

Abstract
The coupling of O-pivaloyl benzhydroxamic acids with donor/acceptor diazo compounds provides iso-indolones in high yield. The reaction tolerates a broad range of benzhydroxamic acids and diazo compounds including substituted 2,2,2-trifluorodiazo ethanes. Mechanistic experiments suggest that C–H activation is turnover limiting and irreversible, while insertion of the diazo compound favors electron deficient substrates.
Multicomponent reactions catalyzed by transition metals represent a powerful approach for the synthesis of heterocycles.1 This concept has been extensively explored in the Rh(III) catalyzed synthesis of nitrogen containing heterocycles mediated by C–H activation.2 A wide swath of unsaturated heterocycles can be accessed though coupling amides, amines, oximes, and anilines with alkynes to access isoquinolones,3 pyridones,3d,4 isoquinolines,5 pyridines,5g,6 indoles,7 and pyrroles.7b,8
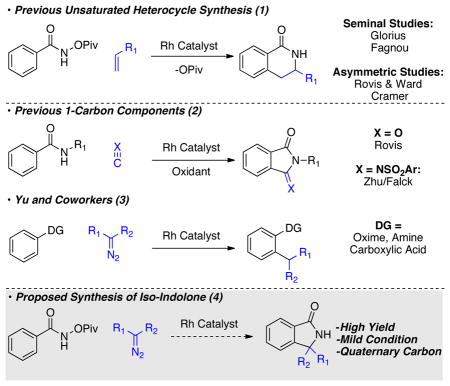
The ability to access nitrogen containing hetero-cycles bearing stereogenic carbons is important due to their prevalence in medicinal targets and natural products.9,10 Recently, Glorius11 and Fagnou3e demonstrated that alkenes can be used to access partially saturated nitrogen containing heterocycles (eq 1). In the presence of an enzyme or chiral cyclopentadiene ligand this reaction can be rendered asymmetric.12 Currently absent from this family of reactivity is the ability to access γ-lactams with the potential to control stereochemistry in the process. The key to achieving this reactivity is identification of a suitable 1-carbon component.
Examples of 1-carbon components in Rh(III) C–H activation reactions are rare. Previously, we found amides could be coupled with carbon monoxide to provide a range of phthalimides in good yield (eq 2).13 Concurrent work by Zhu and Falck found isocyanides to be competent coupling partners in an analogous reaction.14 Carbon monoxide and isocyanides function as 1-carbon components because the carbon has carbenic character. Elegant work by Yu took advantage of the carbenic character of electron deficient diazo-compounds to ortho-functionalize a variety of directing group containing arenes (eq 3).15 Given the importance of γ-lactams, we were interested in using diazo-compounds as 1-carbon components in a cyclization reaction that would provide iso-indolones (eq 4).
We believed a significant barrier to this reactivity would be the need to inhibit protonation of the metal-lacycle. The report by Yu suggests protonation of the rhodacycle occurs rapidly when mono-dentate directing groups are used.15 Fagnou3e and Glorius11 found that use of a bi-dentate directing group precludes β-hydride elimination in the synthesis of dihydroisoquiolones. We imagined that this type of directing group would also be effective toward inhibiting unwanted protonation events. In a set of initial experiments, we chose to explore donor/acceptor diazo-compounds because they are less prone to dimerization while providing interesting products with a stereogenic carbon.
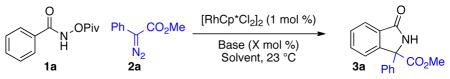
Initial coupling of amide 1a with diazo-ester 2a using super-stoichiometric CsOPiv in MeOH gave the desired product in 24% yield with the remainder of the mass balance being methanolysis of the pivalate ester in the starting material (Table 1, entry 1). A screen of solvents revealed CH3CN to be ideal, providing lactam 3a in 78% yield (Table 1, entry 4). Replacing CsOPiv with CsOAc gave comparable yields (Table 1, entry 5). We were pleased to find that a sub-stoichiometric amount of base did not have a detrimental effect on yield (Table 1, entry 6). Finally, modification of the concentration allowed the desired lactam to be isolated in 81% yield after 150 minutes. The catalyst loading can be decreased to 0.4 mol % when the reaction is performed on gram scale (Table 1, Entry 8). The use of O-methyl hydroxamic acid in place of 1a delivers only 10% yield of the protonated product, but none of the desired product.
