

Abstract
The role of voltage-dependent Ca channels (VDCC) in the membrane permeation of two toxic metals, lead (Pb) and cadmium (Cd), was studied in mammalian cells. Both metals interact with Ca-binding sites, but, while Cd influx appears to occur mainly through the same pathways as Ca, Pb is also rapidly taken up by different passive transport systems. Furthermore, I compared the effect of Cd in two Chinese hamster ovary (CHO) cell lines, a wild-type and a modified cell line, which were permanently transfected with an L-type VDCC. When cultures were subjected to a brief (30–60 min) exposure to 50–100 μM Cd, apoptotic features, metal accumulation, and death were comparable in both cell lines although, in transfected cells, the effect of Cd treatment was partially prevented by nimodipine (VDCC antagonist) and enhanced by BayK8644 (VDCC agonist). Thus, expression of L-type Ca channels is not sufficient to modify Cd accumulation and sensitivity to a toxicological significant extent and while both Cd and Pb can take advantage of VDCC to permeate the membrane, these transport proteins are not the only, and frequently not the most important, pathways of permeation.
1. Introduction
Metals are used by biological systems because of their catalytic versatility, and some of them, namely, sodium (Na), potassium (K), calcium (Ca), and magnesium (Mg) are among the essential nutrients of living cells. In contrast, other metals can be fatal to mammalian cells even in tiny amounts. A common classification tends to sort these potentially toxic metals in to two classes: those required by living organisms as essential micronutrients and those devoid of any biological function and thus potentially toxic even at very low concentration, such as cadmium (Cd), chromium (Cr), mercury (Hg), and lead (Pb). The difference between toxic and nontoxic metals is hard to define. Even micronutrients, such as cobalt (Co), copper (Cu), iron (Fe), manganese (Mn), molybdenum (Mo), and zinc (Zn) can be detrimental to living organisms, when present in excessive levels, and a refined equilibrium between deficient and toxic concentrations has to be maintained. This is particularly important for very specialized tissues, such as the brain, where metals induce oxidative damage and some of the essential micronutrients, such as Fe, Zn, and Cu, have been implicated in etiology and development of different neurological and neurodegenerative diseases [1, 2]. Less obviously, living organism may find use for nonessential toxic metals in extreme condition. An elegant example of unexpected biological function of Cd has been recently reported in marine diatoms [3].
Because most toxic metals have entered the environment as a consequence of historical handcraft and industrial processing, specialized cells might not have developed the intrinsic ability to distinguish between physiological and toxic elements. For example, Cd and Pb represent a threat for mammalian cells, because they are able to replace or mimic essential metals in the first or early step of transport and metabolism, but then are incapable of mediating subsequent vital functions [4, 5]. The toxic effects are then a consequence of both impairment of transport systems and accumulation into the cell. Recent research has gathered an increasing body of evidence for the interaction of toxic metals with intracellular components that lead to cellular injury and defense, but the mechanisms of transport of these metals and metal-containing species across plasma membranes remain to be fully characterized.
Cd and Pb mimic Ca and Zn at their specific sites and bind calmodulin [6, 7], protein kinase C [8–10], and synaptic proteins [11]. These metal ions antagonize Ca influx through voltage-dependent and receptor-operated Ca channels, and these channels are regarded as the major route of their entry into mammalian cells.
Both Cd and Pb are well-known specific blockers of voltage-dependent Ca channels (VDCC), but the mechanism of block is quite different. While Cd binds to the high affinity site inside the VDCC pore [12, 13], Pb competes with Ca at a site external to the VDCC, and the binding is not voltage-dependent [14]. Given these differences in the mechanism of interaction, permeation of these metals through VDCC is also different. VDCCs mediate Cd influx in excitable cells [15–18], including mammalian neurons [19], and have been proposed to participate in Cd uptake also in cells from nonexcitable tissues [20]. In a previous work of my laboratory, we showed that in certain cells Cd permeation occurs mainly through VDCC of the L-type [19, 21]. However, in other studies, the presence of VDCC per se did not seem to enhance sensitivity to Cd; for example, VDCCs expressing (PC12) and nonexpressing (PC18) cells showed comparable LD50 for Cd [18], and the role of this pathway in the induction of cell death appears questionable.
