

SUMMARY
In vitro data suggest that a subgroup of NLR proteins, including NLRP12, inhibits the transcription factor NF-κB, although physiologic and disease-relevant evidence is largely missing. Dysregulated NF-κB activity is associated with colonic inflammation and cancer, and we found Nlrp12-/- mice were highly susceptible to colitis and colitis-associated colon cancer. Polyps isolated from Nlrp12-/- mice showed elevated non-canonical NF-κB activation and increased expression of target genes that were associated with cancer, including Cxcl13 and Cxcl12. NLRP12 negatively regulated ERK and AKT signaling pathways in affected tumor tissues. Both hematopoietic and nonhematopoietic-derived NLRP12 contributed to inflammation, but the latter dominantly contributed to tumorigenesis. The non-canonical NF-κB pathway was regulated upon degradation of TRAF3 and activation of NIK. NLRP12 interacted with both NIK and TRAF3, and Nlrp12-/- cells have constitutively elevated NIK, p100 processing to p52 and reduced TRAF3. Thus, NLRP12 is a checkpoint of noncanonical NF-κB, inflammation and tumorigenesis.
Keywords: Monarch, CLR19.3, Pypaf7, NLR, NIK, TRAF3, CXCL12, CXCL13, Akt, Nr3c1
INTRODUCTION
The nucleotide-binding domain and leucine-rich-repeat-containing (NLR) family of genes have been largely characterized as activators of inflammation. For example, NLRP3, NLRC4, NAIP5 and NLRP6 are essential for inflammasome activation and NOD1 and NOD2 are activators of NF-κB and MAPK transcription factors. Human diseases that are genetically associated with NLRs support the immune activating function of these proteins. While several additional NLR proteins have been characterized in vitro, the in vivo functions for the majority of NLRs remain to be elucidated. Recently, several reports have revealed NLRs that negatively regulate immune activation (Allen et al. 2011;Xia et al. 2011). However, the in vivo relevance associated with the activities of these NLRs is not well defined.
In vitro analysis of NLRP12 (Monarch) (Wang et al. 2002) in human monocytic cell lines suggests that it is a negative regulatory protein that suppresses non-canonical NF-κB activation and p52-dependent chemokine expression (Lich et al. 2007). It also has an effect on canonical NF-kB signaling (Williams et al. 2005), but this effect was modest when RNA interference was used to reduce its expression (Lich et al. 2007). The attenuation of the non-canonical pathway is likely through its ability to associate with NF-κB inducing kinase (NIK), which induces proteasome-dependent degradation of NIK (Lich et al. 2007). Members of the canonical and non-canonical NF-κB family play critical roles in regulating inflammatory and immune responses. NF-κB exists in the cytoplasm in an inactive form held in check by the inhibitor IκB. Proteasome-mediated degradation of IκB is linked to its phosphorylation by the IκB kinase (IKK) complex, IKKα-IKKβ-IKKγ (NEMO). Poly-ubiquitination and degradation of IκB is initiated by its phosphorylation, which results in the release and nuclear translocation of NF-κB to activate various target inflammatory chemokines, cytokines and cell surface proteins.
While in vitro studies in human cell lines have found an important role for NLRP12 as an inhibitor of NF-κB signaling, the physiological evidence is lacking. We tested the function of NLRP12 in gastrointestinal disease, since activated NF-κB is routinely observed in colonic mucosal tissues from patients suffering from inflammatory bowel diseases (IBDs) and dysfunctional NF-κB signaling pathways underlie a diverse range of gastrointestinal diseases. Aberrant noncanonical NF-κB signaling and NIK stabilization have also been found in various cancers (Annunziata et al. 2007;Keats et al. 2007). In addition to dysfunctional NF-κB signaling, polymorphisms in NLRP12 are associated with a spectrum of inflammatory disorders in human populations (Jeru et al. 2008). While the cause of chronic IBD remains to be fully elucidated, it is generally accepted that defects in the intestinal epithelial cell barrier, dysregulated host immune responses and the enteric microflora are significant factors in colitis and colitis-associated colon cancer (CAC)(Rakoff-Nahoum et al. 2004).
Here, we demonstrate that Nlrp12-/- mice were highly susceptible to gastrointestinal inflammation and CAC and showed elevated non-canonical NF-κB signaling. The increase in tumorigenesis was associated with increased NIK-regulated genes that have been associated with cancer (Cxcl12 and Cxcl13), and multiple signaling pathways associated with cancer in the colons of Nlrp12-/- animals. Together, these data implicate NLRP12 as a critical checkpoint during inflammation and tumorigenesis.
RESULTS
NLRP12 Attenuates Experimental Colitis and colonic NIK expression
Previous in vitro studies have shown that NLRP12 functions as a negative regulator of noncanonical NF-κB signaling through inhibition of NIK while the effects on the canonical NF-κB pathway were modest (Lich et al. 2007). The NF-κB family members are regulators of chronic inflammation in the colon and aberrant non-canonical NF-κB signaling and NIK stabilization has been associated with various cancers (Annunziata et al. 2007; Keats et al. 2007). Thus, we sought to determine the effect of NLRP12 deficiency on the development of experimental colitis (EC) in mice. Wild-type and Nlrp12-/- mice were initially challenged in an acute EC model utilizing 5% dextran sulfate sodium (DSS) for 5 days and survival was assessed for 14 days. By day 6, 67% of the Nlrp12-/- mice reached a moribund state that required euthanization and by day 10 all of the Nlrp12-/- mice required euthanization (Figure 1A). This was accompanied by dramatic weight loss among Nlrp12-/- mice (Figure 1B). Weight loss measurements were halted on day 6 due to increased morbidity in the Nlrp12-/- animals (Figure 1A-B). Prior to euthanasia, the clinical parameters that are typically associated with disease progression (weight loss, stool consistency and rectal bleeding) were assessed using a semi-quantitative scoring system to generate clinical scores (Siegmund et al. 2001). Nlrp12-/- mice presented with significantly increased clinical severity compared with the wild-type animals (Figure 1C). These data indicate that NLRP12 serves a protective role during the progression of acute EC. To assess the impact of Nlrp12 loss on NF-κB activity, p65, IκBα, NIK and p52 expression were assayed in ex vivo colons from Nlrp12-/- and wild-type mice over the course of the acute EC model. While we routinely observed subtle mouse-to-mouse differences between the amounts of each of these proteins, in general, we did not observe consistent differences in either pIκBα or pp65 amounts in Nlrp12-/- mice compared to wild type (Figure 1D, top panel). However, NIK and p52 expression was considerably increased under naïve conditions (day 0) and nine days after the initiation of the acute DSS challenge in the Nlrp12-/- mice (Figure 1D, bottom panel). Despite the fact that detection of colonic NIK by was technically challenging, it was elevated in 63% of Nlrp12-/- colons (n=19) compared to controls. In addition to directly assessing NF-κB pathway components, we also evaluated the amounts of IL-1β and TNF-α, which are commonly associated with EC and canonical NF-κB signaling. We observed modest differences that did not achieve statistical significance in the Nlrp12-/- and control mice nine days after the initiation of EC (Figure 1E). It is unlikely that the subtle differences in these cytokines could result in the robust EC susceptibility of the Nlrp12-/- mice. Together, these data suggest that elevated non-canonical NF-κB activity is correlated with increased susceptibility of the Nlrp12-/- mice in the acute colitis model.
Figure 1. NLRP12 attenuates the development of experimental colitis.