Table 1.
Optimization of Reaction Conditions
| |||
|---|---|---|---|
| entrya | base | solvent | yield (%)b |
| 1 | CsOPiv (200 mol %) | MeOH (0.5 M) | 24 |
| 2 | CsOPiv (200 mol %) | EtOH (0.5 M) | 33 |
| 3 | CsOPiv (200 mol %) | TFE (0.5 M) | 59 |
| 4 | CsOPiv (200 mol %) | MeCN (0.5 M) | 78 |
| 5 | CsOAc (200 mol %) | MeCN (0.5 M) | 80 |
| 6 | CsOAc (20 mol %) | MeCN (0.1 M) | 81c |
| 7 | - | MeCN (0.1 M) | 0 |
| 8d | CsOAc (20 mol %) | MeCN (0.1 M) | 75c |
Standard Conditions: 1a (1 equiv), 2a (1 equiv), [RhCp*Cl2]2 (1 mol %).
Yield determined by HPLC.
Isolated Yield.
Catalyst loading (0.4 mol %)
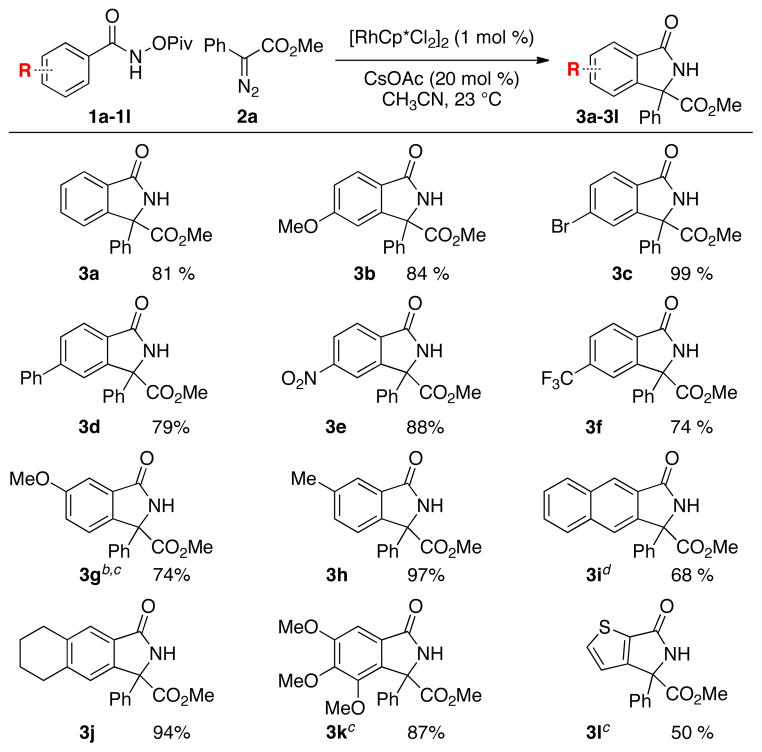
The ideal conditions in hand, the scope of iso-indolone formation was tested with differentially substituted benzhydroxamic acids (Table 2). The reaction affords the desired product with a variety of electronically different para-substituted amides in high yield (Table 2, 3a–3f). Substitution at the meta-position plays an important role in controlling the regioselectivity of the C–H activation event. When meta-methyl benzhydroxamic acid is used (Table 2, 3h), C–H activation occurs exclusively para- to the methyl group presumably to avoid sterically disfavored interactions. However, when a methoxy group is placed at the meta-position (Table 2, 3g), a 1.1:1 ratio of regioisomers is observed. This effect is also observed with naphthyl-amides (Table 2, 3i), where a 2:1 ratio of regioisomers is observed. By comparison, the less electron deficient tetrahydronaphthylene benzhydroxamic acid gives a single regioisomer (Table 2, 3j). Presumably this decrease in regioselectivity can be attributed to the increase in acidity of the ortho C–H bond. Increasing the kinetic acidity of the C–H bond overwhelms the steric bias of the C–H activation to give a mixture of products. The gallic acid derived amide and the heterocyclic substrate also fare well in the reaction, although increased reaction temperatures are required (Table 2, 3k–l).
