

Abstract
Chemical modifications to the DNA and histone protein components of chromatin can modulate gene expression and genome stability. Understanding the physiological impact of changes in chromatin structure remains an important question in biology. As one example, in order to generate antibody diversity with somatic hypermutation and class switch recombination, chromatin must be made accessible for Activation-induced cytidine deaminase (AID)-mediated deamination of cytosines in DNA. These lesions are recognized and removed by various DNA repair pathways, but if not handled properly, can lead to formation of oncogenic chromosomal translocations. In this review, we focus the discussion on how chromatin-modifying activities and -binding proteins contribute to the native chromatin environment for which AID-induced DNA damage is targeted and repaired. Outstanding questions remain regarding the direct roles of histone post-translational modifications and the significance of AID function outside of antibody diversity.
Introduction
Chromatin is the platform for transcription, DNA repair, and recombination. Post-translational modifications (PTMs) of the histone H3, H2B, H2A, and H4 components of chromatin regulate DNA-mediated processes by altering chromatin structure and generating recognition sites for mediating effector protein stabilization (Downs et al., 2007; Felsenfeld and Groudine, 2003; Jenuwein and Allis, 2001; Suganuma and Workman, 2011). The `histone code hypothesis' that a particular histone post-translational modification (PTM) or combination thereof can constitute a code for a cellular action or biological function continues to be tested and has permeated far across the field of DNA repair, from its inception in relation to gene regulation (Downs et al., 2007; Jenuwein and Allis, 2001). The first and still most striking and clear example of how a particular histone modification promotes genome stability came from the observations that mice deficient in the histone variant H2AX, which becomes phosphorylated at serine 139 seconds after ionizing radiation (IR)-induced DNA damage (Bonner et al., 2008), accumulate spontaneous DNA double-strand breaks (DSBs) and develop tumors more rapidly when cell cycle checkpoints are compromised (Bassing et al., 2003; Celeste et al., 2003). In this review, we describe our current understanding of how histone PTMs function in a physiological setting with immunoglobulin class-switch recombination (CSR) as a model. The CSR reaction at the immunolglobulin heavy-chain (Igh) locus can be divided into three general temporal stages including the so-called germ-line transcription step that mediates chromatin accessibility, the targeting and generation of AID-induced DNA damage, and the subsequent repair of CSR-associated DSBs (Figure 1). The two main focus points of this review are to discuss our understanding of how chromatin PTMs function in the accessibility phase as well as in the repair phase of AID-induced DNA damage.
Figure 1. The transcription, DNA damage, and DNA repair phases of CSR.

(A) Organization of the Igh locus in mice, including the antigen recognition V(D)J gene segment in black, the switch (S) regions in blue, the constant (C) region exon segments in green, and the enhancers in orange. The μ, δ, γ3, γ1, γ2b, γ2α, ε and α isotypes correspond to immunoglobulins M, D, G3, G1, G2b, G2a, E, and A. (B) In resting B cells, transcription initiating upstream of the V(D)J gene segment leads to a full-length Igh transcript and initiates from 5'Eì to generate the ì germ-line noncoding sterile switch transcript. However, transcription is completely absent from the downstream cluster of switch regions and constant gene segments. Upon B cell activation during an immune response (or by LPS stimulation in cell culture), germ-line transcription initiates from a switch promoter region. Accessibility of AID to transcribed switch region chromatin targets AID activity to Igh, leading to DSB formation. Synapsis of the two broken switch regions and their repair, mediated by the DNA damage response and NHEJ machineries, promote efficient CSR (to IgG3 in this example) and suppress genomic instability.
Initiating the second wave of antibody diversity with AID
B lymphocytes undergo physiological DNA damage to produce large numbers of antibody molecules that are poised to associate with foreign antigens. This antigen/antibody interaction will subsequently activate pathways for the removal and clearance of pathogens from our body, as part of the adaptive immune response. Each B lymphocyte expresses a single B cell receptor (BCR) that has been assembled by RAG1/2-dependent V(D)J recombination during early B cell development in the bone marrow. When mature naïve IgM-expressing B cells in peripheral lymphoid organs recognize antigen through their BCR and become activated within germinal center structures, they undergo clonal expansion and further diversify their antigen receptors with somatic hypermutation (SHM) and isotype class-switching. The introduction of mutations in rapidly proliferating B cells during SHM ultimately culminates in the production of thousands of B cells expressing slightly different receptors with varying specificity for an antigen, from which the B cell with highest affinity for the antigen can be selected. Isotype class-switching alters antibody effector function, whereby activated B cells swap constant region gene segments of the antibody without altering variable region specificity. This CSR event requires DNA DSB formation and DNA end-joining (Figure 1). Expression of a successfully recombined class-switched Igh gene can help eliminate particular pathogens by activating, for example, phagocytic immune cells (Boboila et al., 2012; Stavnezer et al., 2008).
AID has taken center stage as a B cell-specific factor required both for SHM and CSR. It was first identified as being differentially expressed in a murine B lymphocyte cell line after stimulation to undergo antibody class-switching and was shown to be highly expressed in primary human and murine germinal center B cells from tonsil, lymph node, and spleen (Muramatsu et al., 2007). In addition to expression in germinal center B cells, there are reports that AID is also expressed in oocytes and, albeit at far lower levels, in embryonic stem cells, early embryos, primordial germ cells, testes, and B cell progenitors (Orthwein and Di Noia, 2012). Mutations in the gene encoding AID, Aicda, were found to cause the rare autosomal recessive hyper-IgM syndrome type 2 (HIGM2) that is characterized by the absence of immunoglobulin CSR and somatic hypermutation (Revy et al., 2000). Subsequently, AID−/− mice recapitulated this B cell disease without showing any other noticeable developmental phenotypes (Muramatsu et al., 2000). Interestingly, in an artificial system in which AID is not normally present, expression of AID and a class-switching DNA substrate was shown to be sufficient for class-switch recombination in fibroblasts, suggesting that AID may be the sole B cell specific factor required for initiating CSR (Muramatsu et al., 2007).
During an immune response, peripheral B cells stimulated by antigen and the cytokine milieu become activated and enter the cell cycle; shortly thereafter, the cells begin to express AID and concentrate in germinal center structures of lymph nodes and the spleen (Victora and Nussenzweig, 2012). For SHM, AID is targeted to the variable V(D)J gene segments of the immunoglobulin heavy and light chains and catalyzes cytosine deamination events that lead to increased mutations as a result of error-prone translesion DNA polymerase activity and DNA replication (Di Noia and Neuberger, 2007; Liu and Schatz, 2009). To initiate the CSR reaction, AID gets targeted to the Igh locus downstream of the V(D)J gene segment and initiates DNA lesions that, here, lead to DNA DSB formation (Boboila et al., 2012; Nussenzweig and Nussenzweig, 2010; Stavnezer et al., 2008). For productive CSR, AID-induced DSBs must occur at two switch (S) repeat regions (i.e. Sμ, Sγ3, Sγ1, Sγ2b, Sγ2a, Sε, or Sα in the mouse) that precede participating constant region gene segments (Stavnezer et al., 2008) (Figure 1). Synapsis and DNA repair of the two broken DNA ends are then mediated by protein factors of the DNA damage response (DDR) and the non-homologous end-joining (NHEJ) pathways. This DNA rearrangement process resulting in an orchestrated switch from IgM expression to expression of IgG, IgE, or IgA must be carefully controlled and coordinated in the context of chromatin.
The `accessibility hypothesis' for AID targeting
The lineage specificity of CSR can be explained by AID expression being largely restricted to germinal center B cells. To explain how DNA rearrangements in lymphocytes occur specifically at antigen receptor gene loci, Yancopoulos and Alt put forth the `accessibility hypothesis' in 1985 after observing sterile germ-line transcript initiation only at gene segments undergoing recombination (Yancopoulos and Alt, 1985). Since then, germ-line transcripts coinciding with recombination at a particular gene segment have been observed at all antigen receptor loci and many lines of evidence now support the conclusion that germ-line transcription of an antigen receptor gene segment is an essential feature of the targeting mechanism both for RAG1/2-mediated V(D)J recombination in early developing lymphocytes (Cobb et al., 2006; Krangel, 2009) and for AID-dependent CSR and SHM in mature B cells (Stavnezer et al., 2008). In addition to germ-line expression of sterile non-coding transcripts, the accessibility hypothesis has expanded to encompass the spatial organization and nuclear positioning of antigen receptor loci (Alt et al., 2013; Hewitt et al., 2010; Jhunjhunwala et al., 2009; Johnson et al., 2009). For the purposes of our discussion, we define accessibility as a localized alteration of chromatin structure that facilitates recombination at the locus, and we focus our discussion to how germ-line transcription and histone modifications target AID for CSR.