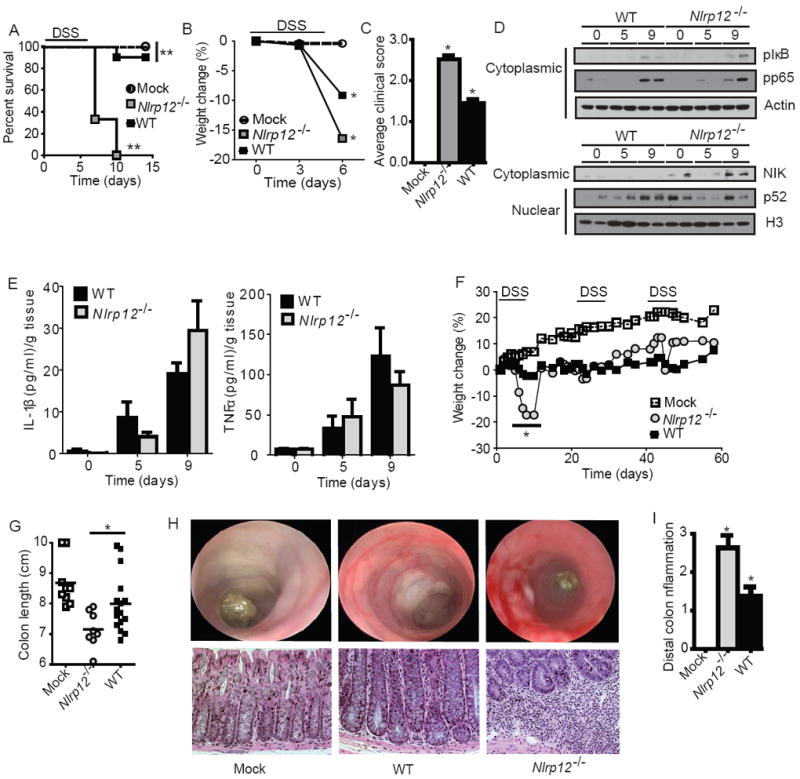
(A-E) Wild-type and Nlrp12-/- mice were challenged with 5% dextran sulfate sodium (DSS) for 5 days and disease progression was assessed daily. (A) Survival and (B) weight loss in Nlrp12-/- and wild-type mice. Due to increased mortality in the Nlrp12-/- mice, weight assessments were halted on Day 6. (C) Composite clinical scores reflecting weight loss, stool consistency and the presence of blood in the stool and/or rectum. (D) pIκBα, pp65 and NIK levels were evaluated in colons harvested from wild type and Nlrp12-/- mice (2 individual mice are shown per time point and all samples were run together on the same gel) prior to the initiation of acute colitis (Day 0) and 9 Days post-colitis initiation. (E) Colon amounts of IL-1β and TNFα were assessed by ELISA from organ culture supernatants. (A-E) WT mock, n = 4; DSS-treated WT, n = 10; Nlrp12-/-, n = 8. (F-I) Mice were treated with 3 rounds of 2.5% DSS for 5 days, followed by 2 weeks of recovery to assess recurring colitis. (F) Weight loss was assessed throughout the recurring DSS model. (G) Colon length from Nlrp12-/- and wild type mice. (H) Disease progression was assessed immediately prior to the third round of DSS (Day 37) via high resolution endoscopy. High resolution endoscopy was performed on 3 animals from each group. Histopathology revealed a considerable amount of crypt loss and immune cell infiltration in Nlrp12-/- mice compared to wild type mice. (I) Histopathology scoring revealed a significant increase in distal colon inflammation in the DSS treated Nlrp12-/- mice compared to the wild type animals. (F-I) WT mock, n = 9; DSS-treated WT, n = 16; Nlrp12-/-, n = 8. Data shown are representative of at least three independent experiments and depict the mean ± SEM. The symbols * and ** indicate P < 0.05 and P < 0.01, respectively, between the DSS-treated WT and Nlrp12-/- mice.
We next assessed the role of NLRP12 in a recurring colitis model, which required three rounds of DSS treatment (2.5%) over 60 days (Neufert et al. 2007). Increased weight loss was observed in all DSS treated mice. Following the first round of DSS treatment, Nlrp12-/- animals exhibited significantly greater weight loss (Figure 1F), which is consistent with the early weight loss observed in the acute model. Both strains recovered following this initial decrease and no other significant differences in weight loss were observed during subsequent DSS administrations. During colitis, shortened colon length is correlated with disease severity. Truncated colons were observed in all DSS treated mice, but Nlrp12-/- colons were significantly shorter than controls (Figure 1G). Disease progression was initially examined via high resolution endoscopy. At the 6 week time point, there was substantially increased inflammation and bleeding in the DSS treated Nlrp12-/- mice (Figure 1H). This was confirmed through histopathology following the completion of the model. Nlrp12-/- mice demonstrated significantly increased distal colon inflammation compared to controls (Figure 1I). Inflammation in the Nlrp12-/- mice was characterized by diffuse and coalescing mucosal inflammation with multi-focal submucosal extensions that included some follicle formation. The inflammation in controls was characterized as small multifocal areas of leukocyte accumulation, which were significantly less severe than the lesions in Nlrp12-/- animals. These data show that NLRP12 functions as a negative in vivo regulator of inflammation during recurring colitis.
Nlrp12-/- Cells Display Enhanced Non-Canonical NF-κB Signaling and MAPK Activation
Previously, NLRP12 was shown to attenuate the non-canonical NF-κB pathway through interactions with NIK in human macrophage cell lines (Lich et al. 2007). The data in Figure 1 also showed enhanced non-canonical NF-κB activation in the colon. To further explore the physiologic regulation of NIK by NLRP12, we investigated primary cells isolated from Nlrp12-/- mice. We chose to evaluate NLRP12 activity in myeloid dendritic cells based on data that found the highest Nlrp12 expression in this cell type (Arthur et al. 2010). To evaluate the ex vivo contribution of NLRP12 in non-canonical NF-κB signaling, dendritic cells were stimulated with TNFα as described by others (Madge and May 2010). In wild type cells, NIK increased during the first two hours post-TNFα stimulation and was not detected five hours after stimulation. However, in Nlrp12-/- cells, there was a sustained increase in NIK expression over the 18 hour TNFα stimulation (Figure 2A). NIK activation results in the degradation of p100 into p52 and Nlrp12-/- cells displayed increased p100 cleavage to p52 in the cytosol (Figure 2B and C). In addition to increased NIK activity, we also detected modest increases of p65 and IκBα in Nlrp12-/- dendritic cells. The ratio of pp65 to p65 was not increased in Nlrp12-/- vs. wild type cells, although the total level of pp65 is increased in the former due to increased p65 expression. The ratio of pIκba to IκBa was increased indicating an effect of NLRP12 on the canonical NF-κB pathway (Figure 2D). However, an extensive analysis of cytokine production from the wild type and Nlrp12-/- cells showed that most cytokines typically associated with canonical NF-κB signaling were not changed (Supplemental Figure 1A-C), agreeing with the modest alteration of canonical NF-κB.
Figure 2. Immune cells isolated from Nlrp12-/- mice have increased non-canonical NF-κB signaling and MAPK activation.
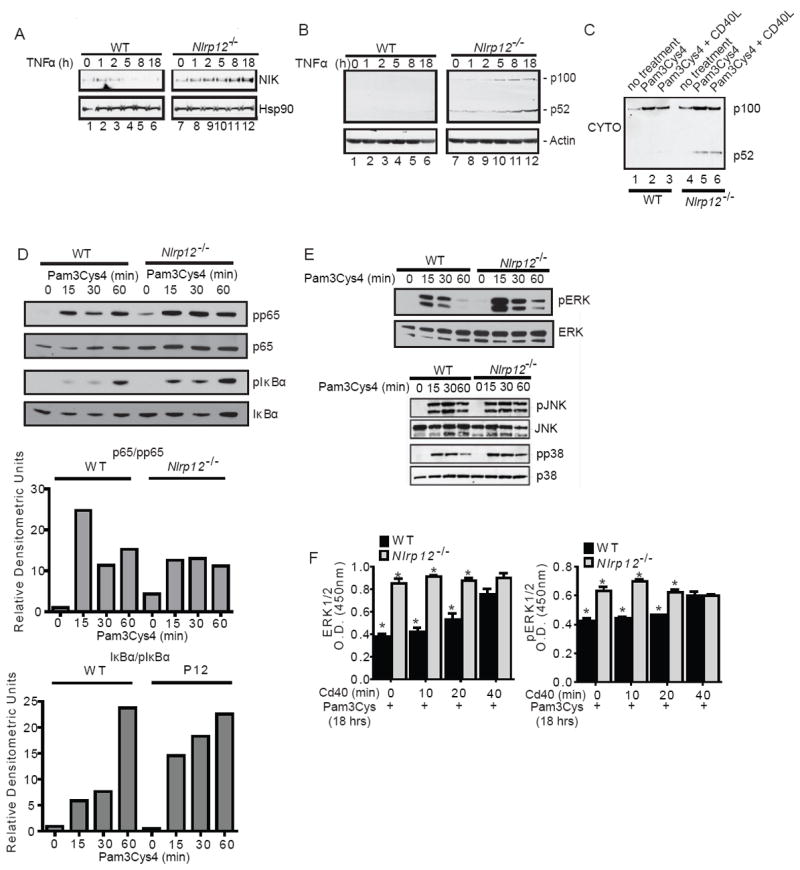
(A) Bone marrow derived dendritic cells were isolated from wild type and Nlrp12-/- mice. Following stimulation with TNFα, NIK levels steadily increased over the 18 hour time course in Nlrp12-/- cells compared to controls. (B) Increased levels of p100 cleavage to p52 were observed in Nlrp12-/- cells over duration of the TNFα challenge. (C) Cells were pretreated with Pam3Cys4 and stimulated with CD40L. Under these conditions, we detected increased p52 levels in the cytosolic fraction in cells isolated from Nlrp12-/- mice. (D) pp65 and pIκBα levels were evaluated in dendritic cells from wild type and Nlrp12-/- mice following Pam3Cys4 and CD40L stimulation. (E) Pam3Cys4 induced the phosphorylation of JNK and p38 in wild type and Nlrp12-/- cells; however, no considerable differences were consistently observed between the two genotypes. A modest increase in the phosphorylation of ERK1/2 was observed in the Nlrp12-/- cells. (F) ERK1/2 and pERK were evaluated by ELISA following Pam3Cys4 stimulation for 18 hours and CD40 stimulation over the time course shown. Primary dendritic cells from 5 independent animals were cultured and pooled to generate the ELISA data. All data shown are representative of at least three independent experiments.