The situation is even more complicated for Pb, whose chemical basis for mimicking Ca is not obvious [4] and which is known to adapt to structurally diverse binding geometries [22]. In cerebellar granule neurons, Pb uses at least three pathways of permeation and besides voltage-dependent calcium channels (VDCC) and NMDA-activated channels, is rapidly taken up through passive transport system [23, 24].
In this paper I will present experimental evidence concerning transport of Pb and Cd across cellular membranes and the relative contribution of VDCC in their uptake.
2. Materials and Methods
2.1. Cell Culture
Cerebellar granule neurons were prepared from 8-day-old Wistar rats as previously described [19, 25], plated on 20 mm poly-L-lysine coated glass coverslips and maintained in basal Eagle's culture medium, supplemented with 10% fetal calf serum, 100 μg/mL gentamicin and 25 mM KCl, in a humidified 95% air/CO2 atmosphere at 37°C. Cultures were treated with 10 μM cytosine arabinoside from day 1 to minimize the proliferation of nonneuronal cells. Experiments were carried out in cultures between 5 and 13 days in vitro.
Wild-type Chinese hamster ovary (CHO) cells were obtained from American Type Culture Collection. CHO stably transfected with cDNA encoding for several subunits of voltage-dependent calcium channels (VDCC) were a generous gift of Franz Hofmann and Norbert Klugbauer (Institut für Pharmakologie und Toxikologie, Münich, Germany). The cells used in this study are termed CHOCα9β 3 α 2/δ4 (abbreviated CHOCα) to indicate that they express the α 1C-b subunit of VDCC, as well as β 3 and α 2/δ4 subunits from smooth muscle [26].
Both wild-type CHO and CHOCα cells were maintained in DMEM medium (Sigma Chemical Co, St. Louis, MO, USA) supplemented with 10% bovine serum, in a 5% CO2 humidified atmosphere at 37°C. For CHOCα cells, the culture medium was routinely supplemented with antibiotic geneticin G418 20 μgr/mL. For the experiments, cells were plated in 12-multiwell trays at a density of 2 × 105 cells/mL.
2.2. Microscopy
Measurement of intracellular Cd and Pb was performed on monolayer cultures plated on glass coverslips. For photometric measurements, cells were incubated with 6 μM of the cell-permeant Fura2-AM ester form of the dye for 45 min at 37°C and then washed several times with standard saline at room temperature, as previously described [19, 23]. Cd and Pb were applied in the absence of Ca and treatment was terminated by the addition of the membrane-permeant metal chelator (N,N,N′,N′-tetrakis-2-pyridyl methyl ethylenediamine, TPEN, 100 μm).
For confocal microscopy, cells were incubated with Oregon Green 488 Bapta-1, a fluorescent indicator, whose fluorescence intensity is increased upon binding Ca, Cd, or Pb. Cells were imaged using a confocal laser scanning microscope Nikon PCM2000 (Nikon Instr., Florence, Italy) with an oil immersion 100 × objective (Na = 1.3), coupled to a 50 μm confocal pinhole condition [27].
2.3. Cd Treatment
Cells were subjected to 30–60 min treatments with 50 or 100 μM CdCl2 in culture medium without serum. When specified, the medium was supplemented with 30 mM KCl to depolarize the cell membrane and with the following dihydropyridines (DHPs) : BayK 8644, which is an L-type VDCC agonist, or nimodipine, a channel antagonist. Stock solutions of nimodipine and BayK (10 mM) were made up in 100% ethanol and diluted in medium to the final concentration. High KCl alone, as well as DHPs (no Cd added), had no effect on cell viability and appearance. After incubation cells were washed three times with PBS containing 1 mM EDTA, and fresh medium containing serum was replaced. Cultures were kept in the incubator for another 18–24 hours before testing. Cell viability in each sample was assessed by trypan-blue exclusion assay. All chemicals, culture media and sera, were from Sigma-Aldrich Italy.