The importance of transcription in targeting the somatic hypermutation activity first came from a genetics experiment showing that insertion of the variable region promoter to a location upstream of the constant region at the Igκ locus promoted hypermutation at the constant region where it normally does not occur (Peters and Storb, 1996). At the Igh locus, all germ-line switch transcripts have the same overall structure with an intronic (I) promoter exon followed by a switch (S) region and a constant (C) region gene segment (Stavnezer et al., 2008) (Figure 2). Activation signals during an immune response direct promoter-driven germ-line transcription and DSB formation to particular switch regions at the Igh locus (Boboila et al., 2012; Stavnezer et al., 2008). Indeed, mouse genetic studies revealed that deletion of Iγ1 abolished CSR to IgG1 (Jung et al., 1993) and that replacement of the Iγ2b promoter and I exon with a neomycin-resistance gene transcribed in the antisense direction abolished CSR to IgG2b (Zhang et al., 1993). The conserved structure of the germ-line transcripts between the different immunoglobulin isotypes suggests that these non-coding RNAs may have a common function in CSR, and elegant mouse genetic studies have suggested that splicing of the I and C exons is required for CSR (Harriman et al., 1996; Hein et al., 1998; Lorenz et al., 1995); nevertheless, the role of the germ-line transcripts and their splicing is still poorly understood.
Figure 2. Transcription-linked histone PTMs and PTIP promote accessibility for AID targeting.

PTMs of active chromatin are found at the μ region of Igh in resting B cells and help to establish a chromatin environment that is permissive for AID targeting. Upon B cell activation, transcription-linked histone PTMs become detectable at downstream switch regions that have been induced to undergo CSR. These inducible histone PTMs at downstream switch regions are dependent on PTIP, at least at the γ3, γ1, and γ2ab regions.
The mechanism for targeting AID to immunoglobulin loci is of great interest given its role in inducing DNA mutations and tumorigenesis (Di Noia and Neuberger, 2007; Pavri and Nussenzweig, 2011; Vuong and Chaudhuri, 2012). Germ-line transcription of immunoglobulin genes paves the way for AID to act on ssDNA, such that AID can induce mutations on both the template and non-template strands, as evidenced from sequencing genomic DNA from B cells undergoing SHM and CSR (Liu and Schatz, 2009; Petersen et al., 2001). The C-terminus of AID appears to be required for CSR while the N-terminus of AID is required for SHM, suggesting that specific interactions may modulate the targeting and/or function of AID (Di Noia and Neuberger, 2007; Xu et al., 2012). In B cells stimulated to undergo CSR, AID physically associates with RNA Pol II (Nambu et al., 2003). Under similar conditions, RNA Pol II localizes at Igh in a special way accumulating from the intronic promoter, through the switch region, and into the constant region for both Sm and the downstream Sg3 switch region (Daniel et al., 2010; Rajagopal et al., 2009; Wang et al., 2009a). This 5' end build up of RNA Pol II at switch regions is independent of AID (Rajagopal et al., 2009; Wang et al., 2009a) and suggests that RNA Pol II stalling may be an intrinsic feature of transcription through the switch regions.
Recent advances have provided additional evidence for how AID activity is linked to transcription by showing that AID directly interacts with Spt5, the RNA exosome, and the PAF elongation complex. RNAi knock-down of any of these factors in the CH12 B cell line leads to reduced chromatin association of AID to Igh and impairs CSR, without affecting germ-line transcription at the switch region (Basu et al., 2011; Pavri and Nussenzweig, 2011; Stanlie et al., 2012; Willmann et al., 2012). Together, these studies lead to a model whereby RNA Pol II stalling facilitates AID recruitment through Spt5 and the RNA exosome, which promotes subsequent targeting of mutations on both DNA strands (Basu et al., 2011; Pavri and Nussenzweig, 2011; Willmann et al., 2012; Yamane et al., 2011). Thus, the process of transcription, or even the transcript itself, may play a direct role in AID targeting, and the function of germ-line transcription may not be limited to simply rendering the target DNA accessible to AID.
Correlating histone modifications with AID localization
Alterations in chromatin structure that are specific consequences of germ-line transcription at Igh may also play a role in promoting and/or stabilizing AID targeting. Using in vitro systems, AID can efficiently target DNA in nucleosomes undergoing transcription and there is evidence that nucleosome stability and positioning can significantly influence AID targeting (Kodgire et al., 2012; Shen et al., 2009). Looking more closely at particular aspects of chromatin structure, multiple studies have correlated localized changes in histone modifications with accessibility for antigen receptor gene recombination. Histone marks including acetylation of H3 and H4 occur at the Igh switch regions in B cells actively undergoing CSR (Chowdhury et al., 2008; Daniel et al., 2010; Kuang et al., 2009; Li et al., 2004; Nambu et al., 2003; Wang et al., 2006; Wang et al., 2009a) but the most clear and direct link has come from the interaction between H3K4me3 and RAG1/2 during V(D)J recombination. The transcription initiation-associated histone H3K4me3 mark (Ruthenburg et al., 2007) can be directly recognized by a PHD domain in RAG2 and this interaction appears to help target and stimulate RAG1/2 activity (Schatz and Swanson, 2011). Indeed, decreases in H3K4me3 result in defective RAG1/2-mediated recombination (Matthews et al., 2007).
Additional clues about the relation between histone marks and accessibility during CSR come from comparison of histone modification profiles of resting and stimulated B cells. The histone modifications and chromatin structure at Sμ, for example, which are present in resting B cells, are sufficient to target AID activity. This was shown by measuring AID-induced mutations at Sμ in resting B cells from mice constitutively expressing AID in B cell progenitors, when it is not normally present (Robbiani et al., 2009) (Figure 2). These data are consistent with H3K4me3 and the transcription elongation-associated H3K36me3 mark at Sμ being detectable and remaining unchanged after stimulation to undergo CSR (Balter et al., 2012; Daniel et al., 2010; Dayal et al., 2011; Wang et al., 2009a). Furthermore, a study from mice lacking the Sμ tandem repeat sequences showed that a shift in H3K4me3 and H3 acetylation patterns correlated with accessibility to the switch regions and suggested that chromatin accessibility is not strictly dependent on the underlying DNA sequence but may instead be controlled by a combination of promoter location, the extent of RNA Pol II association, and histone modifications (Balter et al., 2012; Min et al., 2005).
In contrast to Sμ, H3K4me3, H3K36me3, and RNA Pol II are found at the downstream Igh switch regions only after B cell stimulation (Daniel et al., 2010; Wang et al., 2009a). In activated B cells, these histone marks at the Igh locus are independent of AID (Daniel et al., 2010; Wang et al., 2009a), consistent with their association with transcription rather than with DNA DSBs (Ruthenburg et al., 2007). One particular feature of H3K4me3 at Sμ and the downstream switch regions is that the peak is localized from the promoter-proximal initiator exon to the end of the switch region, although not accumulating as significantly as RNA Pol II (Daniel et al., 2010; Wang et al., 2009a). This is in contrast to the localization pattern genome-wide studies have shown for H3K4me3, with the profile generally being restricted to within 2kb of the TSS (Wang et al., 2009b). Indeed, the peak of H3K4me3 at Igh-γ3 is among the most broad of all H3K4me3 peaks in LPS-stimulated B cells demarcating near 7kb of DNA (Daniel et al., 2010).
Analogous to the RAG2/H3K4me3 co-localization at many sites in the genome of developing lymphocytes undergoing V(D)J recombination (Schatz and Swanson, 2011), AID surprisingly also localizes at many sites in the genome of activated mature B cells, as demonstrated by ChIP-Seq analyses (Yamane et al., 2011). In this report, AID was found to associate with chromatin in the vicinity of nearly 6,000 genes, raising questions as to whether AID has off-target activity or additional functions beyond antibody diversification. Interestingly, histone acetylation and H3K4me marks were significantly enriched at sites of AID localization, as was RNA Pol II and transcriptional activity (Pavri and Nussenzweig, 2011; Yamane et al., 2011). There is also evidence from other groups that SUV39H1-mediated H3K9me3, which normally is associated with gene repression, may function in AID targeting to the Igh locus (Bradley et al., 2006; Chowdhury et al., 2008; Jeevan-Raj et al., 2011; Kuang et al., 2009). Nevertheless, as demonstrated by the widespread AID-induced DNA damage in mice lacking the chromatin-associated DDR factor 53BP1 (as discussed later in the review) (Klein et al., 2011; Yamane et al., 2011), AID activity correlates with an accessible chromatin configuration.