In addition to evaluating NF-κB, we also evaluated components of the MAPK pathway that have been previously associated with colitis and Colitis Associated Cancer (CAC). Following Pam3Cys4 stimulation, the phosphorylation of JNK and p38 occurred at similar amounts in wild type and Nlrp12-/- cells. A modest, but consistent increase in ERK1 and ERK2 phosphorylation was observed in the Nlrp12-/- cells compared with the wild type regardless of stimulation (Figure 2E). To expand upon these findings, we utilized a semi-quantitative ELISA assay to evaluate total ERK1 and 2 and pERK following overnight stimulation with Pam3Cys4 followed by CD40 stimulation over a 40 min time course. We observed a statistically significant increase in both ERK1 and 2 and pERK1 and 2 following overnight Pam3Cys4 stimulation (Figure 2F). The added CD40 stimulation did not augment and actually reduced the difference between control and Nlrp12-/- cells. Together, these data confirm that NF-κB, though predominately non-canonical NF-κB, and ERK pathways are hyperactivated in primary cells isolated from Nlrp12-/- mice.
NLRP12 Functionally Interacts with TRAF3
Previously we showed that NLRP12 can associate with NIK leading to its degradation (Lich et al. 2007). This association was confirmed here using FLAG-tagged NLRP12 (Fg-NLRP12), which co-immunoprecipiated with NIK (Figure 3A). However, another cytoplasmic NLR, NOD2, did not co-immunoprecipiate (Figure 3B). In addition to NIK, we also assessed the interaction between NLRP12 and additional proteins that are important in the non-canonical NF-κB pathway (Razani et al. 2010). Using a bioinformatics approach, we identified multiple TRAF binding motifs in both human and mouse NLRP12 (Fig. 3C). Previous work has shown that in the TRAF2-TRAF3-CIAP1-CIAP2 complex, TRAF3 is the only molecule that directly interacts with NIK, where it induces NIK degradation. Indeed, Traf3-/- cells show increased non-canonical NF-κB activation (Sun 2011). NLRP12 was found to interact with endogenous TRAF3 following overnight stimulation with Pam3Cys4 (Figure 3D). It did not interact with IKKα, which mediates an alternate pathway that regulates NIK stability (Figure 3E)(Razani, et al. 2010). We next assessed the effect of NLRP12 on TRAF3 in primary dendritic cells from Nlrp12-/- and wild type mice that were primed with Pam3Cys4 for 18 hours and stimulated with CD40L. TRAF3 levels were constitutively decreased in cells isolated from Nlrp12-/- mice compared to controls and remained reduced throughout stimulation (Figure 3F). These findings are consistent with several reports that showed that TRAF3 degradation preceded NIK stabilization and p100 processing following engagement of CD40 (Sun 2011;Vallabhapurapu et al. 2008). In contrast, the amount of TRAF6 in Nlrp12-/- and control cells were similar (Figure 3F). Together, our data suggests that NLRP12 maintains the amount of TRAF3, which normally keeps noncanonical NF-κB activation in check.
Figure 3. NLRP12 interacts with and maintains TRAF3 levels.
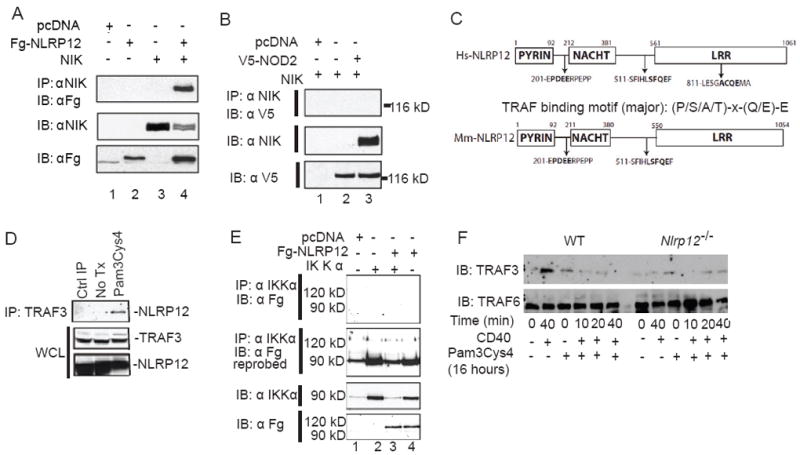
(A) Co-immunoprecipitation of NLRP12 and NIK following co-transfection in HEK293T cells of Fg-NLRP12 and untagged NIK. (B) NOD2 did not co-immunoprecipitate with NIK following co-transfection. (C) Bioinformatics identified multiple TRAF2/3 binding motifs in human (Hs) and murine (Mm) NLRP12. (D) NLRP12 co-immunoprecipitated with TRAF3 following 18hr Pam3Cys4 stimulation in HEK293T cells. (E) NLRP12 did not co-immunoprecipitate with IKKα following overexpression. (F) Levels of TRAF3 and TRAF6 were evaluated by immunoblot following Pam3Cys4 and CD40 stimulation in dendritic cells isolated from wild type and Nlrp12-/- mice. All data shown are representative of at least three independent experiments.
NLRP12 Attenuates Disease Progression during CAC
We next evaluated the role of NLRP12 in the initiation and progression of inflammation driven CAC. Nlrp12-/- mice were subjected to an azoxymethane (AOM)+DSS inflammation-driven colon tumorigenesis model (Figure 4A). The carcinogen AOM, administered at day 1 induces tumorigenesis (Neufert et al. 2007). AOM+DSS treated Nlrp12-/- mice showed increased weight loss compared to controls following the initial and third round of DSS (Figure 4B). Likewise, AOM+DSS treated Nlrp12-/- mice showed significantly increased clinical features during disease progression (Figure 4C). Consistent with the increased morbidity, colons harvested from AOM+DSS treated Nlrp12-/- mice were significantly shortened (Figure 4D).
Figure 4. Nlrp12-/- mice are more susceptible to inflammation driven colon tumorigenesis.
