

Abstract
Antiphospholipid antibody syndrome is defined by the presence of thromboembolic complications and/or pregnancy morbidity in the presence of persistently increased titers of antiphospholipid antibodies. Its clinical presentation can be diverse and any organ can be involved, with a current impact in most surgical and medical specialties. The authors present the case of a 43-year-old man who, over a 13-year period of follow-up, presented with thrombosis of the mesenteric vein, inferior vena cava, and axillary and subclavian veins in a setting where diagnostic and therapeutic options are limited and costly. Through this case report, the authors aim to describe the evolution of this complex pathology, which to date has not been described in the authors’ milieu – probably because of its challenging diagnosis and the limited treatment options available. The authors conclude that clinicians need to have a high index of suspicion of APS in patients who present with a thrombotic episode – clinicians should investigate for the presence of antiphospholipid antibodies, as early diagnosis may influence the course of the disease. Furthermore, resources for the detection of antiphospholipid antibodies should be made readily available in resource-limited settings. Finally, patient education on the importance of drug compliance, periodic monitoring, and prevention of thrombosis is indispensable, especially as mortality could be associated with the effects of vascular thrombosis and/or the effects of bleeding due to anticoagulants.
Keywords: thrombosis, mesenteric venous thrombosis, anticardiolipin antibodies, Budd-Chiari syndrome, Cameroon
Introduction
The term antiphospholipid antibody syndrome (APS) was first coined to denote the clinical association between antiphospholipid antibodies and a syndrome of hypercoagulability.1,2 APS is an autoimmune disease characterized by the presence of thromboembolic complications and/or pregnancy morbidity in the presence of persistently increased titers of antiphospholipid antibodies.3 The most commonly detected subgroups of antiphospholipid antibodies are lupus anticoagulant, anticardiolipin, and anti-beta-2-glycoprotein 1 antibodies. Classification of the antibodies into these groups is based on the biochemical method of detection. The biological function of these antibodies, although not well elucidated, is thought to act as a natural anticoagulant.4 Despite their name, lupus anticoagulant antibodies are associated with thromboembolic events rather than clinical bleeding. Antiphospholipid antibodies can interfere with both pro- and anticoagulant pathways. In vitro, phospholipid surfaces inhibit procoagulant pathways and therefore prolong clotting; in vivo, meanwhile, the microenvironment of cell membranes promotes greater inhibition of anticoagulant pathways and thus also thrombosis.5 Although the pathogenesis of APS is poorly understood, many mechanisms have been postulated, including the binding of antiphospholipid antibodies to endothelial cells, which stimulates an upregulation of adhesion molecules, increasing leucocyte adhesion6 and creating a prothrombotic state.7 Clinically, APS can be very diverse and can occur in a variety of disease states, rendering diagnosis and treatment challenging, especially in resource-limited settings. The authors therefore present the case of a 43-year-old man with multiple thrombotic events and the presence of antiphospholipid antibodies, which, to the best of the authors’ knowledge, is the first time this has been reported in Cameroonians.
Case report
A 43-year-old man presented with a history of fatigue, abdominal pain and discomfort, constipation, nausea, and vomiting of insidious onset over a 1-month period. A diagnosis of subacute intestinal obstruction was made and the patient was conservatively managed with regression of all other symptoms except abdominal pains, for which he was referred to the authors for further reassessment. The patient’s family history was unremarkable, but his personal history showed that 6 years earlier he had undergone an intestinal resection with end-to-end anastomosis of his small intestine, indicated for infarction and gangrene of two-thirds of his small intestine (Figure 1). At that time, intestinal gangrene and infarction were thought to be secondary to mesenteric venous thrombosis, because onset was sudden and there was hemorrhagic infarction of the involved intestinal segment. Furthermore, mesenteric arterial pulses were present at laparotomy. Postoperative recovery was uneventful and the patient was lost to follow-up until the onset of the recent symptoms for which he was referred. It is worth noting that the patient had never smoked and had drunk alcohol sparingly.
Figure 1.