Chromatin modifying activities promoting accessibility for AID
One insight into how chromatin modifying activities may promote accessibility for AID stems from the study of the PTIP protein (Pax transactivation domain-interacting protein), which harbors six BRCT domains and is implicated in both gene expression and the DDR (Daniel and Nussenzweig, 2012; Munoz and Rouse, 2009). The PTIP component of an MLL-like H3K4 methyltransferase complex was shown to be critical for promoting H3K4me3, histone acetylation, and germ-line transcription at Igh switch regions, leading to CSR of multiple isotypes (Daniel et al., 2010). Specifically, primary B cells from B lymphocyte-specific PTIP conditional deletion mice displayed defects in IgG3, IgG2b, and IgG1 class-switching concomitant with loss of RNA Pol II association and initiating transcripts at the γ3, γ2b, and γ1 downstream switch regions, respectively (Daniel et al., 2010). As PTIP stably associates with a subset of MLL-like complexes that contain the MLL3/KMT2C and MLL4/KMT2B methyltransferases and display activity for histone H3K4 (Munoz and Rouse, 2009), the study established that specific chromatin changes may in fact control the accessibility of the Igh locus for CSR (Daniel et al., 2010; Schwab et al., 2011). Moreover, it was recently shown that PTIP also controls accessibility of the T cell receptor α locus during V(D)J recombination (Callen et al., 2012). In both cases, it remains to be determined whether H3K4me3 precedes transcription or vice versa. Thus, the clustered, highly repetitive, and tightly repressed gene segments within the Igh and Tcra loci, which normally lack RNA Pol II and H3K4me3, require PTIP to promote the needed DSB targeting of the locus.
A different study using the CH12 B cell line provided genetic evidence using RNAi knock-down that MLL-like methyltransferase activities are dispensable for Igh germ-line transcription at the α switch region, but are required for H3K4me3, DSB formation, and CSR at the Sα (Begum et al., 2012; Stanlie et al., 2010). From this study, H3K4me3 was proposed to serve as a mark for recruiting the recombinase machinery for CSR independently of its function in transcription (Stanlie et al., 2010). Thus, further investigation is needed to clarify the role(s) of H3K4me3 in CSR.
Processing of AID-induced DNA mutations and DSB formation
As a cytosine deaminase, multiple laboratories have established that AID acts on ssDNA and has an activity about 8-fold higher for cytosine compared to 5-methylcytosine (5mC), with no activity for 5-hydroxymethylcytosine (5hmC) (Di Noia and Neuberger, 2007; Franchini et al., 2012; Nabel et al., 2012). As an epigenetic mark on DNA as opposed to histones, 5mC accounts for about 4% of cytosine bases in the mammalian genome and typically occurs as CpG dinucleotides that can stably silence expression of a gene (Law and Jacobsen, 2010). A potential role for AID in DNA demethylation has been proposed although the biological significance of this activity for B lymphocytes or other cell types remains to be established (Fritz and Papavasiliou, 2010). Furthermore, it remains unclear how methylated CpG motifs in genomic DNA might be sufficiently targeted by deaminases that prefer ssDNA. The available data currently support a model in which AID deaminates cytosine bases in DNA to generate uracil and deaminates 5mC at low levels to generate thymine, both of which lead to dT/dA transitions from dC/dG (Figure 3).
Figure 3. Mutagenic consequences of AID-mediated deamination of cytosine and 5mC if not excised during subsequent cell cycles.
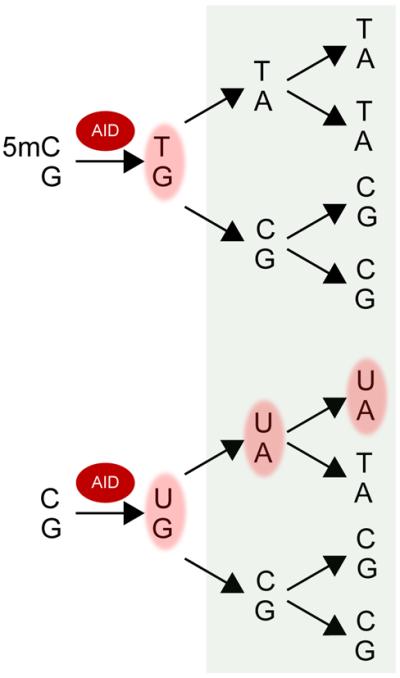
AID catalyzes the deamination of cytosine nearly 10-fold higher than 5mC. Deamination events catalyzed by AID would normally be recognized and excised by the BER and/or MMR machinery, and either repaired in an error-free manner, repaired in an error-prone manner with translesion polymerases, or left unrepaired as a single-strand break. Base pairs shaded in pink are mispaired and would be recognized by repair machineries. Shaded in green to the right are the consequences of an initial DNA lesion that failed to be excised and was transmitted to daughter cells. Note that the consequences are nearly identical.
Great progress has been made to understand the mechanisms explaining how AID-mediated cytosine deamination leads to SHM or CSR (Di Noia and Neuberger, 2007). Powerful genetic evidence for the role of the base excision repair (BER) machinery in processing of AID-induced DNA lesions stems from the identification of mutations in the gene encoding uracil DNA glycosylase, UNG, from a subset of hyper-IgM syndrome patients (Imai et al., 2003). Furthermore, genetic ablation of UNG in mice leads to the detection of uracil in the DNA of immunoglobulin genes, a significant increase in transition mutations at dC/dG pairs without affecting dA/dT pairs, and reduced SHM and CSR (Maul et al., 2011; Rada et al., 2002). These data suggest that uracil excision at Igh-V and switch regions is inhibited by UNG deficiency and that replication over increased dU/dG lesions leads to dT/dA mutations (Figure 3). Mice and cells deficient in the Ape1 apurinic/apyrimidinic endonuclease of the BER pathway have also been shown to display reduced CSR, suggesting that abasic sites on opposite strands may cause single-strand breaks that, when sufficiently close, lead to DSBs (Masani et al., 2013; Stavnezer et al., 2008). In addition, there is evidence from mouse models deficient in MSH2, MSH6, MLH1, MLH3, PMS1, PMS2, or EXO1 that the mismatch repair (MMR) machinery also functions to promote CSR and dA/dT mutations during SHM (Di Noia and Neuberger, 2007; Stavnezer et al., 2008).
Overlapping roles for the BER and MMR pathways in generating antibody diversity were demonstrated by showing that a combined deficiency in both pathways, as shown by Ung−/−Msh2−/− and Ung−/−Msh6−/− mice, leads to a complete ablation of CSR and a complete loss of mutations at dA/dT during Igh-V SHM (Rada et al., 2004; Shen et al., 2006). The mutation spectra observed in Ung−/−Msh2−/− B cells stimulated ex vivo established that AID targets both strands of DNA subsequent to initiation of cytokine-directed germ-line transcription at switch regions (Xue et al., 2006). Moreover, while so-called R loop secondary structures of G-rich sequences in the non-template strands of the switch repeats may enhance CSR as proposed (Yu et al., 2003), they do not appear to be strictly required for AID targeting since V gene segments mutated during SHM are not GC-rich nor were R loops detected at this region in primary B cells (Pavri and Nussenzweig, 2011). These data support the model in which AID-mediated dU/dG mismatches can cause strand-symmetric mutations either by being replicated over during S phase (Figure 3) or by being substrates for BER and MMR pathways. Abasic sites and stretches of ssDNA caused by EXO1-mediated resection around the abasic site could be repaired by low fidelity trans-lesion polymerases creating mutations at both dC/dG and dA/dT pairs leading to the observed SHM spectra or cause staggered single-strand breaks within switch regions that lead to the DSBs required for CSR (Stavnezer et al., 2008). DSBs at Igh are dependent on AID and generated during the G1 phase of the cell cycle, as demonstrated both by immunocytochemistry-FISH analyses of γ-H2AX or Nbs1 foci co-localizing with the Igh locus in B cells actively undergoing CSR (Petersen et al., 2001). However, since AID is expressed throughout the cell cycle and could presumably generate DNA damage in both G1 and S/G2, it remains unclear why the generation of AID-dependent DSBs is restricted to the G1 phase. Moreover, there is no clear evidence whether chromatin modifications are involved in the processing of AID-induced lesions; for example, neither H2AX nor 53BP1 is required for SHM (Nussenzweig and Nussenzweig, 2010).