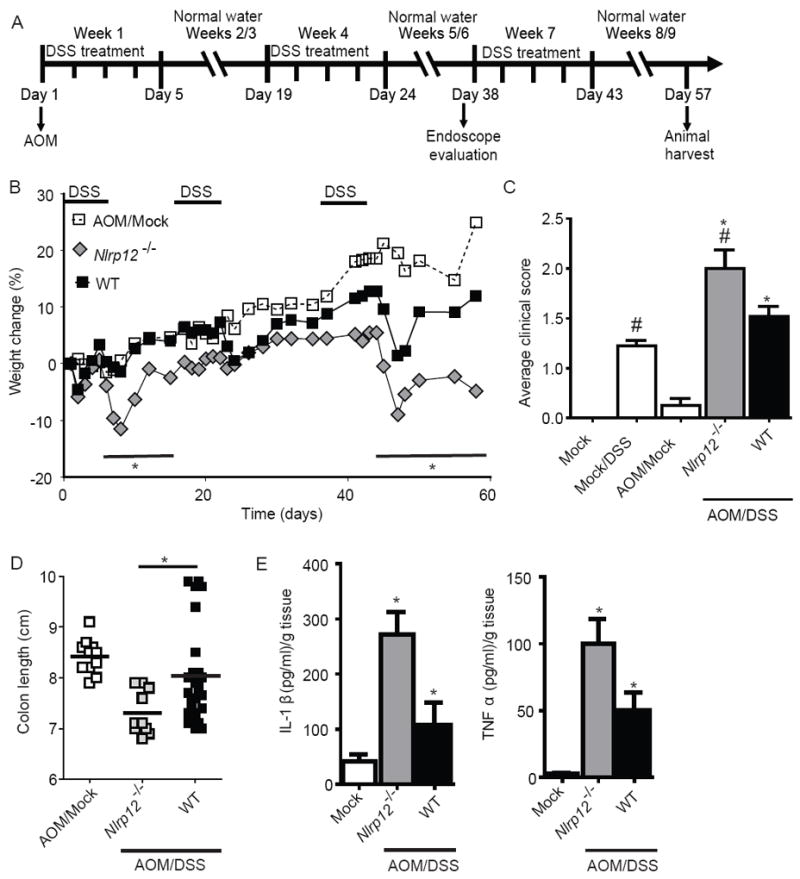
(A) Schematic of the inflammation driven colon tumorigenesis model. Wild type and Nlrp12-/- mice received a single injection of AOM immediately prior to the first DSS administration. The mice were then treated with 3 rounds of 2.5% DSS for 5 days, followed by 2 weeks of recovery. (B) Weight loss was monitored throughout the AOM/DSS model. (C) Composite clinical scores reflecting weight loss, stool consistency and the presence of blood in the stool and/or rectum. (D) Colons removed from AOM/DSS treated Nlrp12-/- mice were significantly truncated compared to similarly treated wild type mice. (A-D) WT mock, n = 4; Mock/DSS WT, n = 4; AOM/Mock WT, n = 11; AOM/DSS Nlrp12-/-, n = 10; AOM/DSS WT, n = 22. (E) Organ cultures were generated to determine the levels of IL-1β and TNFα in the colon. WT mock, n = 4; AOM/DSS Nlrp12-/-, n = 6; AOM/DSS WT, n = 13. For all experiments, data shown are representative of at least three independent experiments and depict the mean ± SEM. The symbols * and # indicate P < 0.05 between the AOM/DSS and DSS only-treated WT mice and the AOM/DSS treated Nlrp12-/- mice, respectively.
A diverse range of proinflammatory cytokines and chemokines are increased during IBD and CAC. To evaluate cytokine production, we generated organ cultures from colons harvested from mice that had completed the CAC model. We observed a significant increase in IL-1β and TNFα levels by ELISA in the organ culture supernatants derived from AOM+DSS treated Nlrp12-/- animals (Figure 4E). These data are consistent with the increased inflammation observed in the Nlrp12-/- mice.
NLRP12 Attenuates Tumorigenesis during CAC
Nlrp12-/- mice showed markedly increased colon inflammation, proinflammatory cytokine production and NIK levels during EC. Previous studies have found that each of these observations is highly correlated with colon tumorigenesis (Sun 2011). In vivo tumorigenesis was assessed during week 6 of the AOM+DSS model utilizing high resolution endoscopy and increased inflammation and extravasation were found in Nlrp12-/- mice compared to controls (Figure 5A). Likewise, the addition of AOM resulted in increased polyps in Nlrp12-/- mice (Figure 5A). Following the completion of the CAC model, we observed macroscopic colon polyps in the distal colons from the majority of AOM/DSS treated mice (Figure 5B). The number and maximal cross-sectional area of macroscopic polyps were greatly increased in Nlrp12-/- mice compared to controls (Figure 5C). In this model, the Histologic Activity Index (HAI) is a composite score of individual assessments of inflammation, crypt atrophy, hyperplasia, dysplasia and the area involved in disease in the mid and distal colon (Meira et al. 2008). AOM/DSS treated Nlrp12-/- mice had significantly enhanced HAI compared to controls (Figure 5D), substantiated by a significant increase in inflammation, hyperplasia and dysplasia with a greater affected area in Nlrp12-/- mice compared to control mice (Figure 5E). Some of these lesions were further characterized as adenocarcinomas in the Nlrp12-/- mice.
Figure 5. NLRP12 attenuates tumorigenesis during CAC.
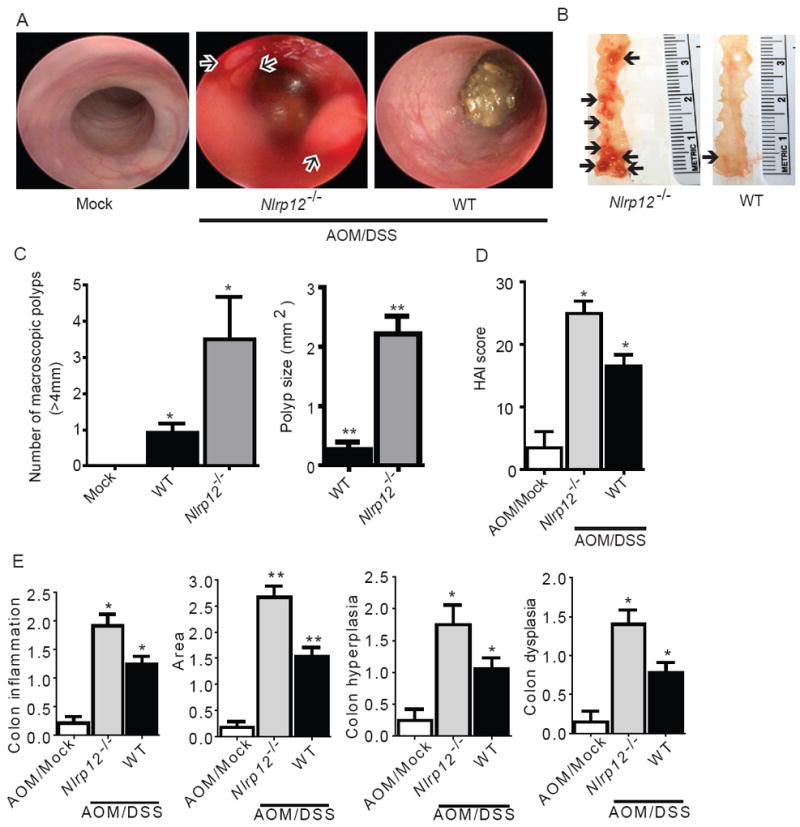
(A) Disease progression was assessed immediately prior to the third round of DSS (Day 37) via high resolution endoscopy. Excessive inflammation and hemorrhaging was observed in the Nlrp12-/- mice. Arrows denote polyp formation in the Nlrp12-/- mice. High resolution endoscopy was performed on 3 animals from each group. (B) Macroscopic polyps (arrows) were identified in the distal and mid colons harvested from Nlrp12-/- and wild type animals. (C) The number and maximal cross-sectional area of macroscopic polyps was quantified. AOM/Mock WT, n = 11; AOM/DSS Nlrp12-/-, n = 10; AOM/DSS WT, n = 22. (D) Histopathologic Activity Index (HAI) score in colons harvested following the completion of the CAC model. (E) Histopathology analysis of colon inflammation, area associated with disease, hyperplasia and dysplasia in the AOM/DSS treated Nlrp12-/- mice. WT mock, n = 5; Mock/DSS WT, n = 3; AOM/Mock WT, n = 3; AOM/DSS Nlrp12-/-, n = 5; AOM/DSS WT, n = 15. For all experiments, data shown are representative of at least three independent experiments and depict the mean ± SEM. The symbols * and ** indicate P < 0.05 and P < 0.01, respectively, between the AOM/DSS-treated WT mice and Nlrp12-/- mice.