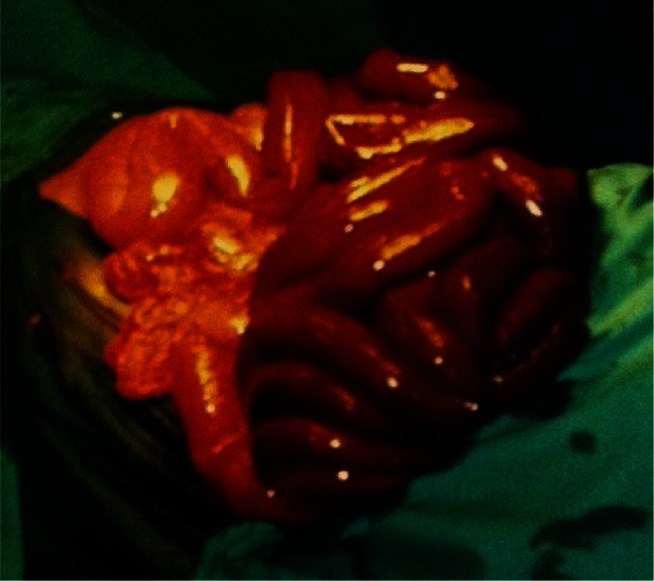
Hemorrhagic infarction of most of the small intestine, as seen at laparotomy.
On clinical examination, the patient looked frail and showed obvious signs of weight loss. His blood pressure was 120/80 mmHg. The most remarkable finding was a non-distended but diffusely tender abdomen. A complete blood count revealed leukocytosis at 12,300/μL, predominantly with neutrophils; a hemoglobin level of 12 g/dL; and a normal platelet count. The patient’s prothrombin time (PT) was 62%, with a prolonged kaolin-cephalin clotting time (KCT) of 41.5 seconds (control, 36 seconds); his blood sugar, blood urea nitrogen, and creatinine levels were all within normal limits. An abdominal computed tomography scan showed moderate dilatation of the inferior vena cava (IVC) with an intraluminal hypodensity consistent with venous thrombosis. The liver appeared hypodense compared with the spleen (Figure 2), with dilated intrahepatic veins that did not appear opacified, even in enhanced images (Figure 3), and there was homogenous splenomegaly. These findings were compatible with Budd-Chiari syndrome (BCS) secondary to IVC thrombosis. The patient was placed on a 10-day course of low-molecular-weight heparin, this in spite of the prolonged PT and KCT. Further anticoagulation was deemed risky because of the patient’s irregularity in respect to follow-up appointments. He was therefore placed on low-dose aspirin and, as there was no local experience with transjugular intrahepatic portosystemic shunting, he was referred to a vascular surgeon in a hospital in Yaoundé, about 150 miles from Douala, for decompressive IVC surgery. Because the cost of the surgery was prohibitive for the patient, he was lost to follow-up for a second time.
Figure 2.
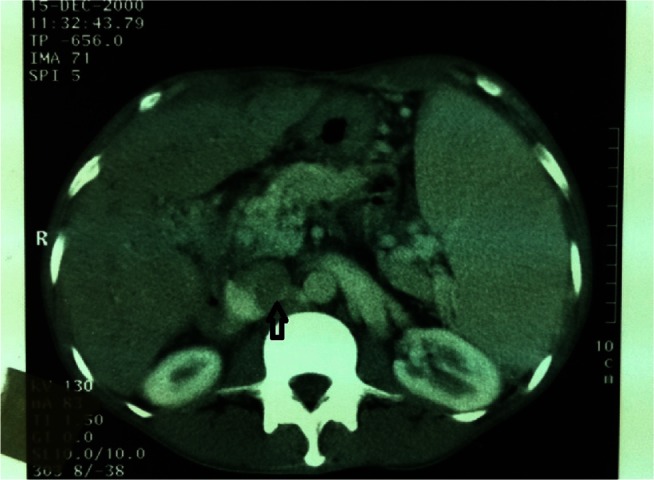
Contrast-enhanced computed tomography scan showing enlarged inferior vena cava with intraluminal clot (indicated by arrow), splenomegaly, and abnormally low enhancement of the liver.
Figure 3.