2.4. Capacitance and Current Measurements by Patch-Clamp Technique
Membrane currents were measured from wild-type CHO and permanently transfected CHOCα cells in whole-cell clamp configuration by a patch-clamp Axopatch amplifier (Molecular Devices Corporation, Union City, CA, USA). Electrodes were manufactured from borosilicate glass capillaries (Hilgenberg GmbH, Malsfeld, Germany) and had resistance of ≈4 MΩ. Voltage stimulation and data acquisition were performed by a PC through a Digidata 1440A interface and Pclamp-10 software (Molecular Devices). Currents were low-pass filtered at 5 kHz and digitized at 10 kHz. Capacitance transients were minimized by analog compensation, and the value obtained by this compensation was taken as an estimate of the cell capacitance. All currents traces were further corrected for leak and residual transients by a computer generated P/4 protocol. The holding potential was −80 mV in all the experiments. Current traces were analyzed with Clampfit-10 and Sigma Plot (Jandel Scientific, Erkrath, Germany) software.
Cells were continuously superfused by gravity flow (10 mL/min) with a solution containing (in mM) NaCl 140, KCl 5.4, CaCl2 1.8, Hepes 5. The pH was 7.4. The internal (pipette) solution contained (in mM): CsCl 20, CsOH 110, Aspartic acid 100, EGTA 5, Hepes 5, with pH adjusted to 7.3 with Trizma base. Calcium currents were recorded in similar external solution containing 130 mM NaCl and 5 mM CaCl2. Wild-type CHO cells do not possess prominent voltage-dependent currents, and CHOCα only contained the L-type VDCC under study; therefore there was no need to antagonize the current through other voltage-dependent channels to resolve Ca currents. The application of modifiers, such as agonist and antagonist DHPs, was accomplished by gravity flow; control ion substitution experiments showed that the external bath was completely changed in 10 sec, which was the maximal stimulation rate in these experiments.
2.5. Absorption Spectroscopy Cd Determination
Cells were exposed to Cd in culture medium without added serum. After treatment, cultures were washed three times with PBS containing 1 mM EDTA and harvested immediately. Viable cells were counted in triplicate in each sample. Cells were then washed again three times with the above buffer, resuspended in distilled water and disrupted by sonication (Sonopuls Ultrasonic Homogenizer, Bandelin) for 2 minutes in an ice bath. The total Cd content of each sample was measured by Flameless Atomic Absorption Spectroscopy (FAAS) using a Perkin Elmer Spectrophotometer (Model 1100 B) equipped with a graphite furnace (Model HGA 700). The Cd content was normalized to the volume occupied by the cells and molarity was calculated (see results).
2.6. Statistical Analysis
Data are presented as mean ± standard error of the mean in at least 3 experiments. Statistical significance was evaluated by Student-Newman-Keuls multiple comparison test (In Stat, GraphPad Software).
3. Results And Discussion
3.1. Influx of Toxic Metals into Mammalian Cells
Influx of toxic divalent metal ions into mammalian cells can be monitored in cells preloaded with specific fluorescent probes, Fura2 [17, 19, 23], or Oregon green. These fluorescent indicators were developed and are predominantly used for determining intracellular calcium levels, but they have relatively high affinity also for other polyvalent ions, including Cd and Pb.
Figure 1 shows how exposure to a solution containing a toxicologically significant concentration of Cd or Pb causes a sizeable increase in Oregon Green fluorescence, similar to that seen following Ca influx [28]. This demonstrates feasibility of this probe as toxic metal influx indicator. In this experiment, while Cd uptake was triggered by depolarization, indicating the essential contribution of VDCC (Figure 1(A) (a)-(b)), neurons rapidly accumulate Pb even in the absence of a specific stimulus (Figure 1(A) (c)-(d)).
Figure 1.