The Pathogenesis in Nlrp12-/- Mice is derived from both Hematopoietic and Non-hematopoietic Sources
Colon tumorigenesis is correlated with intestinal inflammation. Gastrointestinal tumors typically originate from dysplastic epithelial or stem cells and are infiltrated by a variety of lymphoid and myeloid cell types which can potentiate neoplastic transformation and tumorigenesis (Murdoch et al. 2008). To determine the relevant cell compartment responsible for the gastrointestinal inflammation and tumorigenesis in the Nlrp12-/- animals, we generated wild type and Nlrp12-/- chimeric mice using adoptive bone marrow transplantation. After a 6-week reconstitution phase to achieve near-complete chimerism (Supplemental Figure 2A), these animals were subjected to AOM-DSS. Bone marrow chimerism did not affect the phenotypic outcome, in that Nlrp12-/- mice that did (Nlrp12-/- → Nlrp12-/-) or did not receive Nlrp12-/- bone marrow showed greater increases in weight loss compared to wild type mice that did (WT→WT) or did not receive wild type bone marrow grafts (Supplemental Figure 2B-E). Among bone marrow recipients that were treated with AOM+DSS, two trends were observed. Following the first round of DSS, Nlrp12-/- mice that received wild type bone marrow (WT → Nlrp12-/-) and wild type mice that received Nlrp12-/- bone marrow (Nlrp12-/- → WT) showed similar weight loss as Nlrp12-/- → Nlrp12-/-, and all three had significantly more weight loss than WT→WT (Figure 6A). This suggests that during this phase, NLRP12 in both the hematopoietic and non-hematopoietic compartment contributed to disease outcome. During the second round of DSS, we observed significant weight loss in the WT→ Nlrp12-/-, but not the Nlrp12-/- →WT, indicating that during the later stages of this model, the effect of NLRP12 was primarily attributed to the non-hematopoietic compartment (Figure 6A). More detailed HAI scores at the later time point revealed that Nlrp12-/- →Nlrp12-/- and WT→ Nlrp12-/- chimeras demonstrated a significantly increased histopathology compared to the WT→WT and Nlrp12-/- →WT (Figure 6B). The increased HAI scores were associated with a significant increase in colon hyperplasia, dysplasia and area affected, while differences in inflammation were not statistically significant (Figure 6C). A similar pattern was observed with epithelial defects characterized by extensive epithelial tattering, large eroded areas and extensive ulcerations throughout the mid and distal colon (Figure 6D-E). Similar to the non-chimeric mice, Nlrp12-/- →Nlrp12-/- and WT→Nlrp12-/- mice had significantly increased numbers of macroscopic polyps compared to WT→WT and Nlrp12-/- →WT animals (Figure 6F). These results indicate that non-hematopoietic NLRP12 affected tumor outcome. Thus, we evaluated NLRP12 expression in the mouse gastrointestinal system using a publically accessible microarray meta-analysis search engine (http://www.nextbio.com/b/search/ba.nb) as described (Kupershmidt et al. 2010). NLRP12 was found in the gastrointestinal system, including intestinal epithelial cells and the colon (Supplemental Figure 3A). These data suggest an early role for NLRP12 in the AOM/DSS model through both hematopoietic and nonhematopoietic compartments. Later, coincidental with polyp formation, the effect of NLRP12 is derived primarily from the nonhematopoietic compartment. These findings are consistent with the current paradigm, which suggests that intestinal epithelial cell (IEC) derived NF-κB can influence the incidence of colonic tumor, whereas immune-derived NF-κB contributes to inflammation during the early stages of tumorigenesis (Karrasch and Jobin 2008).
Figure 6. NLRP12 mediates the development of CAC through a non-hematopoietic cellular compartment.
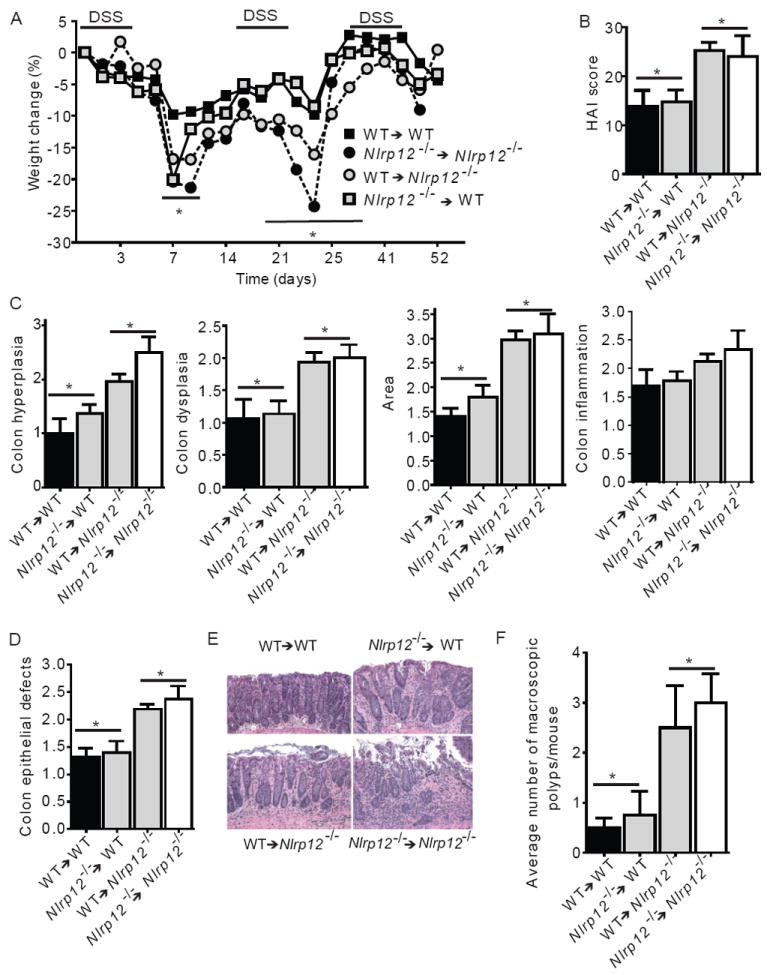
(A) Weight change of indicated bone marrow chimera mice. (B) Composite HAI score. (C) Colon hyperplasia, dysplasia, area involved in disease and inflammation were each individually scored. (D) Nlrp12-/- mice receiving wild type or Nlrp12-/- bone marrow demonstrated a significant increase in colon area affected by disease and defects to the epithelial layer of the colon. (E) Representative histopathology emphasizing defects in the epithelial layer for each group indicated. WT→WT and Nlrp12-/-→WT shows mild to moderate surface layer thinning and tattering. WT→Nlrp12-/- and Nlrp12-/-→Nlrp12-/- shows moderate to severe surface layer attenuation and erosions with multifocal detachments of epithelial islands. (F) The average number of macroscopic polyps observed in the colon. For all experiments, data shown are representative of 3 independent experiments and depict the mean ± SEM. The symbols * and # indicate P < 0.05 between the indicated groups. WT→WT, n = 5; Nlrp12-/-→WT, n = 7; WT→Nlrp12-/-, n = 7; Nlrp12-/-→Nlrp12-/- n = 3.
NLRP12 Attenuates Multiple Pathways Associated with Colon Tumorigenesis
Previous in vitro studies identified two chemokines, CXCL12 (also known as SDF-1) and CXCL13 (also known as BLC), which are known to be regulated by noncanonical NF-κB activation (Bonizzi et al. 2004) and by NLRP12 in cell lines (Lich et al. 2007). Both of these chemokines have been shown to be highly up-regulated in various types of human cancers (Del Grosso et al. 2011; Fushimi et al. 2006;Wharry et al. 2009). We evaluated the expression and protein levels of CXCL12 and CXCL13 in colon samples following either chronic EC or CAC and observed robust increases in their expression in Nlrp12-/- colons following completion of the AOM/DSS treatment (Figure 7A). CXCL12 protein was difficult to detect (data not shown); however, CXCL13 protein was increased >38X in Nlrp12-/- colons over controls (Figure 7B). This increase was only observed in the context of cancer, as neither untreated mice nor mice subjected to chronic DSS showed this difference. These data are consistent with the model that the unchecked non-canonical NF-κB activation in Nlrp12-/- mice led to elevated CXCL12 and CXCL13, which are associated with several cancers. It is worth mentioning that in Supplemental Figure 1B, CXCL12 was not detected in primary dendritic cells. However, this was not unexpected as CXCL12 is predominately expressed by stromal cells and functions in dendritic cell recruitment during tumorigenesis (Orimo et al., 2005).