Repairing AID-induced DNA breaks in the context of chromatin
Once an AID-mediated break is generated during CSR, the joining of a DSB from one switch region, for example Sμ, to a DSB within a downstream switch region requires the NHEJ pathway. How directional joining for productive CSR is imposed, instead of non-physiological inversional rearrangements, remains an open question (Boboila et al., 2012; de Villartay et al., 2003; Nussenzweig and Nussenzweig, 2010). Much of our understanding of the NHEJ pathway stems from its function in V(D)J and class switch recombination. It is now clear that the so-called `classical' NHEJ pathway (C-NHEJ) involving KU70, KU80, DNAPKcs, Artemis, XRCC4, and DNA Ligase IV acts throughout the cell cycle and is essential for completion of V(D)J recombination, as single knock-out mice for these factors display complete blocks in early B and T lymphocyte development (Boboila et al., 2012). This pathway also plays a significant role in CSR, although residual class-switching is observed with genetic deficiency in any of the C-NHEJ factors (Boboila et al., 2012). These and other observations have revealed the existence of `alternative' end-joining (A-EJ) mechanisms (Boboila et al., 2012). While the C-NHEJ pathway typically joins two DSBs together with minimal processing, joins formed in the absence of C-NHEJ usually display short microhomologies that may have guided repair of a DSB after resection of the DNA end by 5–25 nucleotides (Figure 4). Further elucidation of these A-EJ mechanisms clearly warrants additional investigation, particularly since A-EJ has been implicated in the formation of oncogenic translocations found in lymphoid cancer (Gostissa et al., 2011; Zhang and Jasin, 2011). Unlike the error-prone NHEJ pathways, an error-free pathway for DNA repair is homologous recombination (HR), which occurs only in the S/G2 phases of the cell cycle when there exists a sister chromatid or homologous chromosome to use as a template for repair.
Figure 4. The γH2AX/MDC1 and H4K20me/53BP1 interactions suppress DNA end resection and promote repair of CSR-associated breaks by NHEJ.
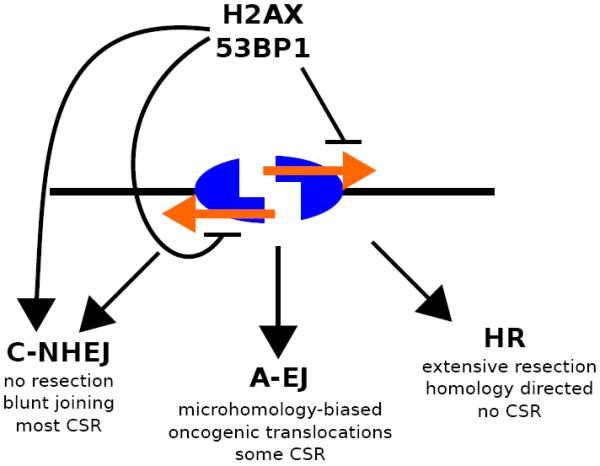
Under normal conditions, these chromatin-mediated interactions facilitate synapsis and repair of two different broken switch regions. These CSR joins mediated by the C-NHEJ pathway show little evidence of resected DSB ends and constitute the majority of successful CSR. In the absence of C-NHEJ, A-EJ mediates some level of CSR but also mediates aberrant joining in the form of chromosomal translocations. In the absence of either H2AX or 53BP1, extensive resection occurs on unrepaired DSBs, thereby inhibiting NHEJ pathways, and ultimately leading to repair of the DSB by HR without class-switching. Orange arrows indicate 5' to 3' DNA end resection of a DSB incurred within an Igh switch region.
Upon DSB formation, the DDR involves PTMs of many proteins to signal for the repair of the DSB; thus far, these include phosphorylation, methylation, acetylation, ubiquitination, sumoylation, and poly(ADP)-ribosylation (Ciccia and Elledge, 2010; Lukas et al., 2011; Polo and Jackson, 2011). For example, the RNF8 and RNF168 ubiquitin ligases initiate a cascade of ubiquitination at sites of DSBs that functions, at least in part, to accumulate 53BP1 and BRCA1 to DNA breaks (Jackson and Durocher, 2013). In addition, while poly(ADP)-ribosylation catalyzed by poly(ADP)-ribosylation polymerases (PARPs) functions in single-strand break repair, this PTM may facilitate DSB repair by transcriptional silencing of the chromatin flanking the damaged sites (Lukas et al., 2011). Protein modules that recognize PTMs can help to target a protein to sites of DNA damage. For example, the BRCT and FHA domains found in several DNA repair proteins are phospho-protein recognition domains (Polo and Jackson, 2011), while the Tudor domain is a methyl-lysine recognition domain (Daniel et al., 2005).
To date, there are two well-established direct protein interactions important for DNA repair during CSR that involve recognition of histone PTMs. The MDC1 protein contains a BRCT domain that associates with the histone variant H2AX phosphorylated at S139 (γ-H2AX) and the tandem Tudor domains of 53BP1 recognize histone H4K20me1 and me2 (Downs et al., 2007). γ-H2AX is a hallmark of DSBs, becoming phosphorylated seconds after IR or laser-induced DNA damage (Bonner et al., 2008). As an integral nucleosomal component of chromatin, γ-H2AX functions as a haploinsufficient tumor suppressor, at least in part, by promoting the accumulation of many DDR factors to sites of DNA damage (Bassing et al., 2003; Celeste et al., 2003). Cells and mice lacking H2AX display general and Igh-associated genomic instability and a mild defect in CSR (Celeste et al., 2002; Franco et al., 2006). 53BP1 also functions as a haploinsufficient tumor suppressor, whose disruption causes a profound defect in CSR (Manis et al., 2004; Reina-San-Martin et al., 2007; Ward et al., 2005; Ward et al., 2004), and strikingly rescues HR, PARP inhibitor sensitivity, and early embryonic lethality of Brca1 nullizygous cells and mice (Bouwman et al., 2010; Bunting et al., 2012; Bunting et al., 2010; Cao et al., 2009). A Tudor domain point mutation that disrupts H4K20 methyl-lysine recognition abrogates chromatin association of 53BP1 and phenocopies complete 53BP1 deletion with respect to all phenotypes tested including CSR (Bothmer et al., 2011). Accumulation of 53BP1 at sites of DNA damage is dependent on the H2AX/MDC1/RNF8/RNF168-mediated ubiquitination pathway; nevertheless, these upstream mediators only display mild defects in CSR (Jackson and Durocher, 2013). These results suggest that the critical function of 53BP1 in CSR is mediated independently from its IR-induced accumulation at DNA breaks; however, this interpretation may not be straightforward since RNF8/RNF168 also mediate the recruitment of BRCA1, which can potentially antagonize and oppose 53BP1.
Current data suggest that the functions of the γ-H2AX/MDC1 and H4K20me/53BP1 interactions during CSR serve at least two similar purposes (Figure 4). As the deficiency of either factor leads to recombination defects between different switch regions without a so-called intra-switch recombination defect between the same switch region, one similar function is to mediate long-range switch recombination of DSBs, which can be nearly 100kbp apart from each other (Bassing et al., 2003; Reina-San-Martin et al., 2007; Reina-San-Martin et al., 2003). Interestingly, while 53BP1 is nearly essential for CSR under normal AID-dependent conditions, only a subtle defect in CSR is observed in 53BP1−/− B cells when DNA breaks at Sμ and Sγ1 are generated with gene-targeted knock-in loxp or I-SceI restriction sites near both switch regions in the absence of AID (Bothmer et al., 2010). This result is consistent with 53BP1−/− cells showing very little evidence of spontaneous genomic instability outside of the Igh locus (Difilippantonio et al., 2008; Franco et al., 2006; Ward et al., 2004) and support the notion that 53BP1 function is not an inherent aspect of repairing a DSB but is instead important for carrying out the CSR reaction with DNA breaks incurred from AID-induced damage. Thus, for productive CSR, long-range recombination between two switch regions must be favored over intra-switch recombination; however, with the exception of involving H2AX and 53BP1, the mechanistic basis remains largely unanswered.