Table 2.
Benzhydroxamic Acid Scopea
|
For standard reaction conditions see Table 1.
Product isolated as a 1.1:1 ratio of regioisomers.
Reaction conducted at 60 °C.
Isolated as a 2:1 ratio of regioisomers.
Table 3.
Scope of Donor/Acceptor Diazo Compoundsa
|
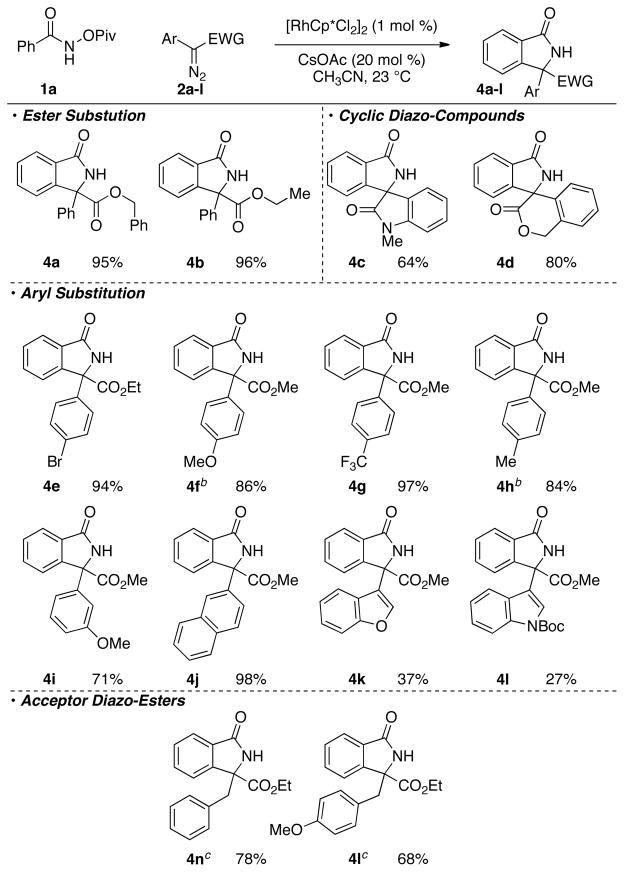
For standard reaction conditions see Table 1.
Reaction conducted at 60 °C.
Reactions conducted at 80 °C.
We moved to explore the scope of diazo-compounds (Table 3). Acceptor diazo-compounds16 dimerize rapidly under the reaction conditions, while acceptor/acceptor diazo compounds are almost completely unreactive, with less than 5% yield observed under these reactions conditions. Donor/acceptor diazo-compounds are broadly tolerated with reasonable scope. Differential substitution on the ester gave the desired product in high yield (Table 3, 4a–b). Cyclic diazo substrates deliver the desired spiro-compounds in high yield under the reaction conditions (Table 3, 4c–d). A wide range of substituents can be tolerated on the aromatic ring.17 While electron poor substrates provide the desired product rapidly under the standard reaction conditions (Table 3, 4e, 4j, 4i), electron rich substrates are far more sluggish and require elevated temperatures and prolonged reaction times (Table 3, 4f, 4h). We were pleased to find that heteroaryl diazo esters also provide product, albeit in lower yield (Table 3, 4k–l). Elevated temperatures allowed for acceptor diazo esters to be incorporated in good yield (Table 3, e3).
While screening electron-withdrawing groups on the donor/acceptor diazo compound, we found that amides and phosphonates provide product in very low yield. In contrast, we were pleased to find the 1-phenyl, 2,2,2-trifluoro diazoethane, as described by Davies and coworkers,18 delivered the desired product in high yield (Chart 1, 5a). Electron deficient aromatic groups on these trifluoroethyl diazo substrates function well in the reaction with high yield and rapid reaction times (Chart 1, 5b, 5d–f). As observed previously, electron rich aromatic groups provide products in substantially lower yield even at high temperatures (Chart 1, 5c). These products contain quaternary carbons with a trifluoromethyl group, a substitution pattern not easily accessed using other methods.19
Chart 1.