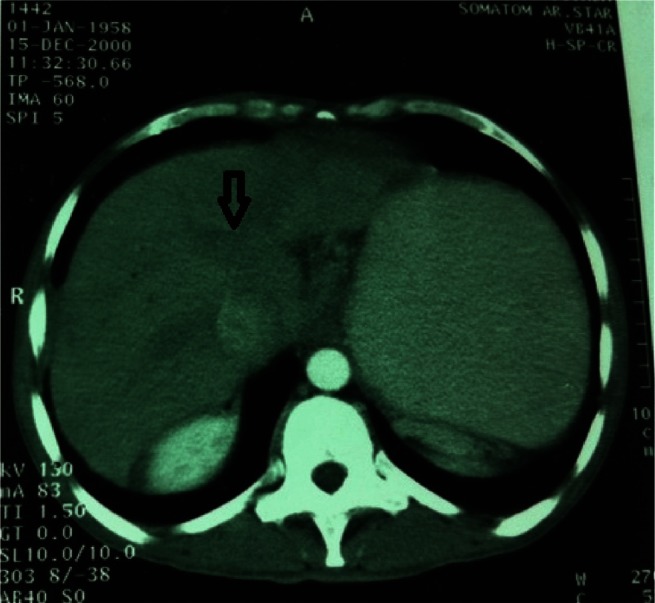
Contrast-enhanced computed tomography scan showing lack of opacification and enlarged intrahepatic veins (indicated by arrow).
The patient reappeared 2 months later with persistent abdominal pains, this time associated with distention of 2 weeks’ duration. On examination, the patient had lost a considerable amount of weight and had pale conjunctivae but no jaundice; there was prominent thoracoabdominal collateral circulation, ascites, and splenomegaly that extended 10 cm below the left costal margin. There was also mild pitting edema of the ankles.
Investigations performed revealed a decrease in the patient’s hemoglobin level to 9.6 g/dL, leucocytes at 7300/μL, normal platelets at 241,000/μL, an erythrocyte sedimentation rate of 20 mm in the first hour, a PT of 41.1%, an international normalized ratio of 1.69, a KCT of 38.5 seconds (control, 30.2 seconds), normal total proteins of 71 g/L but decreased albumin at 28.4 g/L, and almost doubly raised transaminases (serum glutamic oxaloacetic transaminase [aspartate aminotransferase] of 56 IU/L, serum glutamic pyruvic transaminase [alanine aminotransferase] of 64 IU/L). The results of hepatitis B surface antigen, anti–hepatitis C virus, and human immunodeficiency virus serology were negative. The blood urea nitrogen and creatinine levels were within normal limits and the alpha-fetoprotein level was normal. A diagnostic and therapeutic abdominal paracentesis was performed, revealing a transudate on analysis. Grade 3–4 esophageal varices with red signs, portal hypertensive gastropathy, and congestive duodenitis were found on gastroscopy. The patient was placed on oral propranolol to prevent variceal bleeding and spironolactone to deplete his fluid overload. A comparative computed tomography scan showed persistent supra- and infrahepatic IVC thrombosis, splenomegaly, and this time massive ascites (Figure 4).
Figure 4.
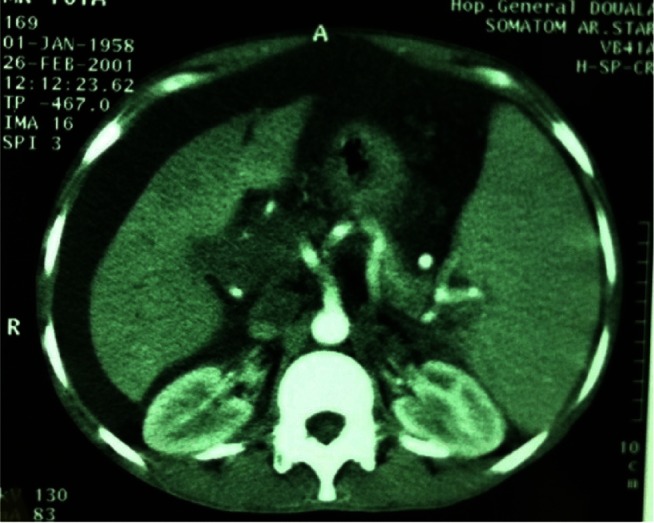
Contrast-enhanced abdominal computed tomography scan showing splenomegaly and ascites.
These findings of portal hypertension complicating hepatic venous thrombosis in a patient with prolonged KCT and a past history of mesenteric venous thrombosis were suggestive of hypercoagulability. In addition, high titers of anticardiolipin antibodies at 52 units (normal range being ,12 units) (a test for immunoglobulin M anticardiolipin antibodies was negative) were consistent with APS. Tests for proteins S and C were normal and a test for lupus anticoagulant antibodies was negative. Steroids were prescribed and once more the patient was lost to follow-up. Six months later, the patient was admitted to hospital for hepatic encephalopathy; this was managed favorably and he was discharged.
