template images and can be analyzed with deformation-based morphometry (DBM) to detect regional changes in brain volume.
BrdU administration, immunohistochemistry and stereological quantification
For the neurogenesis study, all mice received twice daily injections of 100 mg/kg of BrdU (5-bromo-2-deoxyuridine; Sigma) in Dulbecco's phosphate buffered saline solution at 8 h apart for five consecutive days. Animals were perfused at 3 weeks after the last BrdU injection for assessing survival of newly generated cells. BrdU immunostaining was performed as we previously described (15). Briefly, coronal brain sections (35 μm) were incubated in 2 N HCl for 30 min at 37°C and washed in 0.1 m borate buffer (pH 8.5) for 10 min. Endogenous peroxidases were quenched with 0.1% H2O2 for 30 min.
Sections were blocked in 1× TBS containing 3% horse serum and 0.3% Triton X-100 for 1 h, followed by incubation in anti-BrdU antibody (1:300, Accurate Chemical & Scientific Corporation) overnight at 4°C. Sections were then incubated with biotinylated anti-rat IgG for 2 h at room temperature. The immunoreactive product was detected by using the vectastain ABC kit enhancing system (Vector Laboratory, CA, USA) with diaminobenzadine (DAB) as a substrate. Sections were counterstained with Hematoxylin QS (Vector) for stereological quantification. Stereology counting was performed in serial coronal sections on blind-coded slides. The numbers of BrdU-positive cells in the SGZ of the dentate gyrus were assessed by counting all positive cells in sections at the levels of the bregma −1.34 to −3.52 mm by using conventional light microscopy with a 40× objective, and distance between the sections of 350 μm. The ‘optical fractionator’ was used to count BrdU-positive cells in the dentate gyrus. The optical fractionator technique estimates the number of cells by multiplying the sum of cells counted by the reciprocal of the fraction of the region sampled. The stereology investigator software automatically identified and measured profiles.
Protein extraction and western blot analysis
Mouse brain tissues were homogenized and lysed in 9 vol of RIPA buffer (Sigma) containing 50 mm Tris–HCl, pH 8.0, with 150 mm sodium chloride, 1.0% Igepal CA-630 (NP-40), 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and freshly prepared protease inhibitor (1:100, Sigma) and/or phosphatase inhibitor (1:100, sigma). Then samples were centrifuged at 14 000g for 15 min at 4°C and supernatant fractions were collected. Protein concentration was determined by the Micro BCA™ protein assay kit (Pierce Protein Research Products). Soluble proteins were separated by SDS–PAGE and transferred to a nitrocellulose membrane. The membrane was blocked in the presence of BSA or 5% nonfat milk, and then incubated overnight with primary antibodies at 4°C: rabbit polyclonal anti-p-TrkB (TyR 706)(1:200, Santa cruz), rabbit polyclonal anti-TrkB(1:1000, Cell Signaling), rabbit polyclonal anti-p-MAPK(1:1000, Cell Signaling), rabbit polyclonal anti-MAPK(1:1000, cell signaling), rabbit polyclonal anti-DARPP32 (1:2000, Chemicon) and mouse monoclonal anti-beta actin (1:5000, Sigma). The membrane was then exposed for 1 h to HRP-conjugated secondary antibody (1:3000; GE Healthcare) and immunoreactive proteins were visualized using a chemiluminescence-based detection kit according to the manufacturer's protocol (ECL kit; Amersham Corp.).
Statistics
Data are expressed as the mean ± SE. Statistical comparisons between groups were made by one-way ANOVA with Scheffé or Holm-Sidak post-hoc test. Survival data were analyzed by Kaplan–Meier analysis.
FUNDING
This work was supported by NIH NS074196 to W.D. and CHDI Foundation to W.D.