Figure 7. NLRP12 attenuates noncanonical NF-κB regulated chemokines, cancer associated gene expression and specific MAPK family members during CAC.
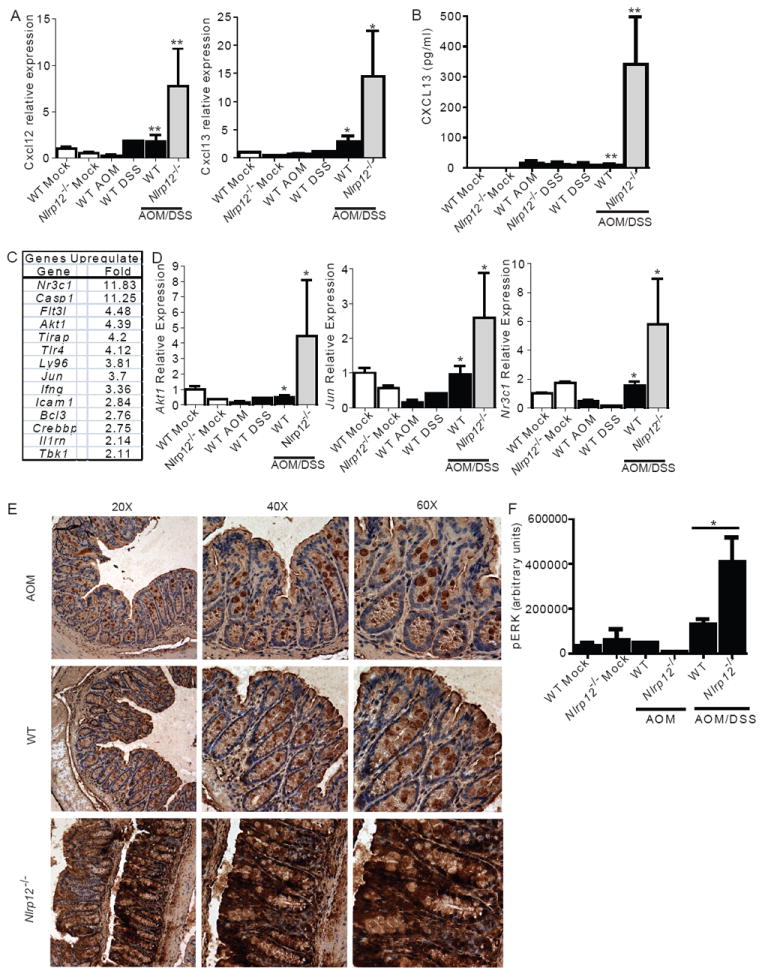
(A) Cxcl12 and Cxcl13 gene expression in colons. (B) Colon levels of CXCL13 were assessed from organ culture supernatants by ELISA. (A-B) WT mock, n = 5; Mock/DSS WT, n = 3; AOM/Mock WT, n = 3; AOM/DSS Nlrp12-/-, n = 5; AOM/DSS WT, n = 15. (C) Macroscopic polyps were removed from wild type and Nlrp12-/- mice and gene expression was assessed using a multiplex gene expression array. Data shown represent genes that were significantly up-regulated, defined as >2 fold increase, in RNA that was pooled from polyps that were microdissected from Nlrp12-/- mice compared to wild type mice. WT, n = 5; Nlrp12-/-, n = 5. (D) Gene expression for Akt1, Jun and Nr3c1 was used to verify the array data using non-pooled RNA collected from the whole colons of additional mice that were not assessed on the array. WT mock, n = 3; Nlrp12-/- mock, n = 3; WT DSS, n = 3; WT AOM, n = 3; AOM/DSS Nlrp12-/-, n = 5; AOM/DSS WT, n = 5. (E) pERK levels were evaluated by immunohistochemistry from paraffin embedded colon sections. (F) pERK levels were evaluated using semiquantitative histopathology image analysis (ImageJ). AOM/Mock WT, n = 3; AOM/DSS Nlrp12-/-, n = 6; AOM/DSS WT, n = 7.
To further evaluate specific genes that are upregulated during colon tumorigenesis, we performed expression profiling of a large subset of cancer associated genes in macroscopic polyps micro-dissected from Nlrp12-/- and wild type mice. There were >3-fold increase of 9 genes, including Akt1 and Jun, and a greater than 2-fold increase in an additional 5 genes in polyps isolated from Nlrp12-/- relative to controls (Figure 7C). Select genes were subsequently verified using non-multiplexed real time PCR on whole colon tissues isolated from individual mice (Figure 7D). Akt, Jun, and Nr3c1 associated with the hyperactivation of cancer pathways were significantly up-regulated in the polyps and colons from Nlrp12-/- mice. We also found genes that were not significantly upregulated, including Nik, Ptgs1 (Cox1) and Ptgs2 (Cox2), but Relb and Nfkb2 transcripts which encode the non-canonical NFkB proteins were increased moderately (Supplemental Figure 4).
In addition to the activation of NF-κB, previous studies have shown that the NOD1 and NOD2 have the ability to modulate MAPK signaling pathways (Kobayashi et al. 2005; Park et al. 2007). Our in vitro data also suggest an effect of NLRP12 on MAPK. Further, non-canonical NF-kB can induce MAPK activation (Dhawan and Richmond 2002). Thus, we evaluated ERK phosphorylation by immunohistochemistry and observed increased pERK staining in colon epithelial cells lining the tops of the crypts and in goblet cells following both EC and CAC models (Figure 7E; Supplemental Figure 4E). However, Nlrp12-/- colon showed robust staining compared to wild type colons, with clear staining observed throughout the crypts (Figure 7E; Supplemental Figure 5). Colon sections were digitally imaged and ERK staining was evaluated using ImageJ software, which confirmed a significant increase in pERK in Nlrp12-/- colons (Figure 7F). These data support the in vitro data in Figure 2, and indicates that NLRP12 serves as a checkpoint for enhanced ERK activation during CAC.
DISCUSSION
Both the canonical and noncanonical NF-κB pathways exert wide-ranging effects on immune regulation, cell proliferation and cell death. Because chronic inflammation and the development of cancer are intimately linked in gastrointestinal disease, extensive research has been focused on dissecting the role of NF-κB and modifiers of NF-κB activity associated with these two diverse biological processes. A recent publication by Zaki et al. suggested the increased inflammation and tumorigenesis observed in the Nlrp12-/- mice was associated with increased canonical NF-κB signaling (Zaki et al., 2011). Consistent with these findings, we also detected a subtle increase in pp65 and pIκBα in vitro. However, our data suggests that NLRP12 is a robust negative regulator of non-canonical NF-κB signaling both in culture and in animals, accompanied by ERK activation and induction of NIK-dependent genes, such as CXCL12 and CXCL13, that are associated with tumorigenesis during gastrointestinal inflammation. It is possible that NLRP12 may indirectly mediate canonical NF-κB signaling through the non-canonical pathway. This hypothesis is supported by previous data, which demonstrated that the noncanonical NF-κB pathway can influence canonical NF-κB signaling through IKKα and TRAF3 dependent mechanisms (Adli et al. 2010; Zarnegar et al. 2008).
In addition to NLRP12, other NLRs have been found to influence the development of colitis and CAC. In humans, mutations in NOD1, NOD2 and NLRP3 have each been shown to contribute to the development of IBD, with NOD2 having the strongest correlation to Crohn’s disease. However, the exact mechanism by which NOD2 affects IBD and CAC remains to be fully elucidated (Sollid and Johansen, 2008). NOD1 and NLRP3 have a significant protective role in mediating EC and CAC in animals (Allen et al. 2010; Chen et al. 2008; Zaki et al. 2010). In both Nod2-/- and Nod1-/- mice, EC and CAC were attenuated following broad spectrum antibiotic treatment, implicating a role for the gut microbiota (Chen et al. 2008). Nlrp3-/- mice are also more sensitive to DSS induced experimental colitis and AOM+DSS induced CAC (Allen et al. 2010; Zaki et al. 2010). In mice lacking the NLR inflammasome adaptor, Pycard (also known as ASC), the mechanism is more clearly attributed to the lack of IL-18 (Dupaul-Chicoine et al. 2010;Salcedo et al. 2010;Zaki et al. 2010).