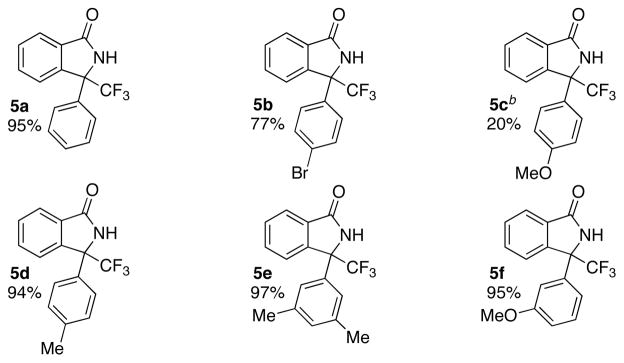
Scope with 1-aryl 2,2,2-trifluoro diazoethanes
aFor standard reaction conditions see Table 1. bReaction conducted at 60 °C.
A series of mechanistic experiments were performed to elucidate the nuances of the reaction (Scheme 1). Competition experiments were used to determine the electronic preference of the reaction. When para-bromo benzamide 1c was run in competition with the unsubstituted amide 1a the reaction favors the electron deficient amide in a 1.9:1 ratio (Scheme 1a). This experiment suggest that C–H activation favors more acid C–H bonds. Indeed, initial rate kinetic isotope studies revealed a KIE value of 2.6 (Scheme 1b). Together these studies suggest that C–H activation occurs via a concerted-metallation deprotonation (CMD) mechanism, and the C–H activation does not proceed through a Wheland intermediate.20 These experiments are consistent with the observation that reactions involving substrates bearing more electron donating groups are sluggish.
Scheme 1.

Mechanistic Experiments
The reversibility of the C–H activation can be determined by removing the diazo compound and running the reaction in the presence of d3-MeCN (Scheme 1c). After 15 minutes less than 1% deuterium incorporation is observed at the ortho-positions, as determined by 1H NMR. After 150 minutes the degree of deuterium incorporation increases to 7%. These results suggest that C–H activation in largely irreversible over the time scale of the reaction, and is consistent with previous reports by Fagnou3e and Glorius.21
A competition experiment was used to determine the reactions preference for electronically different donor/acceptor diazo compounds (Scheme 1d). When para-bromophenyl methyl diazo acetate 2e is run in competition with 2a, the electron deficient substrate is favored 1.6:1 after 15 minutes. These results suggest that migratory insertion or reductive elimination favors the more electron deficient substrate. Since reductive elimination should favor a more electron rich substrate, it is reasonable to surmise that migratory insertion favors an electron deficient diazo substrate because of its increased electrophilicity.
In conclusion, we have developed a new approach to access iso-indolones bearing a tetra-substituted carbon. A wide range of substrates are tolerated, including 1-aryl-2,2,2-trifluoroethyldiazo compounds. Mechanistic studies reveal the reaction to operate under a mechanism similar to that of the synthesis of dihydroisoquinolones.
Supplementary Material
Acknowledgments
Funding Sources
The authors declare no competing financial interest.
We thank NIGMS for generous support of this research (GM80442). T.R. thanks Amgen and Roche for unrestricted support and Johnson Matthey for a generous loan of Rh salts.