Four years on, the patient developed pain and swelling of the left supraclavicular region, with visible subcutaneous collateral veins in the shoulder area and upper anterior chest wall. Doppler ultrasound studies of the patient’s neck veins revealed findings consistent with thrombosis of the left axillary and subclavian veins.
One year following this, the patient presented with sepsis and a gangrenous scrotum. He discharged himself from hospital.
Discussion
This documented case of thrombosis of the mesenteric vein, inferior vena cava, and axillary and subclavian veins in the presence of antiphospholipid antibodies fulfills the Sydney criteria for APS diagnosis, which stipulates cases require the presence of at least one of the clinical criteria (vascular thrombosis and/or pregnancy morbidity) and at least one of the laboratory criteria (detection of lupus anticoagulant, anticardiolipin, or anti-beta-2-glycoprotein 1 antibodies).3 Although the complications associated with vascular thrombosis in the present patient were serious, they seemed to appear one at a time; this is in clear contrast to catastrophic APS, where there are multiple simultaneous vascular occlusions and multiorgan involvement.1
The initial mesenteric vascular ischemia was due to acute venous thrombosis. Six years later, the patient was admitted to hospital with subacute obstruction. Chandra et al8 reported in 2002 a case of subacute intestinal obstruction resulting from ischemic stricture in the proximal small intestine. In hindsight, this is similar to how the patient in the present case presented prior to the overt portal hypertension. At the time, Chandra et al’s8 case was the first reported case of APS presenting with noncirrhotic portal fibrosis and mesenteric venous thrombosis.8 In the presence of an underlying hypercoagulable state, portal hypertension, which results in stagnation of blood in the splanchnic venous bed, predisposes an individual to the development of venous thrombi.
The second major vascular occlusion in the present patient was BCS due to IVC thrombosis. BCS is an uncommon condition induced by thrombotic or nonthrombotic obstruction of hepatic venous outflow. This may occur at any point between hepatic venules and the junction of the IVC and the right atrium of the heart.9,10 BCS is an infrequent complication in APS patients and sometimes can be its first manifestation.11,12 In a prospective study of 22 patients with nontumorous BCS, APS was the second most common cause after myeloproliferative disorders.13
Furthermore, thrombosis of the axillary and subclavian veins – generally termed as upper-extremity deep vein thrombosis – occurs infrequently, thereby constituting less than 11% of all deep venous thromboses.14 Its risk factors include indwelling venous catheters, malignancy, and coagulation defects. Although it occurred several years after the diagnosis of APS, a hypercoagulable state and the absence of anticoagulant treatment were identified as the main risk factors in the present patient.
Finally, the treatment of APS falls under four main areas: (1) thromboprophylaxis with low-dose aspirin,15 (2) prevention of further thrombosis of large vessels, (3) treatment of acute thrombotic event by anticoagulation, and (4) management of pregnancy in association with antiphospholipid antibodies. Management of the present patient involved nutrition (given his remarkable weight loss), the prevention of hepatic encephalopathy through management of chronic liver disease, and the prevention of variceal bleeding. Treatment with prolonged anticoagulation using vitamin K antagonists was not undertaken in the present case for fear of severe complications in a setting of hepatic insufficiency and because the patient was predictably noncompliant. Steroids were hesitantly prescribed because evidence shows that steroids and immunosuppressive agents have disappointing results with regard to thrombosis.16
Conclusion
