

Abstract
Purpose.
To explore the role of triggering receptor expressed on myeloid cells 2 (TREM-2) in Pseudomonas aeruginosa (PA) keratitis.
Methods.
BALB/c mice were routinely infected with PA and evaluated at various postinfection time points for corneal expression of TREM-2, by real-time PCR, Western blot, and flow cytometry. Next, BALB/c and C57BL/6 mice were respectively treated with TREM-2 siRNA or agonistic anti–TREM-2 antibody, to determine the role of TREM-2 in PA keratitis. Bacterial load and neutrophil infiltration were tested by plate count and myeloperoxidase assay, respectively. Th1-/Th2-type and proinflammatory cytokine expression were tested by real-time PCR and ELISA after in vivo and in vitro silencing of TREM-2. Moreover, phosphorylated Akt levels were tested by Western blot in murine macrophages after treatment with agonistic anti–TREM-2 antibody. mRNA levels of proinflammatory cytokines were examined in murine macrophages after TREM-2 activation and lipopolysaccharide stimulation, following pretreatment with inhibitors for PI3K or Akt, to determine whether PI3K/Akt is required in TREM-2–mediated immune modulation. In addition, BALB/c mice were treated with wortmannin and analyzed for bacterial load and proinflammatory cytokine expression.
Results.
TREM-2 expression was elevated in the infected BALB/c corneas at 3 or 5 days postinfection. Silencing of TREM-2 accelerated disease progression by enhancing bacterial load and corneal inflammation, whereas activation of TREM-2 promoted host resistance to PA keratitis. PI3K/Akt signaling is required in the TREM-2–mediated immune modulation, and inhibition of PI3K resulted in worsened disease after PA corneal infection.
Conclusions.
TREM-2 promoted host resistance to PA infection by suppressing corneal inflammation via activation of the PI3K/Akt pathway.
Keywords: inflammation, cornea, bacteria, cytokines
Our study first explored the role of TREM-2 in ocular infectious diseases. We demonstrated that TREM-2 promoted host resistance to Pseudomonas aeruginosa keratitis by suppressing corneal inflammation via the PI3K/Akt signaling pathway, which may hold promise for disease treatment.
Introduction
Contact lens usage increases vulnerability to development of microbial keratitis, which is associated with vision loss and blindness.1–3 The microorganism most often isolated from corneal ulcers induced by contact lens wear is Pseudomonas aeruginosa (PA).4 PA keratitis often progresses rapidly and presents as a suppurative stromal infiltrate with marked mucopurulent exudate.5,6 PA infection also leads to complications that consist of inflammatory epithelial edema, stromal infiltration, and corneal ulceration, which can culminate in significant tissue destruction and loss of function.7 Conventional therapies, such as antibiotic treatment, often fail to control the tissue damage caused by excessive local inflammation, even if viable bacteria are cleared from the cornea.8 Hence, in addition to antibiotic treatment, it is also important to develop new therapeutic modalities to control the inflammatory response.
The host immune response triggered by invading pathogens relies on the innate immune recognition through pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs).7 Activation of PRRs initiates a variety of inflammatory events, including infiltration of inflammatory cells (e.g., polymorphonuclear neutrophils [PMNs] and monocytes/macrophages),6,9–11 production of Th1-type cytokines (e.g., IFN-γ, IL-12, IL-18), Th2-type cytokines (e.g., IL-4, IL-5, IL-10), and proinflammatory cytokines (e.g., TNF-α, macrophage inflammatory protein [MIP]-2, IL-1β).7,12 These PRR-mediated inflammatory responses are necessary for bacterial clearance; however, if uncontrolled, excessive host inflammation also leads to immunopathological tissue damage. Therefore, it is important to precisely balance pro- and anti-inflammatory responses in ocular immune defense.
As a novel PRR family, triggering receptors expressed on myeloid cells (TREMs) have recently emerged as important immune regulators.13–15 There are two major members in the TREM family, TREM-1 and TREM-2, which are mainly expressed on cells derived from the myeloid lineage, such as macrophages and dendritic cells.16 Our previous study demonstrated that TREM-1 enhances corneal inflammation after PA infection by modulating Th1/Th2 responses and TLR signaling.17 However, nothing is known regarding the role of TREM-2 in the eye.
A previous study reports that TREM-2 inhibits the inflammatory response by attenuating macrophage activation in response to lipopolysaccharide (LPS) stimulation.18 Overexpression of TREM-2 in microglia reduces the expression of TNF-α and inducible nitric oxide synthase (iNOS) after culture of these cells with apoptotic neurons.19 And blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis.20 These studies together suggested that TREM-2 functions as a negative regulator in the inflammatory response.
Although TREM-1 and TREM-2 share the same adaptor protein, DNAX activation protein 12 (DAP12), their downstream signals are distinct. TREM-1 functions to amplify the TLR responses in monocytes and neutrophils,13 whereas TREM-2 negatively regulates TLR signaling in macrophages and dendritic cells.18,21 Production of inflammatory cytokines induced by TLR ligands, such as LPS, zymosan, and CpG, are significantly upregulated in macrophages isolated from TREM-2 knockout versus wild-type mice,18 indicating that TREM-2 can counter the TLR-mediated amplification of inflammatory responses. In this regard, activation of TREM-2 may be an effective way to prevent an excessive host inflammatory response and its consequences.
The downstream signaling pathways of TREM-2 are still unclear. A recent study demonstrates that phosphoinositide 3-kinase (PI3K), on interaction with its regulatory subunit p85 and the adaptor molecule DAP12, is recruited to the TREM-2/DAP12 signal complex.22 As a family of lipid kinases, PI3Ks usually activate Akt, resulting in phosphorylation of a variety of downstream substrates, including IκB kinase (IKK),23 mammalian target of rapamycin (mTOR),24 p21,25 and caspase 9.26 Studies have demonstrated that PI3K/Akt signaling can modulate many cellular events, such as cytokine production,27 bacterial elimination,28 cell proliferation,29 and apoptosis.30 However, whether PI3K/Akt is involved in the TREM-2–mediated immune response requires further investigation.
In summary, this study was designed to investigate the role of TREM-2 in PA keratitis. Our data provide compelling evidence that TREM-2 is significantly enhanced in mouse corneas and macrophages after PA infection, and promotes host resistance by suppressing corneal inflammation via activation of PI3K/Akt signaling. To the best of our knowledge, this is the first study to explore the function of TREM-2 in the eye, whose modulation may provide an effective strategy for treatment of microbial keratitis.
Materials and Methods
Mice and Reagents
Eight-week-old female BALB/c and C57BL/6 (B6) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). PA strain 19660 was purchased from the American Type Culture Collection (ATCC, Manassas, VA). Thioglycollate medium and Pseudomonas isolation agar were purchased from BD Difco Laboratories (Sparks, MD). Small interfering RNA (siRNA) for mTREM-2 or appropriate scrambled control were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Primary antibodies (Abs) against TREM-2, phosphorylated Akt (p-Akt), Akt, and β-actin for Western blot were obtained from R&D Systems (Minneapolis, MN), Epitomics (Burlingame, CA), Cell Signaling Technology (Danvers, MA), and Sigma (St. Louis, MO), respectively. Secondary Ab against sheep IgG was obtained from R&D Systems, and secondary Abs to mouse and rabbit IgG were purchased from Bio-Rad (Hercules, CA). Abs for flow cytometry, including anti–TREM-2-phycoerythrin, anti–F4/80-FITC, and anti–Gr-1-allophycocyanin (APC) were obtained from R&D Systems, Invitrogen (Carlsbad, CA), and BD Pharmingen (San Jose, CA), respectively. Isotype controls phycoerythrin-rat IgG2b, FITC-rat IgG2b, and APC-rat IgG2b were purchased from eBioscience (San Diego, CA), Invitrogen, and BD Pharmingen, respectively. LPS from Pseudomonas aeruginosa was obtained from Sigma. ELISA kits for TNF-α, MIP-2, and IL-1β were purchased from R&D Systems, and ELISA kits for IFN-γ and IL-10 were obtained from Multiscience Biotech (Hangzhou, China). PI3K inhibitors LY294002 and wortmannin were purchased from Invitrogen. Akt Inhibitor IX API-59CJ-OMe was purchased from Merck (Darmstadt, Germany) and dimethyl sulfoxide (DMSO) was from Sigma.