Another function for the chromatin-associated γ-H2AX/MDC1 and H4K20me/53BP1 interactions is to limit exonuclease-mediated DNA end resection of CSR-associated breaks, which suppresses DNA repair both by homologous recombination and by alternative end-joining pathways characterized by increased junctional microhomology and formation of chromosomal translocations (Bothmer et al., 2010; Bunting et al., 2010; Gostissa et al., 2011; Helmink et al., 2011). The suppression of DNA end resection by H2AX or 53BP1 has been shown by comparing the number of resected nucleotides observed in CSR-associated joins from mutant and control stimulated B cells, at both the endogenous Igh locus and at an integrated site-specific DNA break (Bothmer et al., 2010; Bunting et al., 2010). Consistent with these data, G1 arrested developing lymphocytes from H2AX or 53BP1 deficient mice have been shown to display increased exonucleolytic processing at V(D)J-associated DNA breaks (Difilippantonio et al., 2008; Helmink et al., 2011). Moreover, inhibition of ATM or CtIP can partially rescue the resection and class-switching defects in H2AX or 53BP1 deficient cells, an activity that appears most apparent in G1 phase cells (Bothmer et al., 2010; Bothmer et al., 2013; Helmink et al., 2011; Yamane et al., 2013). Even more convincing is the dramatic association of the RPA ssDNA-binding protein observed by ChIP-Seq at Igh switch regions undergoing CSR in the absence of H2AX or 53BP1 (Bunting et al., 2012; Yamane et al., 2011; Yamane et al., 2013). While robust γH2AX accumulation can be found at the Igh locus in normal G1 phase B cells undergoing CSR (Petersen et al., 2001), the majority of RPA is recruited to the Igh locus in the S and G2/M phases of the cell cycle (Yamane et al., 2013). Importantly, the extensive DNA end resection observed in H2AX or 53BP1 deficient B cells is not a general phenomenon with all NHEJ mutants, as KU70 deficiency shows only a subtle increase in RPA association at switch regions (Bunting et al., 2012). Thus, unrepaired CSR-associated DSBs in H2AX−/− or 53BP1−/− B cells persist into S phase and their increased exonucleolytic processing appears to contribute, at least to some extent, to the CSR defect. As resection progresses, DSBs may then be repaired by A-EJ for a non-productive intra-switch recombination (also called an internal switch deletion) or by HR from an undamaged homologous template to try CSR in the subsequent G1 phase (Figure 4).
Even though both H2AX or 53BP1 deficiencies show evidence of increased DNA end resection, only 53BP1 deletion can rescue the PARP inhibitor sensitivity observed with Brca1 deficiency (Bothmer et al., 2011). One recent clue as to why deletion of 53BP1, but not H2AX, rescues Brca1 deficiency and displays a much more severe CSR phenotype comes from the finding that phosphorylation of 53BP1 is critical for CSR and blocking resection (Bothmer et al., 2011; Di Virgilio et al., 2013). Phosphorylated 53BP1 stabilizes RIF1 to DSBs, which appears to help ensure that NHEJ predominates over HR (Chapman et al., 2013; Di Virgilio et al., 2013; Escribano-Diaz et al., 2013; Zimmermann et al., 2013). Even so, it still remains largely a mystery why 53BP1 is so important for CSR, when its deficiency only shows a subtle defect in the repair of IR-induced DNA breaks. Knowledge gained from a better understanding of 53BP1 and H2AX in CSR will provide additional insight to their general functions as chromatin-binding factors that maintain genome stability.
A factor that directly interacts with both γ-H2AX and phosphorylated 53BP1 is the 6 BRCT domain-containing PTIP protein (Munoz and Rouse, 2009; Williams et al., 2010; Yan et al., 2011). One of its tandem BRCT domains (BRCT5/6) interacts directly with γ-H2AX (Yan et al., 2011), while the folding of both BRCT3/4 and BRCT5/6 tandom BRCT domains appear to direct interaction with phosphorylated 53BP1 (Gong et al., 2009; Manke et al., 2003; Munoz et al., 2007). IR-induced foci formation of PTIP is dependent on the γ-H2AX/MDC1/RNF8 pathway and is completely abrogated by a single point mutation in BRCT3 of PTIP (Daniel et al., 2010; Gong et al., 2009; Munoz et al., 2007). In stimulated B cells, PTIP accumulation at DSBs was found to contribute to CSR and genome stability independently of its aforementioned role in promoting Igh germ-line transcription at switch regions (Daniel et al., 2010). As such, PTIP is a rare example of a protein that functions both in the transcription and DSB repair phase of a single recombination event (Callen et al., 2012; Daniel et al., 2010). While interaction with γ-H2AX may stabilize PTIP to DNA breaks under conditions still to be determined, RNF8-mediated ubiquitin conjugates appear to amplify 53BP1 which, together with other phosphorylated proteins, accumulate PTIP to sites of DNA damage (Gong et al., 2009; Munoz et al., 2007). As the radiosensitivity phenotype of PTIP-deficient cells is mild, additional elucidation of its potential functions in maintaining genome stability and the significance of its direct interactions with γ-H2AX and phosphorylated 53BP1 warrant further investigation.
Widespread AID-induced breaks and tumorigenesis
Uncontrolled AID activity has been shown to cause genomic instability and tumorigenesis with studies employing genetic disruption or over-expression of AID (Muramatsu et al., 2007; Robbiani and Nussenzweig, 2012). Increased AID expression also correlates with higher levels of autoantibodies in several mouse models of lupus and rheumatoid arthritis (Xu et al., 2012). The data support a model in which B cells incurring AID-induced formation of oncogenic chromosomal translocations do not normally survive unless a DNA damage checkpoint pathway that normally eliminates cells with unrepaired DSBs and oncogenic translocations is impaired (Nussenzweig and Nussenzweig, 2010). Consistent with this model, the genomic instability in H2AX−/− mice does not lead to cancer unless a copy of the p53 apoptotic checkpoint tumor suppressor is disrupted (Bassing et al., 2003; Celeste et al., 2003). Similarly, survival of 53BP1−/− mice is greatly compromised upon p53 deficiency (Nussenzweig and Nussenzweig, 2010). Outside of these H2AX and 53BP1 studies, strong evidence for how chromatin structure or regulators thereof may affect the formation of chromosomal translocations is lacking.
Recent work has explored the extent and nature of AID-induced mutations and DSBs at genes outside of the Igh locus. Genetic experiments with knock-in mice have demonstrated that AID is required for the DSB at c-myc that leads to the c-myc/Igh chromosomal translocation found in Burkitt's lymphoma (Robbiani and Nussenzweig, 2012). In addition to MYC, many other genes have been found to be mutated by AID from analysis of genomic DNA sequences from Ung−/−Msh2−/− B cells (Liu et al., 2008; Pavri and Nussenzweig, 2011; Robbiani and Nussenzweig, 2012; Yamane et al., 2011). Deep sequencing analyses of AID localization in stimulated B cells have also provided unmatched views of AID-induced DNA damage at non-Igh loci (Yamane et al., 2011). It has proposed that off-target AID sites are repaired by HR (Hasham et al., 2010); however, significant association of RPA and RAD51, as markers of HR-mediated repair, were only found at the Igh switch regions undergoing class-switching and not other loci, suggesting that AID recruitment is not sufficient for AID-induced DSB resection (Yamane et al., 2011; Yamane et al., 2013). Thus, while AID promotes somatic hypermutation at many genes (Liu et al., 2008; Muramatsu et al., 2007; Robbiani et al., 2009; Yamane et al., 2011), AID-induced DSBs do not appear to occur frequently or to load significant levels of RPA (Petersen et al., 2001; Yamane et al., 2011). Low levels of AID-induced DNA damage at non-Igh loci have been proposed to be protected from mutations and genomic instability by high fidelity error-free repair mechanisms (Liu et al., 2008; Yamane et al., 2011) and additional investigation is needed to understand how error-prone and error-free DNA repair pathways are targeted to Igh and non-Igh loci, respectively.