Footnotes
ASSOCIATED CONTENT
Detailed experimental procedures, characterization, and mechanistic data. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.(a) D’Souza DM, Muller TJJ. Chem Soc Rev. 2007;36:1095. doi: 10.1039/b608235c. [DOI] [PubMed] [Google Scholar]; (b) Nakamura I, Yamamoto Y. Chem Rev. 2004;104:2127. doi: 10.1021/cr020095i. [DOI] [PubMed] [Google Scholar]
- 2.(a) Satoh T, Miura M. Chem Eur J. 2010;16:11212. doi: 10.1002/chem.201001363. [DOI] [PubMed] [Google Scholar]; (b) Zhu C, Wang R, Falck JR. Chem Asian J. 2012;7:1502. doi: 10.1002/asia.201200035. [DOI] [PubMed] [Google Scholar]; (c) Patureau FW, Wencel-Delord J, Glorius F. Aldrichemica Acta. 2012;45:31. [Google Scholar]
- 3.(a) Guimond N, Gouliaras C, Fagnou K. J Am Chem Soc. 2010;132:6909. doi: 10.1021/ja102571b. [DOI] [PubMed] [Google Scholar]; (b) Mochida S, Umeda N, Hirano K, Satoh T, Miura M. Chem Lett. 2010;39:744. [Google Scholar]; (c) Hyster TK, Rovis T. J Am Chem Soc. 2010;132:10565. doi: 10.1021/ja103776u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Song G, Chen D, Pan CL, Crabtree RH, Li X. J Org Chem. 2010;75:7487. doi: 10.1021/jo101596d. [DOI] [PubMed] [Google Scholar]; (e) Guimond N, Gorelsky SL, Fagnou K. J Am Chem Soc. 2011;133:6449. doi: 10.1021/ja201143v. [DOI] [PubMed] [Google Scholar]; (f) Wang H, Grohmann C, Nimphius C, Glorius F. J Am Chem Soc. 2012;134:19592. doi: 10.1021/ja310153v. [DOI] [PubMed] [Google Scholar]
- 4.(a) Su Y, Zhao M, Han K, Song G, Li X. Org Lett. 2010;12:5462. doi: 10.1021/ol102306c. [DOI] [PubMed] [Google Scholar]; (b) Hyster TK, Rovis T. Chem Sci. 2011;2:1605. doi: 10.1039/C1SC00235J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Guimond N, Fagnou K. J Am Chem Soc. 2009;131:12050. doi: 10.1021/ja904380q. [DOI] [PubMed] [Google Scholar]; (b) Li L, Brennessel WW, Jones WD. J Am Chem Soc. 2008;130:12414. doi: 10.1021/ja802415h. [DOI] [PubMed] [Google Scholar]; (c) Fukutani T, Umeda N, Hirano K, Satoh T, Miura M. Chem Commun. 2009;34:5141. doi: 10.1039/b910198e. [DOI] [PubMed] [Google Scholar]; (d) Too PC, Wang YF, Chiba S. Org Lett. 2010;12:5688. doi: 10.1021/ol102504b. [DOI] [PubMed] [Google Scholar]; (e) Morimoto K, Hirano K, Satoh T, Miura M. Chem Lett. 2011;40:600. [Google Scholar]; (f) Wang YF, Toh KK, Lee JY, Chiba S. Angew Chem Int Ed. 2011;50:5927. doi: 10.1002/anie.201101009. [DOI] [PubMed] [Google Scholar]; (g) Hyster TK, Rovis T. Chem Commun. 2011;47:11846. doi: 10.1039/c1cc15248c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhang X, Chen D, Zhao M, Jia A, Li X. Adv Synth Cat. 2011;353:719. [Google Scholar]
- 6.Too PC, Noji T, Lim YJ, Li X, Chiba S. Synlett. 2011:2789. [Google Scholar]
- 7.(a) Stuart DR, Bertrand-Laperle M, Burgess KMN, Fagnou K. J Am Chem Soc. 2008;130:16474. doi: 10.1021/ja806955s. [DOI] [PubMed] [Google Scholar]; (b) Stuart DR, Alsabeh P, Kuhn M, Fagnou K. J Am Chem Soc. 2010;132:18326. doi: 10.1021/ja1082624. [DOI] [PubMed] [Google Scholar]; (c) Chen J, Song G, Pan CL, Li X. Org Lett. 2010;12:5426. doi: 10.1021/ol1022596. [DOI] [PubMed] [Google Scholar]; (d) Huestis MP, Chan L, Stuart DR, Fagnou K. Angew Chem Int Ed. 2011;50:1338. doi: 10.1002/anie.201006381. [DOI] [PubMed] [Google Scholar]