Ocular Infection and Clinical Examination
The left cornea of 8-week-old female BALB/c mice (Jackson Laboratory) was infected by PA (ATCC 19660), as described previously.31–33 Corneal disease was graded using an established scale: 0, clear or slight opacity partially or fully covering the pupil; +1, slight opacity partially or fully covering the anterior segment; +2, dense opacity partially or fully covering the pupil; +3, dense opacity covering the entire anterior segment; and +4, corneal perforation or phthisis.31 Animals were treated humanely and in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
In Vivo Silencing and Inhibition
In vivo use of siRNA or inhibitor has been previously described by our laboratory34 and others.35 siRNA for mTREM-2 or appropriate scrambled control (Santa Cruz Biotechnology) was injected subconjunctivally (5 μL/mouse at a concentration of 8 μM) into the left eye of BALB/c mice (n = 5/group/time) 1 day before infection and then topically applied onto the infected corneas (5 μL/mouse per time at a concentration of 4 μM, once on the day of infection, and twice on both 1 and 3 days postinfection [p.i.]). Wortmannin (at a concentration of 0.2 mg/mL) or control solvent DMSO/PBS (at a ratio of 1:50) was injected subconjunctivally (5 μL/mouse) into the left eye of BALB/c mice (n = 5/group/time) 1 day before infection.
Real-Time PCR
Total RNA was isolated from individual corneas or cell pellets using TRIzol (Invitrogen) according to the manufacturer's instructions; 1 μg of total RNA was reversely transcribed to produce cDNA, and then amplified using SYBR Green Master Mix (Bio-Rad) following the manufacturer's protocol. Primers for mouse IFN-γ, IL-4, and IL-5 were purchased from SABiosciences (Frederick, MD), and primer sequence for others are listed in the Table. Quantitative real-time PCR reactions were performed using the CFX96 Real-Time PCR System (Bio-Rad). Relative gene expression levels were calculated using the relative standard curve method that compares the amount of target normalized to an endogenous reference, β-actin. Briefly, the mean and SEM values of replicate samples were calculated. Samples were then normalized to β-actin. Results are expressed as the relative mRNA levels between experimental test samples and control samples.
Table.
Nucleotide Sequence of the Specific Primers Used in PCR Amplification
|
Gene |
Primer Sequence (5′–3′) |
Primer Location |
| β-actin | GAT TAC TGC TCT GGC TCC TAG C | F |
| GAC TCA TCG TAC TCC TGC TTG C | R | |
| TREM-2 | GGA GGA CCC TCT AGA TGA CCA AGA | F |
| AGG CCA GGA GGA GAA GAA TGG A | R | |
| IL-1β | CGC AGC AGC ACA TCA ACA AGA GC | F |
| TGT CCT CAT CCT GGA AGG TCC ACG | R | |
| TNF-α | CAC AGA AAG CAT GAT CCG CGA C | F |
| TGC CAC AAG CAG GAA TGA GAA GAG | R | |
| IL-10 | AGC TGG ACA ACA TAC TGC TAA CCG AC | F |
| CTT GAT TTC TGG GCC ATG CTT CTC TG | R | |
| IL-12P40 | GGT CAC ACT GGA CCA AAG GGA CTA TG | F |
| ATT CTG CTG CCG TGC TTC CAA C | R | |
| IL-18 | GCC TGT GTT CGA GGA TAT GAC TGA | F |
| TTC ACA GAG AGG GTC ACA GCC A | R | |
| MIP-2 | TGT CAA TGC CTG AAG ACC CTG CC | F |
| AAC TTT TTG ACC GCC CTT GAG AGT GG | R |
F, forward primer; R, reverse primer.
Western Blot
To detect the corneal expression of TREM-2, whole corneas (n = 5/group/time) were collected and pooled from normal uninfected and infected BALB/c eyes at 1, 3, and 5 days p.i. Pooled corneas were lysed and homogenized using a 1-mL glass tissue homogenizer in lysis buffer containing 1 mM phenylmethylsulfonyl fluoride, 1% (vol/vol) protease inhibitor cocktail (Sigma), and 1 mM dithiothreitol (DTT). For detection of total Akt and p-Akt in macrophages, RAW264.7 cells were washed three times with ice-cold PBS and then lysed in the same lysis buffer. Then, protein concentration of the supernatant was determined by Quick Start Bradford protein assay (Bio-Rad); 20 μg of each sample was loaded, separated on 10% SDS-PAGE, and then transferred to a supported nitrocellulose membrane (Pall Life Sciences, Ann Arbor, MI). After blockage, blots were incubated overnight with the respective primary Abs at 4°C, followed by incubation with appropriate horseradish peroxidase (HRP)-conjugated secondary Abs at room temperature for 1 hour. Finally, blots were visualized with Plus-ECL (PerkinElmer, Shelton, CA) according to the manufacturer's protocol. The intensity of each band was measured using Adobe Photoshop 7.0 software (Adobe Systems, Inc., San Jose, CA) and relative integrated density values (IDV) of each band were calculated by normalizing to the β-actin control.
Hematoxylin-Eosin Staining
Normal uninfected and infected eyes were enucleated (n = 3/group/time) at 5 days p.i. from BALB/c mice, embedded in Tissue-Tek optimal cutting temperature (OCT) compound (Miles, Elkhart, IN), and frozen in liquid nitrogen. Then, 8-μm-thick sections were cut and mounted to glass slides. For histopathology, sections were hematoxylin-eosin (HE) stained as described by others.36 All sections were visualized with a Carl Zeiss microscope (Carl Zeiss, Inc., Oberkochen, Germany).
RAW264.7 Cell Culture and Transient Transfection
Murine macrophage-like RAW264.7 cells (ATCC, TIB-71) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS), 1% penicillin-streptomycin, and 1% L-glutamine (all from Invitrogen) at the permissive temperature of 37°C. According to the manufacturer's instruction, RAW264.7 cells were transiently transfected with 33 nM siRNA for TREM-2 or appropriate scrambled control using Lipofectamine 2000 (Invitrogen). After 24 hours of transfection, cells were washed twice with PBS and then challenged with LPS for 18 hours at a concentration of 1 μg/mL.
Isolation of Peritoneal Macrophages
BALB/c mice were intraperitoneally injected with 1.0 mL of 3% Brewer's thioglycollate medium (BD Difco Laboratories) 5 days before cell harvest. Peritoneal exudate cells were collected by peritoneal lavage with DMEM, stained with trypan blue, and viable cells (>95%) were enumerated using a hemacytometer. Then, cells were seeded (1 × 106/well) in 12-well plates and incubated at 37°C. Nonadherent cells were removed 4 hours later and isolated macrophages (>90% purity) were used for in vitro studies.
Isolation of Murine Bone Marrow–Derived Neutrophils and Macrophages
Bone marrow–derived neutrophils were isolated from BALB/c mice. Marrow cells were flushed from femurs and tibias with ice-cold RPMI-1640, and then rinsed and treated with erythrocyte lysis buffer to remove red blood cells. Resuspended cells were laid on the top of discontinuous Percoll density gradients (52%, 64%, 72%) and centrifuged at 400g for 30 minutes at room temperature. Neutrophils were isolated from the bottom layer (64%–72%), counted, and plated into a 12-well culture plate. Cells were cultured in RPMI-1640 medium supplemented with 10% (vol/vol) FBS, 1% penicillin-streptomycin, and 1% L-glutamine. Trypan blue exclusion indicated that the cell viability was approximately 95%. Cells were stained with APC-conjugated anti-Gr-1 (BD Pharmingen) and the purity of neutrophils was more than 95% as determined by flow cytometry. Isolation of bone marrow-derived macrophages (BMDMs) was done as described previously.37 Then, cells were cultured in DMEM supplemented with 10% FBS and 30% (vol/vol) L-929 conditioned medium as a source of macrophage colony-stimulating factor. BMDMs were obtained as a homogeneous population of adherent cells after 7 days of culture. Cells were stained with Alexa Fluor 488-conjugated anti-F4/80 (Invitrogen) and the purity of isolated BMDM was more than 95%.