ACKNOWLEDGEMENTS
We thank Dr Pamela Talalay for her dedicated editorial advice.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Vonsattel J.P., DiFiglia M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Ross C.A., Tabrizi S.J. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 3.Krobitsch S., Kazantsev A.G. Huntington's disease: from molecular basis to therapeutic advances. Int. J. Biochem. Cell Biol. 2011;43:20–24. doi: 10.1016/j.biocel.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 4.Gil-Mohapel J.M. Screening of therapeutic strategies for Huntington's disease in YAC128 transgenic mice. CNS Neurosci. Ther. 2012;18:77–86. doi: 10.1111/j.1755-5949.2011.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rego A.C., de Almeida L.P. Molecular targets and therapeutic strategies in Huntington's disease. Curr. Drug Target CNS Neurol. Disord. 2005;4:361–381. doi: 10.2174/1568007054546081. [DOI] [PubMed] [Google Scholar]
- 6.Grimbergen Y.A., Roos R.A. Therapeutic options for Huntington's disease. Curr. Opin. Investig. Drugs. 2003;4:51–54. [PubMed] [Google Scholar]
- 7.Bonelli R.M., Wenning G.K., Kapfhammer H.P. Huntington's disease: present treatments and future therapeutic modalities. Int. Clin. Psychopharmacol. 2004;19:51–62. doi: 10.1097/00004850-200403000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Zuccato C., Liber D., Ramos C., Tarditi A., Rigamonti D., Tartari M., Valenza M., Cattaneo E. Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol. Res. 2005;52:133–139. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Zuccato C., Ciammola A., Rigamonti D., Leavitt B.R., Goffredo D., Conti L., MacDonald M.E., Friedlander R.M., Silani V., Hayden M.R., et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 10.Zuccato C., Cattaneo E. Role of brain-derived neurotrophic factor in Huntington's disease. Prog. Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Xie Y., Hayden M.R., Xu B. BDNF overexpression in the forebrain rescues Huntington's disease phenotypes in YAC128 mice. J. Neurosci. 2010;30:14708–14718. doi: 10.1523/JNEUROSCI.1637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giralt A., Carreton O., Lao-Peregrin C., Martin E.D., Alberch J. Conditional BDNF release under pathological conditions improves Huntington's disease pathology by delaying neuronal dysfunction. Mol. Neurodegener. 2011;6:71. doi: 10.1186/1750-1326-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duan W., Guo Z., Jiang H., Ware M., Li X.J., Mattson M.P. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc. Natl Acad. Sci. USA. 2003;100:2911–2916. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duan W., Peng Q., Masuda N., Ford E., Tryggestad E., Ladenheim B., Zhao M., Cadet J.L., Wong J., Ross C.A. Sertraline slows disease progression and increases neurogenesis in N171–82Q mouse model of Huntington's disease. Neurobiol. Dis. 2008;30:312–322. doi: 10.1016/j.nbd.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng Q., Masuda N., Jiang M., Li Q., Zhao M., Ross C.A., Duan W. The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington's disease mouse model. Exp. Neurol. 2008;210:154–163. doi: 10.1016/j.expneurol.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang M., Wang J., Fu J., Du L., Jeong H., West T., Xiang L., Peng Q., Hou Z., Cai H., et al. Neuroprotective role of Sirt1 in mammalian models of Huntington's disease through activation of multiple Sirt1 targets. Nat. Med. 2012;18:153–158. doi: 10.1038/nm.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chao M.V., Hempstead B.L. p75 and Trk: a two-receptor system. Trends Neurosci. 1995;18:321–326. [PubMed] [Google Scholar]
- 18.Jang S.W., Liu X., Yepes M., Shepherd K.R., Miller G.W., Liu Y., Wilson W.D., Xiao G., Blanchi B., Sun Y.E., et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl Acad. Sci. USA. 2010;107:2687–2692. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X., Chan C.B., Jang S.W., Pradoldej S., Huang J., He K., Phun L.H., France S., Xiao G., Jia Y., et al. A synthetic 7,8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J. Med. Chem. 2010;53:8274–8286. doi: 10.1021/jm101206p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mantilla C.B., Ermilov L.G. The novel TrkB receptor agonist 7,8-dihydroxyflavone enhances neuromuscular transmission. Muscle Nerve. 2012;45:274–276. doi: 10.1002/mus.22295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson R.A., Lam M., Punzo A.M., Li H., Lin B.R., Ye K., Mitchell G.S., Chang Q. 7,8-dihydroxyflavone exhibits therapeutic efficacy in a mouse model of Rett syndrome. J. Appl. Physiol. 2012;112:704–710. doi: 10.1152/japplphysiol.01361.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devi L., Ohno M. 7,8-Dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2012;37:434–444. doi: 10.1038/npp.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J., Chua K.W., Chua C.C., Yu H., Pei A., Chua B.H., Hamdy R.C., Xu X., Liu C.F. Antioxidant activity of 7,8-dihydroxyflavone provides neuroprotection against glutamate-induced toxicity. Neurosci. Lett. 2011;499:181–185. doi: 10.1016/j.neulet.2011.05.054. [DOI] [PubMed] [Google Scholar]