In conclusion, clinicians need to have a high index of suspicion of APS in patients who present with a thrombotic episode. Clinicians should investigate for the presence of antiphospholipid antibodies, as early diagnosis may influence the course of the disease. Furthermore, resources for the detection of antiphospholipid antibodies should be made readily available in resource-limited settings so as to test patients with a history of thrombosis or pregnancy complications. Finally, management of APS that involves prevention of thrombosis together with close monitoring of the patient on an anticoagulant could be quite challenging, especially in a setting like the one in this study, where anticoagulants are mostly prescribed by specialist physicians and payment for work-up and medications is exclusively made by the patient. Therefore, patient education on the importance of drug compliance, periodic monitoring, and prevention of thrombosis is indispensable, especially as mortality could be associated with the effects of vascular thrombosis and/or the effects of bleeding due to anticoagulants.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002;346(10):752–763. doi: 10.1056/NEJMra002974. [DOI] [PubMed] [Google Scholar]
- 2.Hughes GR, Harris NN, Gharavi AE. The anticardiolipin syndrome. J Rheumatol. 1986;13(3):486–489. [PubMed] [Google Scholar]
- 3.Devreese K, Peerlinck K, Hoylaerts MF. Thrombotic risk assessment in the antiphospholipid syndrome requires more than the quantification of lupus anticoagulants. Blood. 2010;115(4):870–878. doi: 10.1182/blood-2009-09-244426. [DOI] [PubMed] [Google Scholar]
- 4.Kandiah DA, Krilis SA. Beta 2-glycoprotein I. Lupus. 1994;3(4):207–212. doi: 10.1177/096120339400300401. [DOI] [PubMed] [Google Scholar]
- 5.Esmon NL, Safa O, Smirnov MD, Esmon CT. Antiphospholipid antibodies and the protein C pathway. J Autoimmun. 2000;15(2):221–225. doi: 10.1006/jaut.2000.0407. [DOI] [PubMed] [Google Scholar]
- 6.Del Papa N, Raschi E, Catelli L, et al. Endothelial cells as a target for antiphospholipid antibodies: role of anti-beta 2 glycoprotein I antibodies. Am J Reprod Immunol. 1997;38(3):212–217. doi: 10.1111/j.1600-0897.1997.tb00301.x. [DOI] [PubMed] [Google Scholar]
- 7.Pierangeli SS, Colden-Stanfield M, Liu X, Barker JH, Anderson GL, Harris EN. Antiphospholipid antibodies from antiphospholipid syndrome patients activate endothelial cells in vitro and in vivo. Circulation. 1999;99(15):1997–2002. doi: 10.1161/01.cir.99.15.1997. [DOI] [PubMed] [Google Scholar]
- 8.Chandra S, Dutta U, Das R, Vaiphei K, Nagi B, Singh K. Mesenteric venous thrombosis causing jejunal stricture: secondary to hypercoagulable states and primary portal hypertension. Dig Dis Sci. 2002;47(9):2017–2019. doi: 10.1023/a:1019664627149. [DOI] [PubMed] [Google Scholar]
- 9.Buckley O, O’Brien J, Snow A, et al. Imaging of Budd-Chiari syndrome. Eur Radiol. 2007;17(8):2071–2078. doi: 10.1007/s00330-006-0537-2. [DOI] [PubMed] [Google Scholar]
- 10.Janssen HL, Garcia-Pagan JC, Elias E, Mentha G, Hadengue A, Valla DC, European Group for the Study of Vascular Disorders of the Liver Budd-Chiari syndrome: a review by an expert panel. J Hepatol. 2003;38(3):364–371. doi: 10.1016/s0168-8278(02)00434-8. [DOI] [PubMed] [Google Scholar]
- 11.Asherson RA, Khamashta MA, Ordi-Ros J, et al. The “primary” antiphospholipid syndrome: major clinical and serological features. Medicine (Baltimore) 1989;68(6):366–374. [PubMed] [Google Scholar]
- 12.Espinosa G, Font J, García-Pagan JC, et al. Budd-Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients. Medicine (Baltimore) 2001;80(6):345–354. doi: 10.1097/00005792-200111000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Pelletier S, Landi B, Piette JC, et al. Antiphospholipid syndrome as the second cause of non-tumorous Budd-Chiari syndrome. J Hepatol. 1994;21(1):76–80. doi: 10.1016/s0168-8278(94)80140-1. [DOI] [PubMed] [Google Scholar]
- 14.Joffe HV, Kucher N, Tapson VF, Goldhaber SZ. for Deep Vein Thrombosis FREE Steering Committee. Upper-extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110(12):1605–1611. doi: 10.1161/01.CIR.0000142289.94369.D7. [DOI] [PubMed] [Google Scholar]
- 15.Avcin T, Cimaz R, Meroni PL. Do we need an international consensus statement on classification criteria for the antiphospholipid syndrome in the paediatric population? Lupus. 2001;10(12):897–898. doi: 10.1191/096120301701548427. [DOI] [PubMed] [Google Scholar]
- 16.Edwards CJ, Hughes GR. Hughes syndrome (the antiphospholipid syndrome): 25 years old. Mod Rheumatol. 2008;18(2):119–124. doi: 10.1007/s10165-008-0042-3. [DOI] [PubMed] [Google Scholar]