Flow Cytometry
Flow cytometric analysis was done as described by others.38,39 BMDMs were incubated with phycoerythrin-conjugated anti-TREM-2 (R&D Systems) and Alexa Fluor 488-conjugated anti-F4/80 (Invitrogen) according to the manufacturer's protocol; or stained with isotype control phycoerythrin-conjugated antirat IgG2b (eBioscience) and Alexa Fluor 488-conjugated antirat IgG2a (Invitrogen). Primary neutrophils isolated from bone marrow were incubated with phycoerythrin-conjugated anti–TREM-2 (R&D Systems) and APC-conjugated anti-Gr-1 (BD Pharmingen); or stained with isotype control phycoerythrin-conjugated antirat IgG2b (eBioscience) and APC-conjugated antirat IgG2b (BD Pharmingen). For corneal cells, five corneas were pooled and digested in collagenase type I (Sigma). Cell suspensions were filtered, centrifuged, and resuspended in PBS with 2% BSA. After blocking, cells were distributed into 100-μL samples and incubated with the following Abs: phycoerythrin-conjugated anti–TREM-2 (R&D Systems), Alexa Fluor 488-conjugated anti-F4/80 (Invitrogen), or with isotype control phycoerythrin-conjugated antirat IgG2b (eBioscience) and Alexa Fluor 488-conjugated antirat IgG2a (Invitrogen). Cells were stained for 30 minutes on ice, washed twice with 1 mL FACS buffer, and then resuspended in 1% formaldehyde. Flow cytometry was performed using LSRFortessa Cell Analyzer (BD Biosciences).
Bacterial Plate Counts
Corneas from mTREM-2 versus control siRNA-treated BALB/c mice (at 1 and 5 days p.i.) or corneas from wortmannin versus DMSO/PBS control-treated BALB/c mice (at 5 days p.i.) were collected (n = 5/group/time). The number of viable bacteria was quantitated as described previously.31 Individual corneas were homogenized in normal saline solution containing 0.25% BSA. Serial 10-fold dilutions of the samples were plated on Pseudomonas isolation agar (BD Difco Laboratories) in triplicate and plates were incubated overnight at 37°C. Results are reported as 105 colony forming units (CFU) per cornea ± SEM.
Myeloperoxidase Assay
Infected corneas (n = 5/group/time) from both TREM-2 siRNA and control-treated BALB/c mice were excised at 1 and 5 days p.i., and homogenized in 1 mL of 50 mM phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide (Sigma). Samples were freeze-thawed four times and centrifuged at 16200g for 10 minutes. The supernatant (0.1 mL) was added to 2.9 mL of 50 mM phosphate buffer containing o-dianisidine dihydrochloride (16.7 mg/100 mL; Sigma) and hydrogen peroxide (0.0005%). The change in absorbance at 460 nm was monitored for 5 minutes at 30-second intervals, and the results were expressed as units of myeloperoxidase (MPO) per cornea. One unit of MPO activity is approximately equal to 2 × 105 PMNs.40
ELISA
Corneas from mTREM-2 siRNA- versus control siRNA-treated BALB/c mice (n = 5/group/time) were individually collected at 1 and 5 days p.i. Corneas were homogenized in 0.5 mL PBS with 0.1% Tween-20. All samples were centrifuged at 16200g for 5 minutes and an aliquot of each supernatant was assayed in duplicate for TNF-α, MIP-2, IL-1β, IFN-γ, and IL-10 protein per the manufacturer's instructions. The reported sensitivity of these assays are less than 5.1 pg/mL for TNF-α, less than 1.5 pg/mL for MIP-2, less than 3.0 pg/mL for IL-1β, less than 1.74 pg/mL for IFN-γ, and less than 4.8 pg/mL for IL-10.
Statistical Analysis
The differences in clinical score between TREM-2 siRNA versus control siRNA, or wortmannin versus DMSO/PBS-treated BALB/c mice were tested by the Mann-Whitney U test. Student's t-test or ANOVA was used to determine the statistical significance of other assays. Analysis was performed using Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA). Data were considered significant at P less than 0.05.
Results
TREM-2 Expression in Response to PA Infection
To determine whether TREM-2 mediates ocular inflammation, a well-characterized and accepted murine model of PA keratitis was used to mimic clinical ocular infection. mRNA and protein levels of TREM-2 in normal uninfected and infected BALB/c corneas were tested by real-time PCR and Western blot, respectively. PCR data indicated that TREM-2 was constitutively expressed in normal uninfected BALB/c corneas, whereas after PA infection, TREM-2 mRNA levels were first reduced at 1 day p.i. and then enhanced at 3 and 5 days p.i. (Fig. 1A, all P < 0.001). Western blot data (Fig. 1B) also showed that TREM-2 protein levels were significantly increased at 5 days p.i. (P < 0.001), while no difference was detected at 1 and 3 day p.i., as indicated by relative IDVs (Fig. 1C). Furthermore, flow cytometric analysis demonstrated that TREM-2 expression levels were enhanced in the cells isolated from BALB/c corneas at 3 and 5 days p.i. (Fig. 1D, P < 0.001, P < 0.01, respectively).
Figure 1.
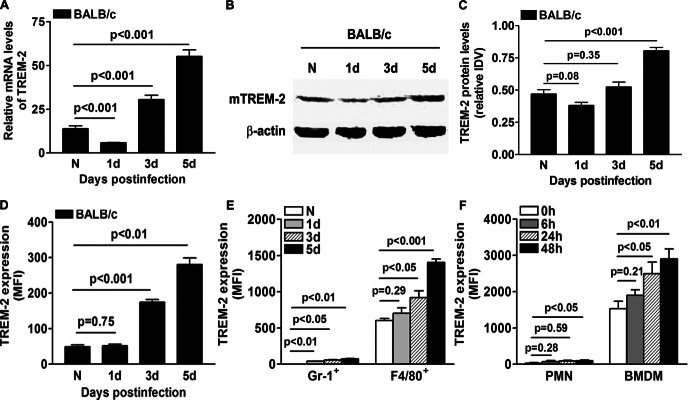
Expression of TREM-2 in response to PA infection. (A) TREM-2 mRNA levels (relative gene expression after normalization to β-actin) were examined in normal uninfected and infected BALB/c corneas at 1, 3, and 5 days p.i. Data are the mean ± SEM and represent three individual experiments each with five animals per time. (B) TREM-2 protein expression levels in BALB/c corneas before and after PA infection were further examined using Western blot. Data for the Western blot represent one of three similar experiments each using five pooled corneas per time. (C) Band intensity was quantitated and normalized to the β-actin control. Data are the mean ± SEM and represent three individual experiments each using five pooled corneas per time. (D, E) Cell suspensions of BALB/c corneas before and at 1, 3, and 5 days p.i. were prepared. TREM-2 expression in normal and infected corneas (D), as well as in neutrophils (Gr-1 positive) and macrophages (F4/80 positive) (E), were analyzed by flow cytometry. Data are mean ± SEM and represent three individual experiments each with five pooled corneas per time. MFI, mean fluorescence intensity. (F) Protein levels of TREM-2 in BMDMs and PMNs isolated from BALB/c mice were examined by flow cytometry before and after PA infection. Data are means ± SEMs (n = 5) and represent three individual experiments.
To further explore the cell source of TREM-2, we analyzed TREM-2 expression on two major infiltrates, neutrophils and macrophages. The results (as indicated by mean fluorescence intensity) showed that TREM-2 was mainly expressed on macrophages in the normal cornea, while no neutrophils were detected before infection. TREM-2 expression was significantly enhanced in neutrophils (Gr-1 positive) at 1, 3, and 5 days p.i. (Fig. 1E, P < 0.01, P < 0.05, P < 0.01, respectively), and elevated in macrophages (F4/80 positive) at 3 and 5 days p.i. (P < 0.05, P < 0.001, respectively). The expression levels of TREM-2 on F4/80-positive cells (macrophages) were approximately 20- to 30-fold higher than that in Gr-1-positive cells (neutrophils) at indicated time points (Fig. 1E). In addition, TREM-2 expression was measured by flow cytometry in BMDMs and neutrophils from BALB/c mice, as well as in A6(1) cells, a corneal epithelial cell line derived from BALB/c mice. The results showed that TREM-2 protein levels were enhanced in neutrophils at 48 hours p.i. (Fig. 1F, P < 0.05) and elevated in BMDM at 24 and 48 hours p.i. (P < 0.05, P < 0.01, respectively). The TREM-2 expression levels in BMDM were approximately 30- to 50-fold higher than that in PMNs at indicated time points (Fig. 1F). In addition, in both uninfected and infected corneal epithelial A6(1) cells, no or little TREM-2 protein expression was detected by flow cytometry (data not shown). These data together suggest that macrophages are the major cell source of TREM-2 expression in the mouse corneas before and after PA infection.