- 24.Andero R., Daviu N., Escorihuela R.M., Nadal R., Armario A. 7,8-dihydroxyflavone, a TrkB receptor agonist, blocks long-term spatial memory impairment caused by immobilization stress in rats. Hippocampus. 2012;22:399–408. doi: 10.1002/hipo.20906. [DOI] [PubMed] [Google Scholar]
- 25.Andero R., Heldt S.A., Ye K., Liu X., Armario A., Ressler K.J. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am. J. Psychiatry. 2011;168:163–172. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J., Peng Q., Li Q., Jahanshad N., Hou Z., Jiang M., Masuda N., Langbehn D.R., Miller M.I., Mori S., et al. Longitudinal characterization of brain atrophy of a Huntington's disease mouse model by automated morphological analyses of magnetic resonance images. Neuroimage. 2010;49:2340–2351. doi: 10.1016/j.neuroimage.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vonsattel J.P., Myers R.H., Stevens T.J., Ferrante R.J., Bird E.D., Richardson E.P., Jr Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 28.de la Monte S.M., Vonsattel J.P., Richardson E.P., Jr Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington's disease. J. Neuropathol. Exp. Neurol. 1988;47:516–525. doi: 10.1097/00005072-198809000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Phillips W., Morton A.J., Barker R.A. Limbic neurogenesis/plasticity in the R6/2 mouse model of Huntington's disease. Neuroreport. 2006;17:1623–1627. doi: 10.1097/01.wnr.0000236855.85962.f6. [DOI] [PubMed] [Google Scholar]
- 30.Phillips W., Morton A.J., Barker R.A. Abnormalities of neurogenesis in the R6/2 mouse model of Huntington's disease are attributable to the in vivo microenvironment. J. Neurosci. 2005;25:11564–11576. doi: 10.1523/JNEUROSCI.3796-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grote H.E., Bull N.D., Howard M.L., van Dellen A., Blakemore C., Bartlett P.F., Hannan A.J. Cognitive disorders and neurogenesis deficits in Huntington's disease mice are rescued by fluoxetine. Eur. J. Neurosci. 2005;22:2081–2088. doi: 10.1111/j.1460-9568.2005.04365.x. [DOI] [PubMed] [Google Scholar]
- 32.Gil J.M., Mohapel P., Araujo I.M., Popovic N., Li J.Y., Brundin P., Petersen A. Reduced hippocampal neurogenesis in R6/2 transgenic Huntington's disease mice. Neurobiol. Dis. 2005;20:744–751. doi: 10.1016/j.nbd.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 33.Tattersfield A.S., Croon R.J., Liu Y.W., Kells A.P., Faull R.L., Connor B. Neurogenesis in the striatum of the quinolinic acid lesion model of Huntington's disease. Neuroscience. 2004;127:319–332. doi: 10.1016/j.neuroscience.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 34.Lazic S.E., Grote H., Armstrong R.J., Blakemore C., Hannan A.J., van Dellen A., Barker R.A. Decreased hippocampal cell proliferation in R6/1 Huntington's mice. Neuroreport. 2004;15:811–813. doi: 10.1097/00001756-200404090-00014. [DOI] [PubMed] [Google Scholar]
- 35.Curtis M.A., Penney E.B., Pearson A.G., van Roon-Mom W.M., Butterworth N.J., Dragunow M., Connor B., Faull R.L. Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc. Natl Acad. Sci. USA. 2003;100:9023–9027. doi: 10.1073/pnas.1532244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zuccato C., Marullo M., Conforti P., MacDonald M.E., Tartari M., Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington's disease. Brain Pathol. 2008;18:225–238. doi: 10.1111/j.1750-3639.2007.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gines S., Bosch M., Marco S., Gavalda N., Diaz-Hernandez M., Lucas J.J., Canals J.M., Alberch J. Reduced expression of the TrkB receptor in Huntington's disease mouse models and in human brain. Eur. J. Neurosci. 2006;23:649–658. doi: 10.1111/j.1460-9568.2006.04590.x. [DOI] [PubMed] [Google Scholar]