While AID-induced DSBs at non-Igh loci do not appear to occur frequently in normal dividing B cells, widespread DSBs caused by AID-induced DNA damage have recently been visualized with certain genetic manipulations. By increasing the mutation load with AID over-expression and increasing DNA end resection with 53BP1 deficiency, AID-induced DNA damage was observed in AIDtg53BP1−/− mice using RPA or RAD51 ChIP-Seq at about 150 genes (Hakim et al., 2012; Yamane et al., 2013). Using deep sequencing technologies, approaches to isolate junctions between a chromosomal DSB introduced at a fixed site and other sequences in primary B cells have revealed that DSBs translocate widely across the B cell genome and prefer to be resolved to a break on the same chromosome. Moreover, translocations were preferentially targeted to transcribed regions, most significantly at 2kb around TSSs (Chiarle et al., 2011; Klein et al., 2011; Zhang et al., 2012), demonstrating that the chromatin environment and/or transcriptional activity are key factors influencing the ability of two DSBs to translocate. Even though AID-dependent SHM at the immunoglobulin variable regions has not been previously detected from B cells stimulated ex vivo (Liu and Schatz, 2009), likely from the result of error-free repair activity (Liu et al., 2008), the sensitivity of translocation sequencing methods has revealed translocation partners fused to the Igh variable region, although their dependency on AID was not investigated (Klein et al., 2011). These sequencing experiments also revealed the surprising identification of translocations, albeit at low levels, at Sμ and Sγ1 in AID−/− B cells, indicating that DSBs at Igh can occur at a low frequency in the absence of AID (Chiarle et al., 2011). Among the translocation `hotspots' to the Igh locus, the number of translocations per hotspot was directly proportional to the amount of chromatin-associated RPA or RAD51 at these regions (Hakim et al., 2012; Yamane et al., 2013). These results not only suggest that Igh translocates to many sites in the genome that incur DSBs but also that, in addition to proximity, frequent DSBs drive recurrent translocations. All together, the data provide indisputable evidence that AID-induced mutation can lead to widespread DSBs and formation of chromosomal translocations and begin to address how the chromatin environment impacts aberrant resolution of DNA breaks.
Other implications for chromatin in AID-independent replication stress
CSR-associated DNA breaks are normally resolved in the G1 phase of the cell cycle, but can persist into the S and G2/M phases and be visualized cytogenetically in metaphase spreads upon disruption of one of a number of DDR or C-NHEJ factors (Boboila et al., 2012; Nussenzweig and Nussenzweig, 2010). A recent study visualized the persistence of the CSR-associated breaks in each cell cycle phase by employing ChIP-Seq of γ-H2AX and RPA from stimulated B cells sorted based on their DNA content (Yamane et al., 2013). The persistence of AID-mediated DNA breaks into S phase allows the possibility of these breaks to aberrantly join with DSBs generated as a result of replication stress. Indeed, many recurrent mutations in B cell lymphoma are not associated with AID activity (Robbiani and Nussenzweig, 2012; Rui et al., 2011); however, genes that are large and highly transcribed are thought to cause problems for the replication machinery and be a source of DSBs that must be properly cared for to suppress tumorigenesis (Helmrich et al., 2011; Prado and Aguilera, 2005). Using an unbiased approach to discover sites of recurrent DNA lesions during early replication through analyses of γ-H2AX, RPA, BRCA1, and SMC5 ChIP-Seq localization data, a new class of early replication fragile sites (ERFS) was recently identified in stimulated B cells (Barlow et al., 2013). ERFS break spontaneously at a low frequency, but are acutely induced upon treatment with hydroxyurea, ATR inhibition, or in response to oncogenic stress (Barlow et al., 2013). While common fragile sites are characterized by being late replicating and within a condensed chromatin structure marked by histone hypoacetylation (Ozeri-Galai et al., 2012), ERFS replicate early, show hallmarks of open chromatin structure, and are gene-rich (Barlow et al., 2013). Although ERFS breakage is AID-independent, some of the regions that are sensitive to replication stress encompass hotspots for AID-induced damage. Cytogenetic examination of metaphase spreads from stimulated AIDtg53BP1−/− B cells, which allow Igh breaks generated in G1 to persist into S phase, led to the identification of a chromosomal translocation between Igh and an ERFS in primary cells (Barlow et al., 2013). Importantly, greater than 50% of common amplifications/deletions observed in human diffuse large B cell lymphoma map to ERFS (Barlow et al., 2013), suggesting that ERFS may be a significant source of genomic instability that act together with AID-induced DNA damage to promote lymphomagenesis.
One ERFS identified using this approach was Bcl-2 (Barlow et al., 2013), an apoptotic regulator located within the so-called major breakpoint region (Mbr) that contains clusters of CpG dinucleotides, adopts a single-stranded non-B-DNA structure, and is translocated to Igh in 70–95% of follicular lymphomas and in 20–30% of diffuse large B cell lymphomas (Raghavan et al., 2004; Tomita, 2011). The observation that CpG dinucleotides can be found within 40–70% of breakpoints at chromosomal translocations in immature human B cell lymphomas recently led to a proposal that DSBs at Bcl-2 and other genes may arise from sequential action of AID and the structure specific nicking activity of the RAG1/2 complex during early B lymphocyte development (Tsai et al., 2008). Similar to Bcl-2, other ERFS are also enriched for CpG dinucleotides (Barlow et al., 2013). As such, it is possible that replication stress further stabilizes the single stranded conformation of the Bcl-2 Mbr (Tsai et al., 2008), which may subsequently be more prone to slippage and collapse during DNA replication.
Thus, AID-induced DNA damage and replication stress can both lead to widespread DSBs that, if not repaired by the NHEJ or HR pathways, can become partners for chromosomal translocations. While chromosomal translocations occur regularly in primary dividing cells, only upon deregulation of cell growth or survival will the translocation lead to tumorigenesis. It is clear that highly transcribed genes displaying H3K4me3, H3ac, and H3K36me3 are prone to breakage and forming translocations (Barlow et al., 2013; Klein et al., 2011), but whether or not the histone marks directly aid in DSB formation or aberrant repair remains unclear. Thus, future work will determine the mechanisms for how chromatin regulates the repair of AID-induced and replication-induced DNA damage.
Concluding remarks
Chromatin biology touches on all facets of developmental and disease biology and the DNA repair field is no exception. The ever-increasing number of proteins implicated in the DNA damage response continues to call for detailed genetic studies to clearly determine which proteins and PTMs are physiologically relevant and which may potentially be targeted for therapeutics. With respect to accessibility for AID, whether a specific chromatin-mediated activity is required for this DNA rearrangement event remains to be clarified. Moreover, whether or not germ-line transcripts are the cause or the effect of chromatin changes at antigen receptor loci remains unclear. With respect to recombination and repair of Igh-associated DNA breaks, it is clear that histone H2AX and methyl-lysine recognition by 53BP1 play critical roles within the context of chromatin; however, we have only scratched the surface with much still to be learned. For example, we understand next to nothing about the roles of so many of the other myriad histone PTMs (Tan et al., 2011). Since multiple genes encode most histones, generating useful mammalian models with a single amino acid point mutation in a histone has not been feasible. Instead, we have the technological means now to explore this exciting avenue of research with detailed mechanistic studies in physiologically relevant model systems using deep sequencing and proteomics, which promise to reveal new insight. Along the way, we will also find out whether AID has additional functions beyond antibody diversification.