In Vivo Study of TREM-2
Because TREM-2 expression in the infected corneas paralleled disease progression, next in vivo studies were designed to determine the role of TREM-2 in disease severity/onset of PA keratitis. BALB/c mice were treated with mTREM-2 siRNA versus control siRNA by subconjunctival injection together with topical application. Clinical score data (Fig. 2A) showed that TREM-2–silenced mice exhibited enhanced disease levels at 3 and 5 days p.i. (P < 0.01, P < 0.001, respectively). Representative photographs taken with a slit lamp of the infected corneas at 5 days p.i. in BALB/c mice treated with scrambled control (Fig. 2B) versus TREM-2 siRNA (Fig. 2C) are provided. Treatment with TREM-2 siRNA resulted in dense opacity covering the entire anterior segment (grade = +3, as shown in Fig. 2C) or perforation (grade = +4, data not shown), and overall more inflammation than scrambled control treatment (grade = +1/+2, as shown in Fig. 2B) at 5 days p.i. These results demonstrated that silencing of TREM-2 shifted BALB/c mice from a resistant to a susceptible phenotype in response to PA corneal infection.
Figure 2.
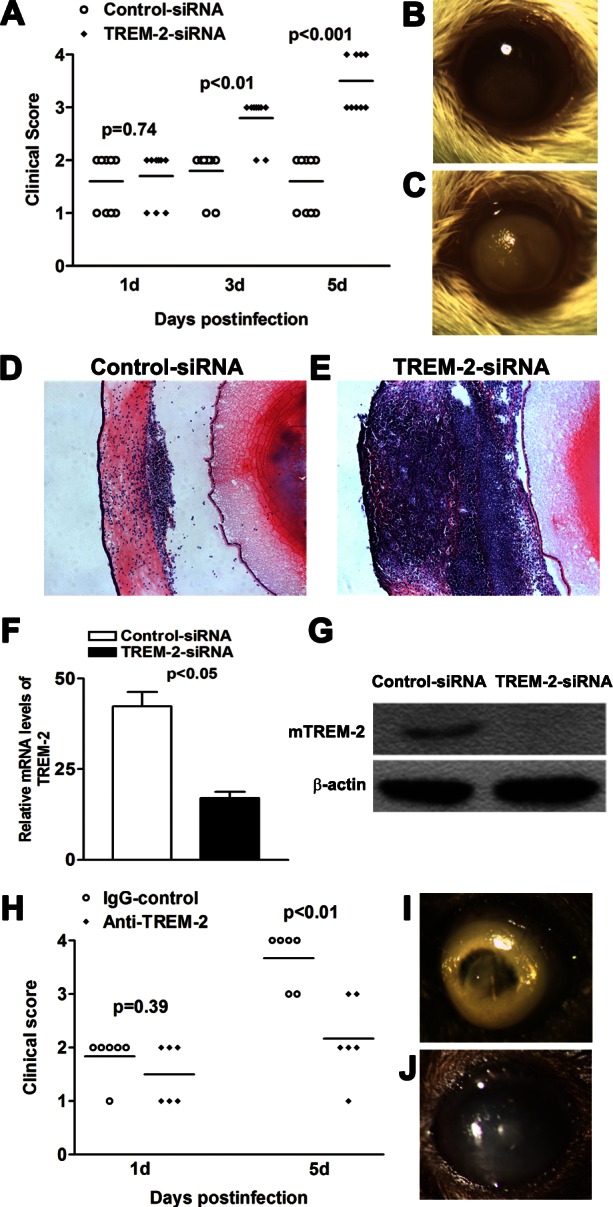
In vivo studies of TREM-2. (A–G) BALB/c mice were subconjunctivally injected with TREM-2 siRNA or scrambled control, and then infected with PA routinely. Clinical scores (A) indicated statistically significant differences at 3 and 5 days p.i. between TREM-2–silenced and control groups. Representative photographs of infected eyes at 5 days p.i. displayed more opacity in TREM-2 siRNA- (C) versus control- (B) treated mice. Magnification ×10. HE staining was used to examine the histopathology of infected eyes in both TREM-2–silenced (E) and control-treated (D) mice. Magnification ×100. Images shown are representative of three individual experiments each with three mice per group. (F) mRNA levels of TREM-2 in TREM-2–silenced versus control-treated corneas were examined at 5 days p.i. Data are the mean ± SEM and represent two individual experiments each with five animals per group. (G) Protein levels of TREM-2 in TREM-2–silenced versus control-treated corneas were examined by Western blot at 5 days p.i. Data represent one of three similar experiments each using five pooled corneas per group. (H–J) B6 mice were subconjunctivally injected with agonistic anti–TREM-2 Ab or isotype control, and then infected with PA routinely. Clinical scores (H) indicated statistically significant differences at 5 days p.i. between TREM-2–activated and control groups. Representative photographs of infected eyes at 5 days p.i. displayed less opacity in TREM-2-activated (J) versus control-treated (I) mice. Magnification ×10.
To further confirm these data, we enucleated the infected eyes at 5 days p.i. from TREM-2–silenced versus control-treated BALB/c mice for histopathology. TREM-2–silenced BALB/c mice (Fig. 2E) exhibited a more swollen cornea, numerous infiltrating cells in the corneal stroma and anterior chamber, and tissue destruction after PA infection. In contrast, the infected corneas of BALB/c mice treated with control siRNA (Fig. 2D) were less swollen, displayed a relatively intact epithelium, and fewer infiltrated inflammatory cells in the corneal stroma and anterior chamber. In addition, real-time PCR and Western blot data demonstrated that at 5 days p.i., both mRNA (Fig. 2F, P < 0.05) and protein levels of TREM-2 (Fig. 2G) were decreased in the TREM-2–silenced versus control-treated corneas, confirming the efficacy of silencing.
To further confirm the role of TREM-2 in PA keratitis, susceptible B6 mice were subconjunctivally injected with agonistic anti–TREM-2 Ab versus isotype control IgG, and then infected with PA routinely. Clinical score data showed that TREM-2–activated mice displayed less disease severity at 5 days p.i. (P < 0.05), indicating that TREM-2 promotes host resistance against PA corneal infection. TREM-2–activated B6 mice displayed less severe disease after PA corneal infection (representative photograph is shown in Fig. 2J, grade = +2), while most of the control-treated B6 corneas showed a dense opacity covering the anterior chamber (Fig. 2I, grade = +3), or corneal perforation (grade = +4, data not shown).
Silencing of TREM-2 Enhanced Bacterial Load and PMN Infiltration After PA Infection
Because both bacterial virulence and host inflammation contributed to the disease pathogenesis of PA keratitis, we further assessed the effect of TREM-2 silencing on bacterial load and PMN infiltration. Bacterial plate counts were used to detect viable bacteria in the infected corneas of TREM-2–silenced versus control-treated mice at 1 and 5 days p.i. The results showed that silencing of TREM-2 elevated bacterial load at 5 days p.i. (Fig. 3A, P < 0.001), whereas no change was shown between the two groups at 1 day p.i. Moreover, PMN infiltration (Fig. 3B, shown as MPO activity) was enhanced in TREM-2–silenced mouse corneas at 1 and 5 days p.i. (both P < 0.001). In addition, silencing of TREM-2 significantly downregulated the mRNA levels of cathelicidin-related antimicrobial peptide (CRAMP) and NADPH oxidase (NOX2), an enzyme for reactive oxygen species (ROS) generation (Fig. 3C, both P < 0.05), suggesting that TREM-2 is required in both oxygen-independent and oxygen-dependent bacterial killing.
Figure 3.