- 38.Greengard P., Allen P.B., Nairn A.C. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- 39.Fienberg A.A., Hiroi N., Mermelstein P.G., Song W., Snyder G.L., Nishi A., Cheramy A., O'Callaghan J.P., Miller D.B., Cole D.G., et al. DARPP-32: regulator of the efficacy of dopaminergic neurotransmission. Science. 1998;281:838–842. doi: 10.1126/science.281.5378.838. [DOI] [PubMed] [Google Scholar]
- 40.van Dellen A., Welch J., Dixon R.M., Cordery P., York D., Styles P., Blakemore C., Hannan A.J. N-Acetylaspartate and DARPP-32 levels decrease in the corpus striatum of Huntington's disease mice. Neuroreport. 2000;11:3751–3757. doi: 10.1097/00001756-200011270-00032. [DOI] [PubMed] [Google Scholar]
- 41.Hickey M.A., Kosmalska A., Enayati J., Cohen R., Zeitlin S., Levine M.S., Chesselet M.F. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington's disease mice. Neuroscience. 2008;157:280–295. doi: 10.1016/j.neuroscience.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ivkovic S., Polonskaia O., Farinas I., Ehrlich M.E. Brain-derived neurotrophic factor regulates maturation of the DARPP-32 phenotype in striatal medium spiny neurons: studies in vivo and in vitro. Neuroscience. 1997;79:509–516. doi: 10.1016/s0306-4522(96)00684-7. [DOI] [PubMed] [Google Scholar]
- 43.Ivkovic S., Ehrlich M.E. Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J. Neurosci. 1999;19:5409–5419. doi: 10.1523/JNEUROSCI.19-13-05409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stroppolo A., Guinea B., Tian C., Sommer J., Ehrlich M.E. Role of phosphatidylinositide 3-kinase in brain-derived neurotrophic factor-induced DARPP-32 expression in medium size spiny neurons in vitro. J. Neurochem. 2001;79:1027–1032. doi: 10.1046/j.1471-4159.2001.00651.x. [DOI] [PubMed] [Google Scholar]
- 45.Li Y., Yui D., Luikart B.W., McKay R.M., Rubenstein J.L., Parada L.F. Conditional ablation of brain-derived neurotrophic factor-TrkB signaling impairs striatal neuron development. Proc. Natl Acad. Sci. USA. 2012;109:15491–15496. doi: 10.1073/pnas.1212899109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simpson J.M., Gil-Mohapel J., Pouladi M.A., Ghilan M., Xie Y., Hayden M.R., Christie B.R. Altered adult hippocampal neurogenesis in the YAC128 transgenic mouse model of Huntington disease. Neurobiol. Dis. 2011;41:249–260. doi: 10.1016/j.nbd.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 47.Kandasamy M., Couillard-Despres S., Raber K.A., Stephan M., Lehner B., Winner B., Kohl Z., Rivera F.J., Nguyen H.P., Riess O., et al. Stem cell quiescence in the hippocampal neurogenic niche is associated with elevated transforming growth factor-beta signaling in an animal model of Huntington disease. J. Neuropathol. Exp. Neurol. 2010;69:717–728. doi: 10.1097/NEN.0b013e3181e4f733. [DOI] [PubMed] [Google Scholar]
- 48.Gil-Mohapel J., Simpson J.M., Ghilan M., Christie B.R. Neurogenesis in Huntington's disease: can studying adult neurogenesis lead to the development of new therapeutic strategies. Brain Res. 2011;1406:84–105. doi: 10.1016/j.brainres.2011.06.040. [DOI] [PubMed] [Google Scholar]
- 49.Li Y., Luikart B.W., Birnbaum S., Chen J., Kwon C.H., Kernie S.G., Bassel-Duby R., Parada L.F. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. 2008;59:399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bergami M., Berninger B., Canossa M. Conditional deletion of TrkB alters adult hippocampal neurogenesis and anxiety-related behavior. Commun. Integr. Biol. 2009;2:14–16. doi: 10.4161/cib.2.1.7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X., Chan C.B., Qi Q., Xiao G., Luo H.R., He X., Ye K. Optimization of a small tropomyosin-related kinase B (TrkB) agonist 7,8-dihydroxyflavone active in mouse models of depression. J. Med. Chem. 2012;55:8524–8537. doi: 10.1021/jm301099x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Massa S.M., Yang T., Xie Y., Shi J., Bilgen M., Joyce J.N., Nehama D., Rajadas J., Longo F.M. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Invest. 2010;120:1774–1785. doi: 10.1172/JCI41356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmid D.A., Yang T., Ogier M., Adams I., Mirakhur Y., Wang Q., Massa S.M., Longo F.M., Katz D.M. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J. Neurosci. 2012;32:1803–1810. doi: 10.1523/JNEUROSCI.0865-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng Y., Peng Q., Hou Z., Aggarwal M., Zhang J., Mori S., Ross C.A., Duan W. Structural MRI detects progressive regional brain atrophy and neuroprotective effects in N171–82Q Huntington's disease mouse model. Neuroimage. 2011;56:1027–1034. doi: 10.1016/j.neuroimage.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]