- 8.Rakshit S, Patureau FW, Glorius F. J Am Chem Soc. 2010;132:9585. doi: 10.1021/ja104305s. [DOI] [PubMed] [Google Scholar]
- 9.McGrath NA, Brichacek M, Njardarson JT. J Chem Ed. 2010;87:1348. [Google Scholar]
- 10.(a) Augner D, Gerbino DC, Slavov N, Neudorfl JM, Schmalz HG. Org Lett. 2011;13:5374. doi: 10.1021/ol202271k. [DOI] [PubMed] [Google Scholar]; (b) Nozawa Y, Yamamoto K, Ito M, Sakai N, Mizoue K, Mizobe F, Hanada K. J Antibiot. 1997;50:635. doi: 10.7164/antibiotics.50.635. [DOI] [PubMed] [Google Scholar]; (c) Comins D, Schilling S, Zhang Y. Org Lett. 2005;7:95. doi: 10.1021/ol047824w. [DOI] [PubMed] [Google Scholar]; (d) Stuk TL, Assink BK, Bates RC, Erdman DT, Fedij V, Jennings SM, Lassig JA, Smith RJ, Smith TL. Org Process Res Dev. 2003;7:851. [Google Scholar]; (e) Belliotti TR, Brink WA, Kesten SR, Rubin JR, Wustrow DJ, Zoski KT, Whetzel SZ, Corbin AE, Pugsley TA, Heffner TG, Wise LD. Bioorg Med Chem Lett. 1998;8:1499. doi: 10.1016/s0960-894x(98)00252-2. [DOI] [PubMed] [Google Scholar]
- 11.Rakshit S, Grohmann C, Besset T, Glorius F. J Am Chem Soc. 2011;133:2350. doi: 10.1021/ja109676d. [DOI] [PubMed] [Google Scholar]
- 12.(a) Hyster TK, Knorr L, Ward TR, Rovis T. Science. 2012;338:500. doi: 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ye B, Cramer N. Science. 2012;338:504. doi: 10.1126/science.1226938. [DOI] [PubMed] [Google Scholar]
- 13.Du Y, Hyster TK, Rovis T. Chem Commun. 2011;47:12074. doi: 10.1039/c1cc15843k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu C, Xie W, Falck JR. Chem Eur J. 2011;17:12591. doi: 10.1002/chem.201102475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan WW, Lo SF, Zhao Z, Yu WY. J Am Chem Soc. 2012;134:13565. doi: 10.1021/ja305771y. [DOI] [PubMed] [Google Scholar]
- 16.Davies has defined diazo compounds as donor, acceptor or both; see: Morten D, Davies HML. Chem Soc Rev. 2011;40:1857.
- 17.Donor/acceptor diazo compounds bearing alkenes as the donor group underwent pyrazole formation; see: Manning JR, Davies HML. Org Synth. 2007;84:334.
- 18.Denton JR, Sukumaran D, Davies HML. Org Lett. 2007;9:2625. doi: 10.1021/ol070714f. [DOI] [PubMed] [Google Scholar]
- 19.For select leading references, see: Guo Y, Zhao X, Zhang D, Murahashi S-i. Angew Chem Int Ed. 2009;48:2047. doi: 10.1002/anie.200805852.Mizuta S, Galicia-Lopez O, Engle KM, Verhoog S, Wheel-house K, Rassias G, Gouverneur V. Chem Eur J. 2012;18:8583. doi: 10.1002/chem.201201707.Shibata N, Suzuki S, Furukawa T, Kawai H, Tokunaga E, Yuan Z, Cahard D. Adv Synth Cat. 2011;353:2037.
- 20.(a) Lapointe D, Fagnou K. Chem Lett. 2010;39:1118. [Google Scholar]; (b) Wheland GW. J Am Chem Soc. 1942;64:900. [Google Scholar]; (c) Davies DL, Donald SMA, Al-Duaij O, Macgregor SA, Polleth M. J Am Chem Soc. 2006;128:4210. doi: 10.1021/ja060173+. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Glorius F. Angew Chem Int Ed. 2012;51:7318. doi: 10.1002/anie.201201273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.