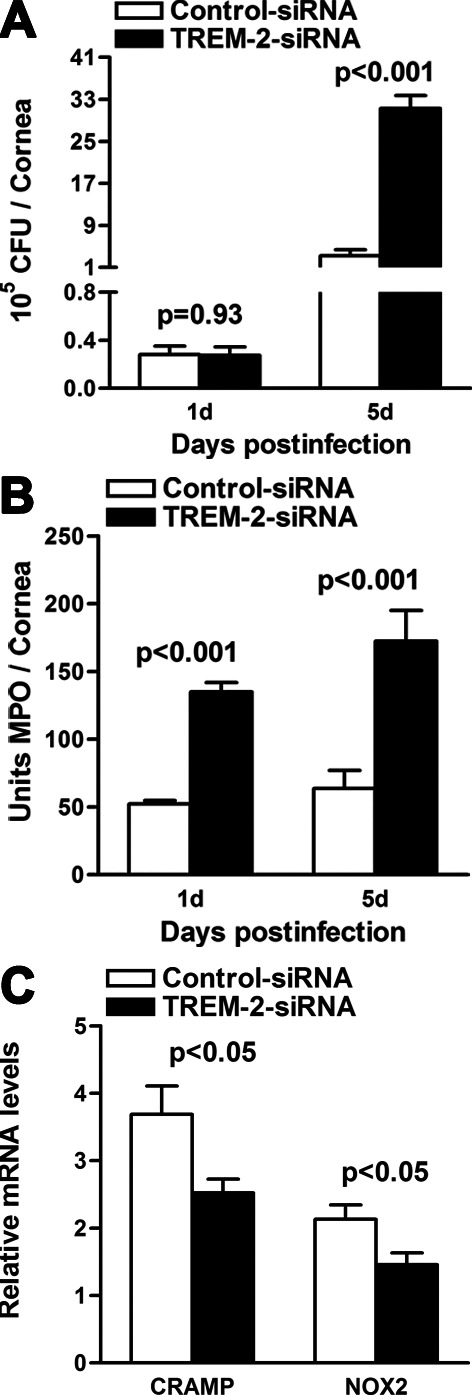
Silencing of TREM-2 enhanced bacterial load and PMN infiltration after PA infection. Bacterial load (A) was enhanced in TREM-2 siRNA versus control-treated corneas at 5 days p.i. The number of infiltrated PMNs as detected by MPO assay (B) was elevated in the corneas at 1 and 5 days p.i. (C) mRNA levels of CRAMP and NOX2 were decreased in TREM-2–silenced versus control-treated corneas at 5 days p.i. Data are the means ± SEMs and represent three individual experiments each with five animals per group per time per assay.
Silencing of TREM-2–Modulated Production of Th1- and Th2-Type Cytokines in PA Keratitis
To further ascertain the mechanism by which TREM-2 modulated the inflammatory response, mRNA and protein levels of selected Th1- and Th2-type cytokines were analyzed by real-time PCR and ELISA in the infected corneas of TREM-2– versus control siRNA-treated BALB/c mice. At 1 and 5 days p.i., silencing of TREM-2 significantly upregulated the mRNA levels of Th1-type cytokines, including IFN-γ (Fig. 4A, both P < 0.05), IL-12 (Fig. 4B, P < 0.001, P < 0.01, at 1 and 5 days p.i., respectively), and IL-18 (Fig. 4C, P < 0.05, P < 0.01, at 1 and 5 days p.i., respectively), but downregulated the mRNA levels of Th2-type cytokines, such as IL-4 (Fig. 4D, both P < 0.05), IL-5 (Fig. 4E, P < 0.001, P < 0.05, at 1 and 5 days p.i., respectively), and IL-10 (Fig. 4F, both P < 0.05), when compared with control treatment. Furthermore, silencing of TREM-2 significantly upregulated protein levels of IFN-γ (Fig. 4G, both P < 0.05), but downregulated protein levels of IL-10 (Fig. 4H, both P < 0.05) at 1 and 5 days p.i. These results indicated that silencing of TREM-2 promoted Th1- versus Th2-type cytokine responses in PA keratitis.
Figure 4.

Expression of Th1-/Th2-type cytokines after in vivo silencing of TREM-2. (A–F) Real-time PCR data demonstrated that at 1 and 5 days p.i., silencing of TREM-2 upregulated the expression of Th1-type cytokines, including IFN-γ, AIL-12 (B), and IL-18 (C), but downregulated the expression of Th2-type cytokines, including IL-4 (D), IL-5 (E), and IL-10 (F) in the PA-infected BALB/c corneas, when compared with control treatment. (G, H) ELISA data demonstrated that at 1 and 5 days p.i., silencing of TREM-2 upregulated IFN-γ expression but downregulated IL-10 expression (H). Data are the mean ± SEM and represent three individual experiments each with five animals per group per time.
Silencing of TREM-2 Enhanced the Production of Proinflammatory Cytokines in PA Keratitis
We also tested mRNA and protein expression levels of proinflammatory cytokines by real-time PCR and ELISA, respectively. At 1 and 5 days p.i., silencing of TREM-2 significantly enhanced the mRNA levels of TNF-α (Fig. 5A, P < 0.01, P < 0.05, at 1 and 5 days p.i., respectively), MIP-2 (Fig. 5B, P < 0.05, P < 0.01, at 1 and 5 days p.i., respectively), and IL-1β (Fig. 5C, P < 0.05, P < 0.01, at 1 and 5 days p.i., respectively), when compared with controls. Moreover, ELISA data also showed higher protein expression levels of TNF-α (Fig. 5D, P < 0.001, P < 0.01, at 1 and 5 days p.i., respectively), MIP-2 (Fig. 5E, P < 0.001, P < 0.01, at 1 and 5 days p.i., respectively), and IL-1β (Fig. 5F, P < 0.001, P < 0.01, at 1 and 5 days p.i., respectively) in TREM-2–silenced versus control-treated BALB/c corneas at 1 and 5 days p.i. These data suggested that silencing of TREM-2 enhanced corneal inflammation in response to PA ocular infection.
Figure 5.

Expression of proinflammatory cytokines after in vivo silencing of TREM-2. Real-time PCR data demonstrated that at 1 and 5 days p.i., silencing of TREM-2 enhanced the mRNA levels of proinflammatory cytokines, including TNF-α (A), MIP-2 (B), and IL-1β (C) in the PA-infected BALB/c mouse cornea, compared with control treatment. ELISA data further demonstrated that at both 1 and 5 days p.i., protein expression levels of TNF-α (D), MIP-2 (E), and IL-1β (F) were enhanced in TREM-2–silenced versus control-treated BALB/c mouse cornea. Data are the means ± SEMs and represent three individual experiments each with five animals per group per time per assay.
Production of Th1-/Th2-Type and Proinflammatory Cytokines After In Vitro Silencing of TREM-2
In addition to in vivo murine models, we also tested the role of TREM-2 on the inflammatory response by using in vitro culture of murine macrophages. RAW 264.7 cells (a macrophage cell line derived from BALB/c mice) were transfected with TREM-2 siRNA or scrambled control for 24 hours, and then challenged with LPS derived from PA for 18 hours at a concentration of 1 μg/mL. PCR data showed that silencing of TREM-2 significantly upregulated Th1-type cytokines IFN-γ, IL-12, and IL-18 (Fig. 6A, P < 0.05, P < 0.01, and P < 0.01, respectively), and proinflammatory cytokines TNF-α, MIP-2, and IL-1β (Fig. 6C, P < 0.05, P < 0.01, and P < 0.01, respectively), but downregulated Th2-type cytokines IL-4, IL-5, and IL-10 (Fig. 6B, all P < 0.05) in RAW264.7 cells after LPS stimulation.
Figure 6.

Expression of Th1/Th2 and proinflammatory cytokines after in vitro silencing of TREM-2. PCR data demonstrated that in LPS-stimulated RAW264.7 cells, silencing of TREM-2 increased expression of Th1 cytokines, such as IFN-γ, IL-12, and IL-18 (A) and proinflammatory cytokines, such as TNF-α, MIP-2, and IL-1β (C), but decreased expression of Th2 cytokines, including IL-4, IL-5, and IL-10 (B), when compared with control treatment. Data are the means ± SEMs (n = 5) and represent three individual experiments.