Inflammation is a dynamic process that requires molecular brakes or checkpoints to prevent an overwhelming response that can result in damage to the host. Although several reports suggest that some NLRs function to attenuate inflammation, this idea has yet to gain traction because in vivo evidence has been lacking (Ting et al. 2010). In the present study, cells and colons from Nlrp12-/- mice have enhanced activation of NF-κB, most robustly in the non-canonical NF-κB pathway and this is consistent with more severe EC and CAC. Activation of the canonical pathway is more moderate and showed a great degree of variation between animals. The ability of NLRP12 to interact with NIK and attenuate the generation of p52 is of particular importance in light of recent studies that have implicated dysregulated non-canonical NF-κB signaling in various cancers. For example, NIK stabilization is associated with inactivating mutations in TRAF2, TRAF3, cIAP1 and cIAP2 and has been extensively characterized in multiple myeloma, while gain-of-function mutations in NIK are associated with this malignancy (Annunziata et al. 2007; Keats et al. 2007). In addition to multiple myeloma, increased levels of NIK have been associated with several other hematological malignancies and solid tumors (Thu and Richmond 2010). While NIK detection is considered technically challenging, we were able to detect its elevation in vivo, as well as the elevation of chemokines that have previously been found to be regulated by the non-canonical NF-κB pathway and by NLRP12 (Bonizzi et al. 2004). In CAC, we identified robust amounts of CXCL13 (and Cxlc13 expression) and increased expression of Cxlc12 in all of the Nlrp12-/- mice. These findings are significant as these chemokines are strongly associated with various types of cancers in human populations, including colorectal tumorigenesis (Del Grosso et al. 2011;Fushimi et al. 2006). The finding that these chemokines were only dysregulated in the CAC model and not the colitis models further implicates their contribution to the increased tumorigenesis observed in the Nlrp12-/- mice. In addition to the non-canonical NF-κB pathway, our findings also indicate that NLRP12 directly or indirectly contributes to the hyperactivation of ERK and cancer associated signaling pathways, such as Akt and Jun.
The common hypothesis that inflammation promotes tumorigenesis is exemplified during the development of CAC. Our previous analysis of Nlrp12-/- mice showed that they have an attenuated inflammatory response during the development of contact hypersensitivity, which was associated with a defect in dendritic cell and neutrophil migration (Arthur et al. 2010). However, we were not able to define the underlying molecular mechanism in this previous study. Thus, it is likely that NLRP12, similar to other NLRs and pathogen recognition receptor proteins, might display distinct tissue-specific functions. In the context of colitis and CAC, this work shows that NLRP12 functions as a negative regulator of the NF-κB pathway through its interaction and regulation of NIK and TRAF3, and as a checkpoint of critical pathways associated with inflammation and inflammation-associated tumorigenesis.
EXPERIMENTAL PROCEDURES
Experimental Animals
All studies were conducted in accordance with the IACUC guidelines of UNC Chapel Hill and NIH Guide for the Care and Use of Laboratory Animals. Nlrp12+/- mice have been described (Arthur et al. 2010). All experiments were conducted with male mice housed under SPF conditions that were age matched and backcrossed for at least 9 generations onto the C57Bl/6 background.
Induction of Colitis and Inflammation-Driven Tumor Progression
The induction of acute colitis was achieved by exposing the mice to one cycle of 5% DSS (MP Biomedicals). The induction of recurring colitis was achieved by exposing the mice to 3 cycles of 2.5% DSS (Neufert et al., 2007). For CAC, mice were given 1 i.p. injection (10 mg/kg body weight) of the mutagen AOM (Sigma Aldrich), followed by 3 DSS exposures (2.5%). Mice were sacrificed and characterized at specific time points throughout the course of the DSS or AOM/DSS challenge or when the animals were moribund.
Disease progression was determined by body weight changes, the presence of rectal bleeding and stool consistency. Each parameter was scored and averaged to generate a semi-quantative clinical score, as described (Siegmund et al. 2001). Body weight was assessed at least 5 days per week throughout the course of the experiment. Stool consistency and rectal bleeding were each assessed, 1 day and 4 days after each DSS treatment. We utilized visual inspection and a Hemoccult Immunochemical Fecal Occult Blood Test (Beckman Coulter) to sample for blood in the stool. Colon length was measured upon completion of each study.
Endoscopic Tumor Investigation
Tumor growth and progression was evaluated in situ by mini-endoscopy, as previously described (Uronis et al., 2007) using a Coloview system (Karl Storz Veterinary Endoscope). The endoscopy was performed 6 weeks after the initiation of the colitis and colitis associated cancer models and each session was digitally recorded. Mice were fasted overnight to avoid fecal matter obstructions. We preformed real time evaluations of colons (~3-4cm) that included the area from the anal verge to the splenic flexure.
Macroscopic Polyp Analysis and Histopathology
The entire colon was removed, flushed, and opened longitudinally to assess macroscopic polyp formation as previously described (Allen et al. 2010). Polyps were identified by a trained investigator using a dissecting microscope (10X magnification). For histopathology, the colons were swiss rolled and fixed in 10% buffered formalin. Paraffin embedded colons were sectioned at 5 μm, processed for H&E staining, and evaluated by an experienced veterinary pathologist (A.B.R.) who was blinded to genotype and treatment and scored as described (Meira et al. 2008).
Colon Organ Culture and Proinflammatory Mediator Assessments
Organ cultures were generated to assess cytokine levels, as previously described (Greten et al. 2004). The distal most section of colon was isolated and cut into 1 cm2 sections. These strips were washed with PBS containing penicillin/streptomycin, and the wet weight of each section was recorded. The isolated colon sections were incubated overnight in non-supplemented RPMI media containing penicillin/streptomycin. Cell free supernatants were harvested and assayed via ELISA.
Western Blots and NF-κB Assessments from colon tissue
The colon tissue was processed into cytosolic and nuclear fractions using the NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Scientific) according to the manufacturer’s instructions. A total of 30 μg of protein from each sample was separated by SDS-PAGE using 4-12% NuPAGE® Bis-Tris gels (Invitrogen), transferred and incubated overnight with the respective antibodies (Supplemental Experimental Procedures).
Expression Profiling
Total RNA was isolated from either polyps or polyp-free adjacent tissue following the completion of the CAC model. RNA from polyps isolated from 5 different animals for each genotype was pooled together prior to the cDNA reaction. Pooled samples were analyzed using RT2 Profiler PCR Arrays (SABiosystems) following the manufacturers protocols or by Taqman rtPCR analysis (ABI). Array results were confirmed on total RNA isolated from distal colon sections from individual mice that were exposed to AOM/DSS independently of those used for array profiling.
Co-immunoprecipitation
Detailed protocols for cell culture, co-immunoprecipitation and antibodies used have been previously published (Lich et al., 2007)(Supplemental Experimental Procedures). Wild type and Nlrp12-/- dendritic cells were isolated and cultured under standard conditions. Cells were lysed in 1% RIPA buffer for western blotting. For MAPK activation assessments, primary dendritic cells were plated and assayed using a cell-based ERK1/2 ELISA sampler kit (RayBio). For HEK293T cells, cells were plated and transfected using FuGene 6 (Roche). For immunoprecipitations, the cell lysates were prepared as previously described (Williams et al., 2005). All plasmids were used at equal molar concentrations, unless otherwise stated. Cell lines stably expressing Ha-Monarch-1 or shRNA targeting Monarch-1 have been described (Lich et al., 2007; Williams et al., 2005).
Statistical Analysis
All data are presented as the mean +/- standard error of the mean (SEM). Complex data sets were analyzed by Analysis Of Variance (ANOVA), which was followed by either Tukey-Kramer HSD or Newman-Keuls Post Test for multiple comparisons. Single data points were assessed by the Student’s two-tailed t-test. The product limit method of Kaplan and Meier was utilized to generate the survival curves, which were compared by using the log rank test. A p-value that was less than 0.05 was considered statistically significant for all data sets.
Supplementary Material
HIGHLIGHTS.