Acknowledgments
We thank Rafael Casellas and Joan Yuan for critical reading of the manuscript and apologize for not being able to cite more primary literature because of space limitations. Work in the laboratory of J.A.D. is supported by the Novo Nordisk Foundation and the Danish Council for Independent Research in Medical Sciences. Work in the laboratory of A.N. is supported by the Intramural Research Program of the National Institutes of Health, the National Cancer Institute, and the Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152:417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balter BB, Ciccone DN, Oettinger MA, Selsing E. Mice lacking Smu tandem repeats maintain RNA polymerase patterns but exhibit histone modification pattern shifts linked to class switch site locations. Molecular immunology. 2012;52:1–8. doi: 10.1016/j.molimm.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow JH, Faryabi RB, CallÈn E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun H-W, McKinnon P, Wright G, et al. Identification of Early Replicating Fragile Sites that Contribute to Genome Instability. Cell. 2013 doi: 10.1016/j.cell.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum NA, Stanlie A, Nakata M, Akiyama H, Honjo T. The histone chaperone Spt6 is required for activation-induced cytidine deaminase target determination through H3K4me3 regulation. The Journal of biological chemistry. 2012;287:32415–32429. doi: 10.1074/jbc.M112.351569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Advances in immunology. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nature reviews. Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207:855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Rommel PC, Gazumyan A, Polato F, Reczek CR, Muellenbeck MF, Schaetzlein S, Edelmann W, Chen PL, Brosh RM, Jr., et al. Mechanism of DNA resection during intrachromosomal recombination and immunoglobulin class switching. J Exp Med. 2013;210:115–123. doi: 10.1084/jem.20121975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature structural & molecular biology. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SP, Kaminski DA, Peters AH, Jenuwein T, Stavnezer J. The histone methyltransferase Suv39h1 increases class switch recombination specifically to IgA. J Immunol. 2006;177:1179–1188. doi: 10.4049/jimmunol.177.2.1179. [DOI] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. BRCA1 Functions Independently of Homologous Recombination in DNA Interstrand Crosslink Repair. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Faryabi RB, Luckey M, Hao B, Daniel JA, Yang W, Sun HW, Dressler G, Peng W, Chi H, et al. The DNA damage- and transcription-associated protein paxip1 controls thymocyte development and emigration. Immunity. 2012;37:971–985. doi: 10.1016/j.immuni.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell. 2009;35:534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol Cell. 2013 doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury M, Forouhi O, Dayal S, McCloskey N, Gould HJ, Felsenfeld G, Fear DJ. Analysis of intergenic transcription and histone modification across the human immunoglobulin heavy-chain locus. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15872–15877. doi: 10.1073/pnas.0808462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb RM, Oestreich KJ, Osipovich OA, Oltz EM. Accessibility control of V(D)J recombination. Advances in immunology. 2006;91:45–109. doi: 10.1016/S0065-2776(06)91002-5. [DOI] [PubMed] [Google Scholar]
- Daniel JA, Nussenzweig A. Roles for histone H3K4 methyltransferase activities during immunoglobulin class-switch recombination. Biochimica et biophysica acta. 2012;1819:733–738. doi: 10.1016/j.bbagrm.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, Pray-Grant MG, Grant PA. Effector proteins for methylated histones: an expanding family. Cell Cycle. 2005;4:919–926. doi: 10.4161/cc.4.7.1824. [DOI] [PubMed] [Google Scholar]
- Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, Filsuf D, Chen HT, Gazumyan A, Yamane A, et al. PTIP Promotes Chromatin Changes Critical for Immunoglobulin Class Switch Recombination. Science. 2010 doi: 10.1126/science.1187942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayal S, Nedbal J, Hobson P, Cooper AM, Gould HJ, Gellert M, Felsenfeld G, Fear DJ. High resolution analysis of the chromatin landscape of the IgE switch region in human B cells. PloS one. 2011;6:e24571. doi: 10.1371/journal.pone.0024571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villartay JP, Fischer A, Durandy A. The mechanisms of immune diversification and their disorders. Nature reviews. Immunology. 2003;3:962–972. doi: 10.1038/nri1247. [DOI] [PubMed] [Google Scholar]
- Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annual review of biochemistry. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. Rif1 Prevents Resection of DNA Breaks and Promotes Immunoglobulin Class Switching. Science. 2013 doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JA, Nussenzweig MC, Nussenzweig A. Chromatin dynamics and the preservation of genetic information. Nature. 2007;447:951–958. doi: 10.1038/nature05980. [DOI] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol Cell. 2013 doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- Franchini DM, Schmitz KM, Petersen-Mahrt SK. 5-methylcytosine DNA demethylation: more than losing a methyl group. Annual review of genetics. 2012;46:419–441. doi: 10.1146/annurev-genet-110711-155451. [DOI] [PubMed] [Google Scholar]
- Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21:201–214. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Fritz EL, Papavasiliou FN. Cytidine deaminases: AIDing DNA demethylation? Genes Dev. 2010;24:2107–2114. doi: 10.1101/gad.1963010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Cho YW, Kim JE, Ge K, Chen J. Accumulation of Pax2 transactivation domain interaction protein (PTIP) at sites of DNA breaks via RNF8-dependent pathway is required for cell survival after DNA damage. The Journal of biological chemistry. 2009;284:7284–7293. doi: 10.1074/jbc.M809158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostissa M, Alt FW, Chiarle R. Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu Rev Immunol. 2011;29:319–350. doi: 10.1146/annurev-immunol-031210-101329. [DOI] [PubMed] [Google Scholar]
- Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature. 2012;484:69–74. doi: 10.1038/nature10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriman GR, Bradley A, Das S, Rogers-Fani P, Davis AC. IgA class switch in I alpha exon-deficient mice. Role of germline transcription in class switch recombination. The Journal of clinical investigation. 1996;97:477–485. doi: 10.1172/JCI118438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, Wilpan RY, He Y, King BL, Mills KD. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nature immunology. 2010;11:820–826. doi: 10.1038/ni.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein K, Lorenz MG, Siebenkotten G, Petry K, Christine R, Radbruch A. Processing of switch transcripts is required for targeting of antibody class switch recombination. J Exp Med. 1998;188:2369–2374. doi: 10.1084/jem.188.12.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Tubbs AT, Dorsett Y, Bednarski JJ, Walker LM, Feng Z, Sharma GG, McKinnon PJ, Zhang J, Bassing CH, et al. H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature. 2011;469:245–249. doi: 10.1038/nature09585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Tora L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell. 2011;44:966–977. doi: 10.1016/j.molcel.2011.10.013. [DOI] [PubMed] [Google Scholar]
- Hewitt SL, Chaumeil J, Skok JA. Chromosome dynamics and the regulation of V(D)J recombination. Immunological reviews. 2010;237:43–54. doi: 10.1111/j.1600-065X.2010.00931.x. [DOI] [PubMed] [Google Scholar]
- Imai K, Catalan N, Plebani A, Marodi L, Sanal O, Kumaki S, Nagendran V, Wood P, Glastre C, Sarrot-Reynauld F, et al. Hyper-IgM syndrome type 4 with a B lymphocyte-intrinsic selective deficiency in Ig class-switch recombination. The Journal of clinical investigation. 2003;112:136–142. doi: 10.1172/JCI18161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Durocher D. Regulation of DNA Damage Responses by Ubiquitin and SUMO. Mol Cell. 2013;49:795–807. doi: 10.1016/j.molcel.2013.01.017. [DOI] [PubMed] [Google Scholar]
- Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F, Alt FW, Losson R, Reina-San-Martin B. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med. 2011;208:1649–1660. doi: 10.1084/jem.20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jhunjhunwala S, van Zelm MC, Peak MM, Murre C. Chromatin architecture and the generation of antigen receptor diversity. Cell. 2009;138:435–448. doi: 10.1016/j.cell.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K, Reddy KL, Singh H. Molecular pathways and mechanisms regulating the recombination of immunoglobulin genes during B-lymphocyte development. Advances in experimental medicine and biology. 2009;650:133–147. doi: 10.1007/978-1-4419-0296-2_11. [DOI] [PubMed] [Google Scholar]
- Jung S, Rajewsky K, Radbruch A. Shutdown of class switch recombination by deletion of a switch region control element. Science. 1993;259:984–987. doi: 10.1126/science.8438159. [DOI] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodgire P, Mukkawar P, North JA, Poirier MG, Storb U. Nucleosome stability dramatically impacts the targeting of somatic hypermutation. Molecular and cellular biology. 2012;32:2030–2040. doi: 10.1128/MCB.06722-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krangel MS. Mechanics of T cell receptor gene rearrangement. Current opinion in immunology. 2009;21:133–139. doi: 10.1016/j.coi.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang FL, Luo Z, Scharff MD. H3 trimethyl K9 and H3 acetyl K9 chromatin modifications are associated with class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5288–5293. doi: 10.1073/pnas.0901368106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature reviews. Genetics. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Luo Z, Scharff MD. Differential regulation of histone acetylation and generation of mutations in switch regions is associated with Ig class switching. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15428–15433. doi: 10.1073/pnas.0406827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- Liu M, Schatz DG. Balancing AID and DNA repair during somatic hypermutation. Trends in immunology. 2009;30:173–181. doi: 10.1016/j.it.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Lorenz M, Jung S, Radbruch A. Switch transcripts in immunoglobulin class switching. Science. 1995;267:1825–1828. doi: 10.1126/science.7892607. [DOI] [PubMed] [Google Scholar]
- Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nature cell biology. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nature immunology. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Molecular and cellular biology. 2013 doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews AG, Kuo AJ, Ramon-Maiques S, Han S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, Gramlich HS, Schatz DG, Woodgate R, Wilson DM, 3rd, et al. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nature immunology. 2011;12:70–76. doi: 10.1038/ni.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min IM, Rothlein LR, Schrader CE, Stavnezer J, Selsing E. Shifts in targeting of class switch recombination sites in mice that lack mu switch region tandem repeats or Msh2. J Exp Med. 2005;201:1885–1890. doi: 10.1084/jem.20042491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz IM, Jowsey PA, Toth R, Rouse J. Phospho-epitope binding by the BRCT domains of hPTIP controls multiple aspects of the cellular response to DNA damage. Nucleic acids research. 2007;35:5312–5322. doi: 10.1093/nar/gkm493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz IM, Rouse J. Control of histone methylation and genome stability by PTIP. EMBO Rep. 2009;10:239–245. doi: 10.1038/embor.2009.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Nagaoka H, Shinkura R, Begum NA, Honjo T. Discovery of activation-induced cytidine deaminase, the engraver of antibody memory. Advances in immunology. 2007;94:1–36. doi: 10.1016/S0065-2776(06)94001-2. [DOI] [PubMed] [Google Scholar]
- Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, Zhang Y, Kohli RM. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nature chemical biology. 2012;8:751–758. doi: 10.1038/nchembio.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, Yokota Y, Shimizu A. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 2003;302:2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141:27–38. doi: 10.1016/j.cell.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A, Di Noia JM. Activation induced deaminase: how much and where? Seminars in immunology. 2012;24:246–254. doi: 10.1016/j.smim.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Ozeri-Galai E, Bester AC, Kerem B. The complex basis underlying common fragile site instability in cancer. Trends in genetics : TIG. 2012;28:295–302. doi: 10.1016/j.tig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Pavri R, Nussenzweig MC. AID targeting in antibody diversity. Advances in immunology. 2011;110:1–26. doi: 10.1016/B978-0-12-387663-8.00005-3. [DOI] [PubMed] [Google Scholar]
- Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4:57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F, Aguilera A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. The EMBO journal. 2005;24:1267–1276. doi: 10.1038/sj.emboj.7600602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Current biology : CB. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- Raghavan SC, Swanson PC, Wu X, Hsieh CL, Lieber MR. A non-BDNA structure at the Bcl-2 major breakpoint region is cleaved by the RAG complex. Nature. 2004;428:88–93. doi: 10.1038/nature02355. [DOI] [PubMed] [Google Scholar]
- Rajagopal D, Maul RW, Ghosh A, Chakraborty T, Khamlichi AA, Sen R, Gearhart PJ. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med. 2009;206:1237–1244. doi: 10.1084/jem.20082514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B, Chen J, Nussenzweig A, Nussenzweig MC. Enhanced intra-switch region recombination during immunoglobulin class switch recombination in 53BP1−/− B cells. European journal of immunology. 2007;37:235–239. doi: 10.1002/eji.200636789. [DOI] [PubMed] [Google Scholar]
- Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J Exp Med. 2003;197:1767–1778. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, McBride KM, Klein IA, Stone G, Eisenreich TR, et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani DF, Nussenzweig MC. Chromosome Translocation, B Cell Lymphoma, and Activation-induced Cytidine Deaminase. Annual review of pathology. 2012 doi: 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- Rui L, Schmitz R, Ceribelli M, Staudt LM. Malignant pirates of the immune system. Nature immunology. 2011;12:933–940. doi: 10.1038/ni.2094. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annual review of genetics. 2011;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- Schwab KR, Patel SR, Dressler GR. Role of PTIP in class switch recombination and long-range chromatin interactions at the immunoglobulin heavy chain locus. Molecular and cellular biology. 2011;31:1503–1511. doi: 10.1128/MCB.00990-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Poirier MG, Allen MJ, North J, Lal R, Widom J, Storb U. The activation-induced cytidine deaminase (AID) efficiently targets DNA in nucleosomes but only during transcription. J Exp Med. 2009;206:1057–1071. doi: 10.1084/jem.20082678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U. Somatic hypermutation and class switch recombination in Msh6(−/−)Ung(−/−) double-knockout mice. J Immunol. 2006;177:5386–5392. doi: 10.4049/jimmunol.177.8.5386. [DOI] [PubMed] [Google Scholar]
- Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:22190–22195. doi: 10.1073/pnas.1016923108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanlie A, Begum NA, Akiyama H, Honjo T. The DSIF subunits Spt4 and Spt5 have distinct roles at various phases of immunoglobulin class switch recombination. PLoS genetics. 2012;8:e1002675. doi: 10.1371/journal.pgen.1002675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JE, Schrader CE. Mechanism and Regulation of Class Switch Recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annual review of biochemistry. 2011;80:473–499. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita N. BCL2 and MYC dual-hit lymphoma/leukemia. Journal of clinical and experimental hematopathology : JCEH. 2011;51:7–12. doi: 10.3960/jslrt.51.7. [DOI] [PubMed] [Google Scholar]
- Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–1142. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- Vuong BQ, Chaudhuri J. Combinatorial mechanisms regulating AID-dependent DNA deamination: interacting proteins and post-translational modifications. Seminars in immunology. 2012;24:264–272. doi: 10.1016/j.smim.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Whang N, Wuerffel R, Kenter AL. AID-dependent histone acetylation is detected in immunoglobulin S regions. J Exp Med. 2006;203:215–226. doi: 10.1084/jem.20051774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med. 2009a;206:1817–1830. doi: 10.1084/jem.20081678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Schones DE, Zhao K. Characterization of human epigenomes. Curr Opin Genet Dev. 2009b;19:127–134. doi: 10.1016/j.gde.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Difilippantonio S, Minn K, Mueller MD, Molina JR, Yu X, Frisk CS, Ried T, Nussenzweig A, Chen J. 53BP1 cooperates with p53 and functions as a haploinsufficient tumor suppressor in mice. Molecular and cellular biology. 2005;25:10079–10086. doi: 10.1128/MCB.25.22.10079-10086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, et al. 53BP1 is required for class switch recombination. The Journal of cell biology. 2004;165:459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, Russell P. gammaH2A binds Brc1 to maintain genome integrity during S-phase. The EMBO journal. 2010;29:1136–1148. doi: 10.1038/emboj.2009.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmann KL, Milosevic S, Pauklin S, Schmitz KM, Rangam G, Simon MT, Maslen S, Skehel M, Robert I, Heyer V, et al. A role for the RNA pol II-associated PAF complex in AID-induced immune diversification. J Exp Med. 2012;209:2099–2111. doi: 10.1084/jem.20112145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nature reviews. Immunology. 2012;12:517–531. doi: 10.1038/nri3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue K, Rada C, Neuberger MS. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2−/− ung−/− mice. J Exp Med. 2006;203:2085–2094. doi: 10.1084/jem.20061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, Casellas R. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nature immunology. 2011;12:62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane A, Robbiani DF, Resch W, Bothmer A, Nakahashi H, Oliveira T, Rommel PC, Brown EJ, Nussenzweig A, Nussenzweig MC, et al. RPA Accumulation during Class Switch Recombination Represents 5'–3' DNA-End Resection during the S-G2/M Phase of the Cell Cycle. Cell reports. 2013;3:138–147. doi: 10.1016/j.celrep.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Shao Z, Li F, Niu L, Shi Y, Teng M, Li X. Structural basis of gammaH2AX recognition by human PTIP BRCT5-BRCT6 domains in the DNA damage response pathway. FEBS letters. 2011;585:3874–3879. doi: 10.1016/j.febslet.2011.10.045. [DOI] [PubMed] [Google Scholar]
- Yancopoulos GD, Alt FW. Developmentally controlled and tissue-specific expression of unrearranged VH gene segments. Cell. 1985;40:271–281. [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature immunology. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- Zhang J, Bottaro A, Li S, Stewart V, Alt FW. A selective defect in IgG2b switching as a result of targeted mutation of the I gamma 2b promoter and exon. The EMBO journal. 1993;12:3529–3537. doi: 10.1002/j.1460-2075.1993.tb06027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nature structural & molecular biology. 2011;18:80–84. doi: 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 Regulates DSB Repair Using Rif1 to Control 5' End Resection. Science. 2013 doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]