TREM-2 Inhibited Proinflammatory Cytokine Production via a PI3K/Akt Signaling Pathway
To further evaluate which signaling pathway is involved in TREM-2–mediated immune regulation, murine macrophage-like RAW264.7 cells were incubated with agonistic anti–TREM-2 antibody, which can activate TREM-2 signaling by cross-linking ligation. Western blot data showed that protein levels of p-Akt were significantly upregulated at 10 and 15 minutes after treatment with agonistic anti–TREM-2 antibody (Fig. 7A), suggesting the involvement of Akt in the downstream signaling of TREM-2 activation. To further confirm the role of PI3K/Akt signaling in the TREM-2–modulated immune response, peritoneal macrophages from BALB/c mice were pretreated with inhibitors for PI3K or Akt, and then stimulated with agonistic anti–TREM-2 antibody together with LPS stimulation. Although activation of TREM-2 decreased production of proinflammatory cytokines, such as TNF-α, MIP-2, and IL-1β in LPS-stimulated BALB/c peritoneal macrophages (Fig. 7B, all P < 0.01), pretreatment with Akt inhibitor IX API-59CJ-OMe (Fig. 7C), PI3K inhibitors LY294002 (Fig. 7D), or wortmannin (Fig. 7E) restored TREM-2–mediated downregulation of these proinflammatory cytokines in BALB/c peritoneal macrophages. These data indicated that TREM-2 suppressed the production of proinflammatory cytokines via activating the PI3K/Akt signaling pathway.
Figure 7.

TREM-2 suppressed production of proinflammatory cytokines via PI3K/Akt signaling pathway. (A) Phosphorylated and total protein levels of Akt in RAW264.7 cells were examined by Western blot before and after treatment with agonistic TREM-2 antibody. Activation of TREM-2 increased protein levels of p-Akt at 10 and 15 minutes posttreatment, with no changes detected in the protein levels of total Akt. Data for the Western blot represent one of three similar experiments. (B) Treatment with agonistic TREM-2 antibody significantly downregulated mRNA levels of TNF-α, MIP-2, and IL-1β in BALB/c peritoneal macrophages after LPS stimulation, although pretreatment with Akt inhibitor IX API-59CJ-OMe (C), PI3K inhibitors LY294002 (D), or wortmannin (E) restored the TREM-2–mediated downregulation of proinflammatory cytokines. Data are the means ± SEMs (n = 5) and represent three individual experiments.
Inhibition of PI3K Accelerated Disease Progression After PA Infection
Because in vitro data suggested that TREM-2 suppressed the production of proinflammatory cytokines via activating the PI3K/Akt signaling pathway, we then examined whether PI3K had a role in disease progression. BALB/c mice were subconjunctivally injected with the PI3K inhibitor wortmannin versus DMSO/PBS control. Clinical score data showed that treatment with wortmannin significantly enhanced disease levels at 5 days p.i. (P < 0.01), whereas no difference was detected between the two groups at 1 day p.i. (Fig. 8A). At 5 days p.i., corneas of wortmannin-treated mice exhibited either dense opacity covering the entire anterior segment (grade = +3, as shown in Fig. 8C), or corneal perforation (grade = +4, data not shown), and overall more inflammation than control-treated corneas (grade = +2/+3, as shown in Fig. 8B). Furthermore, treatment with wortmannin resulted in elevated bacterial load at 5 days p.i. (Fig. 8D, P < 0.05) and enhanced mRNA levels of TNF-α, MIP-2, and IL-1β (Fig. 8E, P < 0.01, P < 0.05, P < 0.01, respectively), compared with controls. These data indicated that PI3K/Akt signaling is required in the ocular immune defense response to PA infection.
Figure 8.
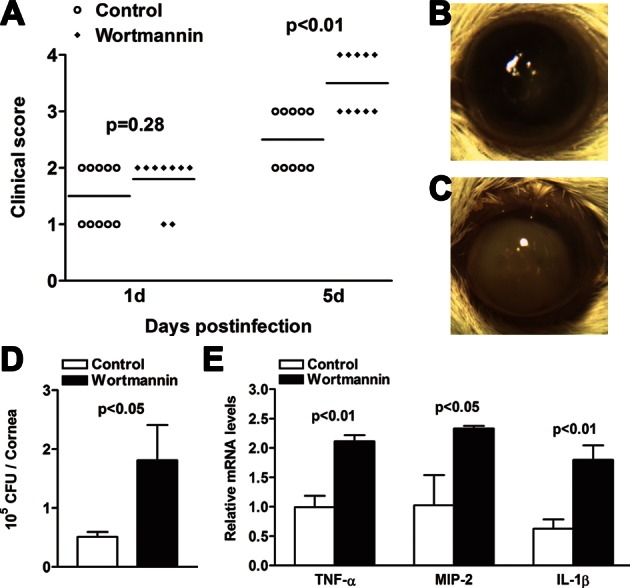
Inhibition of PI3K accelerated disease progression after PA infection. (A) Clinical scores indicated significant differences at 5 days p.i. between wortmannin and control group. Representative photographs of infected eyes at 5 days p.i. displayed more opacity in wortmannin- (C) versus control- (B) treated mice. Magnification ×10. Bacterial load (D) was enhanced in wortmannin- versus control-treated corneas at 5 days p.i. Real-time PCR data (E) demonstrated that at 5 days p.i., treatment with wortmannin enhanced the mRNA levels of proinflammatory cytokines, including TNF-α, MIP-2, and IL-1β in the PA-infected BALB/c corneas, compared with control treatment. Data are the means ± SEMs and represent three individual experiments each with five animals per group per time.
Discussion
TREM-2 is a novel cell surface receptor that is expressed on immature dendritic cells, microglia, and osteoclasts, as well as on a variety of primary macrophages and macrophage cell lines.41 Studies have demonstrated that TREM-2 may function as a negative regulator of TLR signaling in the immune response.21 However, nothing is known regarding the role of TREM-2 in the eye.
In the present study, TREM-2 expression (mRNA and protein) levels were significantly enhanced at later time points p.i. (3 or 5 days p.i.), whereas at 1 day p.i., TREM-2 mRNA expression was slightly decreased in the infected BALB/c cornea, and no difference in TREM-2 protein expression was detected. We further used flow cytometry to analyze TREM-2 in two major infiltrates, PMNs and macrophages. Both in vivo and in vitro data showed that TREM-2 expression levels in PMNs are much lower than those in macrophages, either with or without PA infection, indicating that the macrophage is the major cell type that expresses TREM-2. Previous studies have demonstrated that during PA keratitis, PMN influx occurs in the early time p.i. (usually begins at 1 day p.i.), whereas macrophages infiltrate into the infected corneas later,7 which may explain the TREM-2 upregulation detected at later time points. We also observed that before infection, TREM-2 is expressed on some F4/80+ cells, whereas no Gr-1+ cells were detected, indicating that local resident macrophages are the major cell source of TREM-2 expression in the normal cornea.
The PA-induced TREM-2 expression in vivo and in vitro suggests a potential association of this molecule with PA keratitis. Our studies demonstrated that silencing of TREM-2 increased the production of IFN-γ, IL-12, and IL-18, but decreased the expression of IL-4, IL-5, and IL-10 in PA-infected BALB/c mouse corneas, indicating that TREM-2 promoted host resistance against PA infection by enhancing a Th2-type response. Studies have demonstrated that in the C57BL/6 mice, which are Th1 responders, PA corneal infection often causes a rapid disease progression and leads to corneal perforation at 5 days p.i.,17,42 whereas in BALB/c mice, which are Th2 responders, PA challenge usually results in less disease severity, and the infected corneas often heal at 5 days p.i.31,33 Thus, the balance of Th1- and Th2-type responses is critical in determining the disease outcome of PA keratitis.6,7,43,44
Furthermore, our study demonstrated that silencing of TREM-2 enhanced production of proinflammatory cytokines, such as TNF-α, MIP-2, and IL-1β, in the infected corneas, indicating that TREM-2 acts as an anti-inflammatory PRR in the eye. Interestingly, after PA infection, TNF-α protein expression in the cornea was very low, whereas MIP-2 and IL-1β protein levels were much higher. This phenomenon was also observed in the susceptible B6 murine model of PA keratitis, as reported by our laboratory previously.17 Both MIP-2 and IL-1β are important chemokines to recruit neutrophils,45 thus the high expression of MIP-2 and IL-1β would likely contribute to increased neutrophil infiltration, as indicated by the MPO data. Although these inflammatory cytokines and cells are beneficial to the antibacterial response, excessive inflammatory infiltration and upregulated proinflammatory cytokine production also will lead to immunopathological tissue damage and corneal perforation.