Nlrp12-/- mice are significantly more susceptible to colitis-associated colon cancer
NLRP12 is a negative regulator NIK, CXCL12, CXCL13, ERK and AKT in vivo
Hematopoietic and non-hematopoietic compartments contribute to Nlrp12-/- phenotype
NLRP12 functionally interacts with and alters the balance of NIK and TRAF3
Acknowledgments
The authors thank the Center for Gastrointestinal Biology and Disease (CGIBD) for providing core and technical support (P30DK34987). We also thank Dr. John Bertin and Millenium Pharmaceuticals for providing the Nlrp12-/- mice. In addition, we would like to acknowledge Drs. Eda Holl, Kelly Roney, Marcus Mühlbauer and Hendrick vanDeventer for technical assistance. This work is supported by: R21CA131645 and CCFA (B.K.D); CA156330, AI077437, AI067798 and UNC-CH UCRF (J.P.Y.T.); AR007416, the American Cancer Society and K01DK092355 (I.C.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adli M, Merkhofer E, Cogswell P, Baldwin AS. IKKalpha and IKKbeta each function to regulate NF-kappaB activation in the TNF-induced/canonical pathway. PLoS One. 2010 Feb 25;5(2):e9428. doi: 10.1371/journal.pone.0009428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity. 2011;34:854–865. doi: 10.1016/j.immuni.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JP. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045–1056. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JC, Lich JD, Ye Z, Allen IC, Gris D, Wilson JE, Schneider M, Roney KE, O’Connor BP, Moore CB, et al. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J Immunol. 2010;185:4515–4519. doi: 10.4049/jimmunol.1002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M. Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB:p52 dimers. EMBO J. 2004;23:4202–4210. doi: 10.1038/sj.emboj.7600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Shaw MH, Redondo G, Nunez G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 2008;68:10060–10067. doi: 10.1158/0008-5472.CAN-08-2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan P, Richmond A. J Biol Chem. 2002;277:7920–7928. doi: 10.1074/jbc.M112210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Grosso F, Coco S, Scaruffi P, Stigliani S, Valdora F, Benelli R, Salvi S, Boccardo S, Truini M, Croce M, et al. Role of CXCL13-CXCR5 crosstalk between malignant neuroblastoma cells and Schwannian stromal cells in neuroblastic tumors. Mol Cancer Res. 2011;9:815–823. doi: 10.1158/1541-7786.MCR-10-0367. [DOI] [PubMed] [Google Scholar]
- Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, Meunier C, Turbide C, Gros P, Beauchemin N, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. 2010;32:367–378. doi: 10.1016/j.immuni.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Fushimi T, O’Connor TP, Crystal RG. Adenoviral gene transfer of stromal cell-derivedfactor-1 to murine tumors induces the accumulation of dendritic cells and suppresses tumor growth. Cancer Res. 2006;66:3513–3522. doi: 10.1158/0008-5472.CAN-05-1493. [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Jéru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, Grimprel E, Landman-Parker J, Hentgen V, Marlin S, McElreavey K, Sarkisian T, Grateau G, Alnemri ES, Amselem S. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A. 2008 Feb 5;105(5):1614–9. doi: 10.1073/pnas.0708616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karrasch T, Jobin C. NF-kappaB and the intestine: friend or foe? Inflamm Bowel Dis. 2008;14:114–124. doi: 10.1002/ibd.20243. [DOI] [PubMed] [Google Scholar]
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, Wall GD, Wisotzkey R, Alag S, Akhtari S, Ronaghi M. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One. 2010 Sep 29;5(9) doi: 10.1371/journal.pone.0013066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, Taxman DJ, Ting JP. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J Immunol. 2007;178:1256–1260. doi: 10.4049/jimmunol.178.3.1256. [DOI] [PubMed] [Google Scholar]
- Madge LA, May MJ. Classical NF-kappaB activation negatively regulates noncanonical NF-kappaB-dependent CXCL12 expression. J Biol Chem. 2010;285:38069–38077. doi: 10.1074/jbc.M110.147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline JL, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–2525. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- Neerincx A, Lautz K, Menning M, Kremmer E, Zigrino P, Hosel M, Buning H, Schwarzenbacher R, Kufer TA. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J Biol Chem. 2010;285:26223–26232. doi: 10.1074/jbc.M110.109736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc. 2007;2:1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Park JH, Kim YG, Shaw M, Kanneganti TD, Fujimoto Y, Fukase K, Inohara N, Nunez G. Nod1/RICK and TLR signaling regulate chemokine and antimicrobial innate immune responses in mesothelial cells. J Immunol. 2007;179:514–521. doi: 10.4049/jimmunol.179.1.514. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Razani B, Zarnegar B, Ytterberg AJ, Shiba T, Dempsey PW, Ware CF, Loo JA, Cheng G. Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci Signal. 2010;3:ra41. doi: 10.1126/scisignal.2000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, Wang E, Ma W, Haines D, O’HUigin C, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207:1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A. 2001;98:13249–13254. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollid LM, Johansen FE. Animal models of inflammatory bowel disease at the dawn of the new genetics era. PLoS Med. 2008;5:e198. doi: 10.1371/journal.pmed.0050198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxman DJ, Holley-Guthrie EA, Huang MT, Moore CB, Bergstralh DT, Allen IC, Lei Y, Gris D, Ting JP. The NLR adaptor ASC/PYCARD regulates DUSP10, mitogen-activated protein kinase (MAPK), and chemokine induction independent of the inflammasome. J Biol Chem. 2011;286:19605–19616. doi: 10.1074/jbc.M111.221077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thu YM, Richmond A. NF-kappaB inducing kinase: a key regulator in the immune system and in cancer. Cytokine Growth Factor Rev. 2010;21:213–226. doi: 10.1016/j.cytogfr.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JP, Duncan JA, Lei Y. How the noninflammasome NLRs function in the innate immune system. Science. 2010 Jan 15;327(5963):286–90. doi: 10.1126/science.1184004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uronis JM, Herfarth HH, Rubinas TC, Bissahoyo AC, Hanlon K, Threadgill DW. Flat colorectal cancers are genetically determined and progress to invasion without going through a polypoid stage. Cancer Res. 2007;67:11594–11600. doi: 10.1158/0008-5472.CAN-07-3242. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. NatImmunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Manji GA, Grenier JM, Al-Garawi A, Merriam S, Lora JM, Geddes BJ, Briskin M, DiStefano PS, Bertin J. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem. 2002 Aug 16;277(33):29874–80. doi: 10.1074/jbc.M203915200. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ, Kitani A, Strober W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118:545–559. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharry CE, Haines KM, Carroll RG, May MJ. Constitutive non-canonical NFkappaB signaling in pancreatic cancer cells. Cancer Biol Ther. 2009 Aug;8(16):1567–76. doi: 10.4161/cbt.8.16.8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, Kurtz S, Coffield VM, Accavitti-Loper MA, Su L, et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem. 2005;280:39914–39924. doi: 10.1074/jbc.M502820200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, Yang X, Hong J, Songyang Z, Chen ZJ, et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity. 2011;34:843–853. doi: 10.1016/j.immuni.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Fuss IJ, Watanabe T, Asano N, Davey MP, Rosenbaum JT, Strober W, Kitani A. NOD2 transgenic mice exhibit enhanced MDP-mediated down-regulation of TLR2 responses and resistance to colitis induction. Gastroenterology. 2007;133:1510–1521. doi: 10.1053/j.gastro.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Lich JD, Moore CB, Duncan JA, Williams KL, Ting JP. ATP binding by monarch-1/NLRP12 is critical for its inhibitory function. Mol Cell Biol. 2008;28:1841–1850. doi: 10.1128/MCB.01468-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. 2010;185:4912–4920. doi: 10.4049/jimmunol.1002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, Green DR, Lamkanfi M, Kanneganti TD. The NOD-Like Receptor NLRP12 Attenuates Colon Inflammation and Tumorigenesis. Cancer Cell. 2011;20(5):649–60. doi: 10.1016/j.ccr.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnegar B, Yamazaki S, He JQ, Cheng G. Control of canonical NF-kappaB activation through the NIK-IKK complex pathway. Proc Natl Acad Sci U S A. 2008;105(9):3503–8. doi: 10.1073/pnas.0707959105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.