It is worth pointing out that bacterial virulence is another main factor of tissue damage and corneal perforation. In the present study, silencing of TREM-2 increased bacterial load in the PA-infected BALB/c mouse corneas, which also contributes to the corneal destruction and disease progression. However, this elevation of bacterial burden in the TREM-2 silenced mouse cornea seems to be contrary to the upregulated PMN infiltration, which would be anticipated to kill more bacteria.7 These paradoxical findings between the increased PMN infiltration and increased CFU were also observed in other studies on PA keratitis,46,47 and may be explained by the following reasons. First, although PMNs function as important phagocytes, in turn, bacterial infection also induces host cells to produce more chemokines, such as IL-1β and MIP-2, to recruit more PMNs infiltrated into the infected cornea. As we know, every force leads to an equal and opposite counter force. It is common that more bacterial infection (increased CFUs) leads to more PMN infiltration in order to clear bacteria. Second, the excessive PMN infiltration results in stromal destruction and tissue damage, which may provide nutritional components to favor bacterial growth. Third, our data showed that silencing of TREM-2 decreased the production of CRAMP and NOX2 in the infected cornea, indicating that TREM-2 is also involved in both oxygen-dependent and oxygen-independent microbicidal systems, which may be another reason for the increased CFU in TREM-2 silenced corneas.
Besides using the in vivo murine model, we also tested the role of TREM-2 in mouse peritoneal macrophages and macrophage cell line RAW264.7 cells. Studies have demonstrated that depletion of macrophages in susceptible C57BL/6 mice decreased bacterial clearance, delayed PMN infiltration, aggravated tissue damage, and increased onset/severity of PA keratitis,7 indicating the importance of macrophages in determining the disease outcome of PA infection. Our in vitro studies indicated that TREM-2 inhibited the production of Th1-type and proinflammatory cytokines but elevated Th2-type cytokine expression in mouse macrophages after LPS challenge, which further confirmed the anti-inflammatory role of TREM-2 in vivo.
In addition, our study demonstrated that phosphorylated Akt levels were enhanced in TREM-2–activated versus control-treated macrophages, whereas pretreatment of PI3K or Akt inhibitors restored the TREM-2–mediated downregulation of proinflammatory cytokines, indicating that PI3K/Akt signaling is required in TREM-2–mediated immune regulation. We further demonstrated that treatment with PI3K inhibitor wortmannin accelerated the disease progression of PA keratitis in BALB/c mice, and enhanced the bacterial load, as well as proinflammatory cytokine production, indicating that PI3K signaling is required in the ocular immune defense against PA infection. This observation is consistent with some studies showing that intraperitoneal injection with wortmannin increased serum levels of IL-1β, TNF-α, and IL-6 and led to susceptibility to sepsis,48 but different from other reports showing that wortmannin inhibited inflammation in dextran sulphate sodium-induced colitis.49 Although the in vivo effects of wortmannin are still controversial, substantial evidence has shown that PI3K/Akt activation promotes the internalization of PA strains PAO1 and PAK by epithelial cells,50–52 and suppresses a proinflammatory response through negatively regulating TLR signaling.53 It is also reported that PI3K/Akt is critical in modulating the activities of macrophages, such as the balance of classical activation (M1-polarized macrophages) versus alternative activation (M2-polarized macrophages).54–56 Studies have demonstrated that M1/M2 polarization can influence the balance between Th1- and Th2-type responses.57 M1-polarized macrophages often secrete a large amount of proinflammatory cytokines, causing tissue injury, whereas M2-polarized macrophages promote healing and repair.54 In this regard, the PI3K/Akt-dependent macrophage polarization may also contribute to TREM-2–mediated host resistance.
In summary, our in vivo and in vitro studies provide substantial evidence that TREM-2 was significantly enhanced in mouse corneas after PA infection, and promoted host resistance against infection by reducing corneal inflammation via a PI3K/Akt signaling pathway. Collectively, the data may hold promise for alternative clinical treatment of PA keratitis and other infectious diseases.
Acknowledgments
Supported by grants from the National Natural Science Foundation of China (U0832006, 81172811, 31200662, 81261160323), the Open Project Grant at State Key Laboratory of Ophthalmology (Zhongshan Ophthalmic Center), Guangdong Innovative Research Team Program (No. 2009010058, No. 2011Y035), Specialized Research Fund for the Doctoral Program of Higher Education of China (20100171110047), Guangdong Natural Science Foundation (10251008901000013, S2012040006680), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (No. 2009), National Science and Technology Key Projects for Major Infectious Diseases (2013ZX10003001), and National Institutes of Health Grants R01 EY019021 and P30 EY04068.
Disclosure: M. Sun, None; M. Zhu, None; K. Chen, None; X. Nie, None; Q. Deng, None; L.D. Hazlett, None; Y. Wu, None; M. Li, None; M. Wu, None; X. Huang, None
References
- 1. Markoulli M, Papas E, Cole N, Holden B. Corneal erosions in contact lens wear. Cont Lens Anterior Eye. 2012; 35: 2–8 [DOI] [PubMed] [Google Scholar]
- 2. Szczotka-Flynn LB, Pearlman E, Ghannoum M. Microbial contamination of contact lenses, lens care solutions, and their accessories: a literature review. Eye Contact Lens. 2010; 36: 116–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Patel A, Hammersmith K. Contact lens-related microbial keratitis: recent outbreaks. Curr Opin Ophthalmol. 2008; 19: 302–306 [DOI] [PubMed] [Google Scholar]
- 4. Pinna A, Usai D, Sechi LA, Molicotti P, Zanetti S, Carta A. Detection of virulence factors in Pseudomonas aeruginosa strains isolated from contact lens-associated corneal ulcers. Cornea. 2008; 27: 320–326 [DOI] [PubMed] [Google Scholar]
- 5. Willcox MD. Pseudomonas aeruginosa infection and inflammation during contact lens wear: a review. Optom Vis Sci. 2007; 84: 273–278 [DOI] [PubMed] [Google Scholar]
- 6. Hazlett LD. Pathogenic mechanisms of P. aeruginosa keratitis: a review of the role of T cells, Langerhans cells, PMN, and cytokines. DNA Cell Biol. 2002; 21: 383–390 [DOI] [PubMed] [Google Scholar]
- 7. Hazlett LD. Corneal response to Pseudomonas aeruginosa infection. Prog Retin Eye Res. 2004; 23: 1–30 [DOI] [PubMed] [Google Scholar]
- 8. Engel LS, Callegan MC, Hobden JA, Reidy JJ, Hill JM, O'Callaghan RJ. Effectiveness of specific antibiotic/steroid combinations for therapy of experimental Pseudomonas aeruginosa keratitis. Curr Eye Res. 1995; 14: 229–234 [DOI] [PubMed] [Google Scholar]
- 9. McClellan SA, Huang X, Barrett RP, van Rooijen N, Hazlett LD. Macrophages restrict Pseudomonas aeruginosa growth, regulate polymorphonuclear neutrophil influx, and balance pro- and anti-inflammatory cytokines in BALB/c mice. J Immunol. 2003; 170: 5219–5227 [DOI] [PubMed] [Google Scholar]
- 10. Kernacki KA, Barrett RP, McClellan S, Hazlett LD. MIP-1alpha regulates CD4+ T cell chemotaxis and indirectly enhances PMN persistence in Pseudomonas aeruginosa corneal infection. J Leukoc Biol. 2001; 70: 911–919 [PubMed] [Google Scholar]
- 11. Kernacki KA, Barrett RP, Hobden JA, Hazlett LD. Macrophage inflammatory protein-2 is a mediator of polymorphonuclear neutrophil influx in ocular bacterial infection. J Immunol. 2000; 164: 1037–1045 [DOI] [PubMed] [Google Scholar]
- 12. Kernacki KA, Goebel DJ, Poosch MS, Hazlett LD. Early cytokine and chemokine gene expression during Pseudomonas aeruginosa corneal infection in mice. Infect Immun. 1998; 66: 376–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001; 410: 1103–1107 [DOI] [PubMed] [Google Scholar]
- 14. Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. 2003; 3: 445–453 [DOI] [PubMed] [Google Scholar]
- 15. Lagler H, Sharif O, Haslinger I, et al. TREM-1 activation alters the dynamics of pulmonary IRAK-M expression in vivo and improves host defense during pneumococcal pneumonia. J Immunol. 2009; 183: 2027–2036 [DOI] [PubMed] [Google Scholar]
- 16. Klesney-Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol. 2006; 7: 1266–1273 [DOI] [PubMed] [Google Scholar]
- 17. Wu M, Peng A, Sun M, et al. TREM-1 amplifies corneal inflammation after Pseudomonas aeruginosa infection by modulating Toll-like receptor signaling and Th1/Th2-type immune responses. Infect Immun. 2011; 79: 2709–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Turnbull IR, Gilfillan S, Cella M, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006; 177: 3520–3524 [DOI] [PubMed] [Google Scholar]
- 19. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005; 201: 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piccio L, Buonsanti C, Mariani M, et al. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. 2007; 37: 1290–1301 [DOI] [PubMed] [Google Scholar]
- 21. Ito H, Hamerman JA. TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur J Immunol. 2012; 42: 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. 2010; 3:ra 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999; 401: 86–90 [DOI] [PubMed] [Google Scholar]
- 24. Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999; 344: 427–431 [PMC free article] [PubMed] [Google Scholar]
- 25. Lawlor MA, Rotwein P. Insulin-like growth factor-mediated muscle cell survival: central roles for Akt and cyclin-dependent kinase inhibitor p21. Mol Cell Biol. 2000; 20: 8983–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998; 282: 1318–1321 [DOI] [PubMed] [Google Scholar]
- 27. Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003; 24: 358–363 [DOI] [PubMed] [Google Scholar]
- 28. Buntru A, Kopp K, Voges M, Frank R, Bachmann V, Hauck CR. Phosphatidylinositol 3′-kinase activity is critical for initiating the oxidative burst and bacterial destruction during CEACAM3-mediated phagocytosis. J Biol Chem. 2011; 286: 9555–9566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006; 7: 606–619 [DOI] [PubMed] [Google Scholar]
- 30. Chen KF, Yeh PY, Yeh KH, Lu YS, Huang SY, Cheng AL. Down-regulation of phospho-Akt is a major molecular determinant of bortezomib-induced apoptosis in hepatocellular carcinoma cells. Cancer Res. 2008; 68: 6698–6707 [DOI] [PubMed] [Google Scholar]
- 31. Huang X, Du W, Barrett RP, Hazlett LD. ST2 is essential for Th2 responsiveness and resistance to Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci. 2007; 48: 4626–4633 [DOI] [PubMed] [Google Scholar]
- 32. Wu M, McClellan SA, Barrett RP, Hazlett LD. Beta-defensin-2 promotes resistance against infection with P. aeruginosa. J Immunol. 2009; 182: 1609–1616 [DOI] [PubMed] [Google Scholar]
- 33. Huang X, Hazlett LD, Du W, Barrett RP. SIGIRR promotes resistance against Pseudomonas aeruginosa keratitis by down-regulating type-1 immunity and IL-1R1 and TLR4 signaling. J Immunol. 2006; 177: 548–556 [DOI] [PubMed] [Google Scholar]
- 34. Huang X, Barrett RP, McClellan SA, Hazlett LD. Silencing Toll-like receptor-9 in Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci. 2005; 46: 4209–4216 [DOI] [PubMed] [Google Scholar]
- 35. Peng LH, Shen W, Yong W, Lu L, Liu L. Effects of AMD3100 subconjunctival injection on alkali burn induced corneal neovascularization in mice. Int J Ophthalmol. 2011; 4: 44–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weihua Z, Makela S, Andersson LC, et al. A role for estrogen receptor beta in the regulation of growth of the ventral prostate. Proc Natl Acad Sci U S A. 2001; 98: 6330–6335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc. 2008; 2008: pdb.prot5080 [DOI] [PubMed] [Google Scholar]
- 38. Sun Y, Karmakar M, Roy S, et al. TLR4 and TLR5 on corneal macrophages regulate Pseudomonas aeruginosa keratitis by signaling through MyD88-dependent and -independent pathways. J Immunol. 2010; 185: 4272–4283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roy S, Sun Y, Pearlman E. Interferon-gamma-induced MD-2 protein expression and lipopolysaccharide (LPS) responsiveness in corneal epithelial cells is mediated by Janus tyrosine kinase-2 activation and direct binding of STAT1 protein to the MD-2 promoter. J Biol Chem. 2011; 286: 23753–23762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Williams RN, Paterson CA, Eakins KE, Bhattacherjee P. Quantification of ocular inflammation: evaluation of polymorphonuclear leucocyte infiltration by measuring myeloperoxidase activity. Curr Eye Res. 1982; 2: 465–470 [DOI] [PubMed] [Google Scholar]
- 41. Sharif O, Knapp S. From expression to signaling: roles of TREM-1 and TREM-2 in innate immunity and bacterial infection. Immunobiology. 2008; 213: 701–713 [DOI] [PubMed] [Google Scholar]
- 42. McClellan SA, Zhang Y, Barrett RP, Hazlett LD. Substance P promotes susceptibility to Pseudomonas aeruginosa keratitis in resistant mice: anti-inflammatory mediators downregulated. Invest Ophthalmol Vis Sci. 2008; 49: 1502–1511 [DOI] [PubMed] [Google Scholar]
- 43. Hazlett LD. Bacterial infections of the cornea (Pseudomonas aeruginosa). Chem Immunol Allergy. 2007; 92: 185–194 [DOI] [PubMed] [Google Scholar]
- 44. Hazlett LD, McClellan S, Kwon B, Barrett R. Increased severity of Pseudomonas aeruginosa corneal infection in strains of mice designated as Th1 versus Th2 responsive. Invest Ophthalmol Vis Sci. 2000; 41: 805–810 [PubMed] [Google Scholar]
- 45. Gupta S, Feng L, Yoshimura T, Redick J, Fu SM, Rose CE Jr. Intra-alveolar macrophage-inflammatory peptide 2 induces rapid neutrophil localization in the lung. Am J Respir Cell Mol Biol. 1996; 15: 656–663 [DOI] [PubMed] [Google Scholar]
- 46. Zhou Z, Wu M, Barrett RP, McClellan SA, Zhang Y, Hazlett LD. Role of the Fas pathway in Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci. 2010; 51: 2537–2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hazlett LD, McClellan SA, Barrett RP, et al. IL-33 shifts macrophage polarization, promoting resistance against Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci. 2010; 51: 1524–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Williams DL, Li C, Ha T, et al. Modulation of the phosphoinositide 3-kinase pathway alters innate resistance to polymicrobial sepsis. J Immunol. 2004; 172: 449–456 [DOI] [PubMed] [Google Scholar]
- 49. Dai C, Zheng CQ, Meng FJ, Zhou Z, Sang LX, Jiang M. VSL#3 probiotics exerts the anti-inflammatory activity via PI3k/Akt and NF-kappaB pathway in rat model of DSS-induced colitis. Mol Cell Biochem. 2013; 374: 1–11 [DOI] [PubMed] [Google Scholar]
- 50. Kierbel A, Gassama-Diagne A, Mostov K, Engel JN. The phosphoinositol-3-kinase-protein kinase B/Akt pathway is critical for Pseudomonas aeruginosa strain PAK internalization. Mol Biol Cell. 2005; 16: 2577–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kierbel A, Gassama-Diagne A, Rocha C, et al. Pseudomonas aeruginosa exploits a PIP3-dependent pathway to transform apical into basolateral membrane. J Cell Biol. 2007; 177: 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sana TG, Hachani A, Bucior I, et al. The second type VI secretion system of Pseudomonas aeruginosa strain PAO1 is regulated by quorum sensing and Fur and modulates internalization in epithelial cells. J Biol Chem. 2012; 287: 27095–27105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002; 277: 32124–32132 [DOI] [PubMed] [Google Scholar]
- 54. Whyte CS, Bishop ET, Ruckerl D, et al. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J Leukoc Biol. 2011; 90: 845–854 [DOI] [PubMed] [Google Scholar]
- 55. MacKinnon AC, Farnworth SL, Hodkinson PS, et al. Regulation of alternative macrophage activation by galectin-3. J Immunol. 2008; 180: 2650–2658 [DOI] [PubMed] [Google Scholar]
- 56. Weisser SB, McLarren KW, Voglmaier N, et al. Alternative activation of macrophages by IL-4 requires SHIP degradation. Eur J Immunol. 2011; 41: 1742–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000; 164: 6166–6173 [DOI] [PubMed] [Google Scholar]