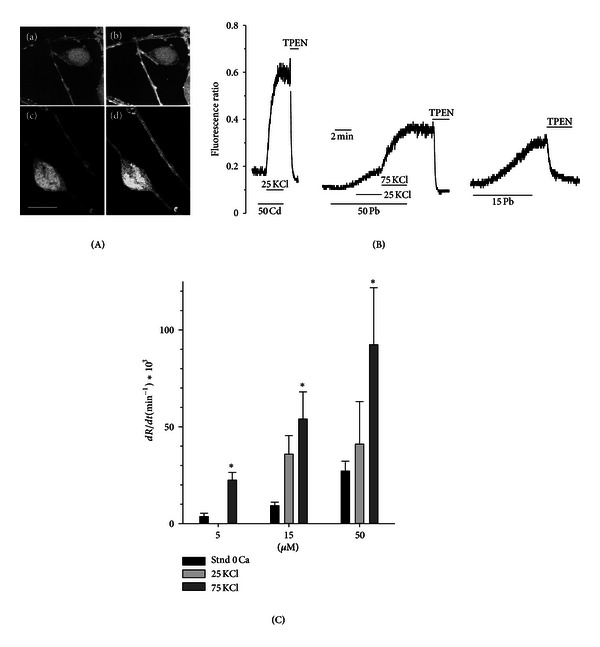
Uptake of Cd and Pb in cerebellar granule neurons measured by fluorescent dyes. (A) Confocal microscopy images of Cd (a, b) and Pb (c, d) uptake by cerebellar granule cells preloaded with the divalent metal-sensitive dye Oregon Green. In (a) and (c), neurons were bathed in a physiological saline. In (b) they have been superfused with a solution containing 0 Ca, 30 mM KCl and 100 μM Cd Cl2, which caused the dye fluorescence to increase significantly. In (d), external solution contained 0 Ca and Pb 15 μM, which permeates through the neuron membrane even in the absence of a depolarizing stimulus and also caused an increase in the dye fluorescence. Bar = 10 μM. (B) Real-time recording of the influx of Cd and Pb in Fura-2 loaded cerebellar granule neuron and the effect of membrane depolarization. Cells were treated with the metals in nominal Ca-free solution. From left to right: time course of the fluorescence ratio, R = E340/E380, following application of 50 μM Cd and membrane depolarization (25 mM KCl) and application of 15–50 μM Pb in resting conditions and increasing depolarizations (25 and 75 mM KCl). (C) Summary of the results obtained in basal and in depolarizing solutions (25 and 75 mM KCl) with different doses of Pb. The time course of R(t) was approximated by a straight line and the slope dR/dt was calculated in each case, as a measure of Pb influx. Data are mean ± SEM in 4 experiments and *indicates significantly different from basal (no depolarization) for each dose of Pb (P < 0.05).
Figure 1(B) shows real-time measurements of Cd and Pb influx in Fura2-loaded neurons [19, 23]. The influx was quantified by measuring the fluorescence emission ratio R (E380/E340), and membranes were depolarized by increasing concentration of external KCl [25]. In the left panel, the fluorescent ratio, E340/E380, increased negligibly when 50 μM Cd was applied in resting condition, but the rate of rise, dR/dt, was enhanced by >60 times following depolarization with 25 KCl. In contrast, Pb (15–50 μM) determined a sizeable increase of R even in the absence of any specific stimulus that opens VDCC, and dR/dt was increased by 5 times from the resting level with 75 mM KCl (Figure 1(C)). Fast uptake of Pb in the absence of any specific stimulus appears to be a peculiar feature of neurons and may be linked to the specific lipidic composition of the neuronal membranes, while the role of VDCC is likely to significant, but not essential [23].
3.2. Role of VDCC in Cd-Induced Mortality
As VDCC channels, and in particular those of L-type [15, 19] play a prominent role in uptake of Cd by excitable cells, the involvement of these channels in Cd toxicity was investigated further. Tests of mortality, chromatin condensation, and DNA fragmentation showed that a pulsed Cd treatment induces delayed apoptotic cell death in VDCC-containing insulinoma cells and that nimodipine protects against Cd-induced apoptosis and necrosis in these cells, supporting a specific involvement of L-type VDCC [21]. The same treatments were largely harmless in VDCC-free HeLa cell cultures, in which neither death nor DNA fragmentation was observed. Therefore, excitable cells appear more susceptible than nonexcitable epithelial-like cells to Cd accumulation. However, it is not clear whether the mere presence of VDCC can make cells more vulnerable to toxic metal injury. To answer this question we have compared the effects of Cd in two cell types that are virtually identical, except for the expression of VDCC in one of the two. This approach is made possible by the availability of a CHO cell line that has been permanently transfected with VDCC α 1, β, and α 2/δ subunits, CHOCα9β 3 α 2/δ 4 cells [26, 29]. Prominent voltage-dependent Ca currents were recorded from the permanently transfected cell line, while such currents are completely absent in wild-type CHO (Figure 2(a)). The currents are larger with Ba as charge carrier and are sensitive to DHP modulation (Figure 2(b)), as expected for an L-type Ca channel.
Figure 2.
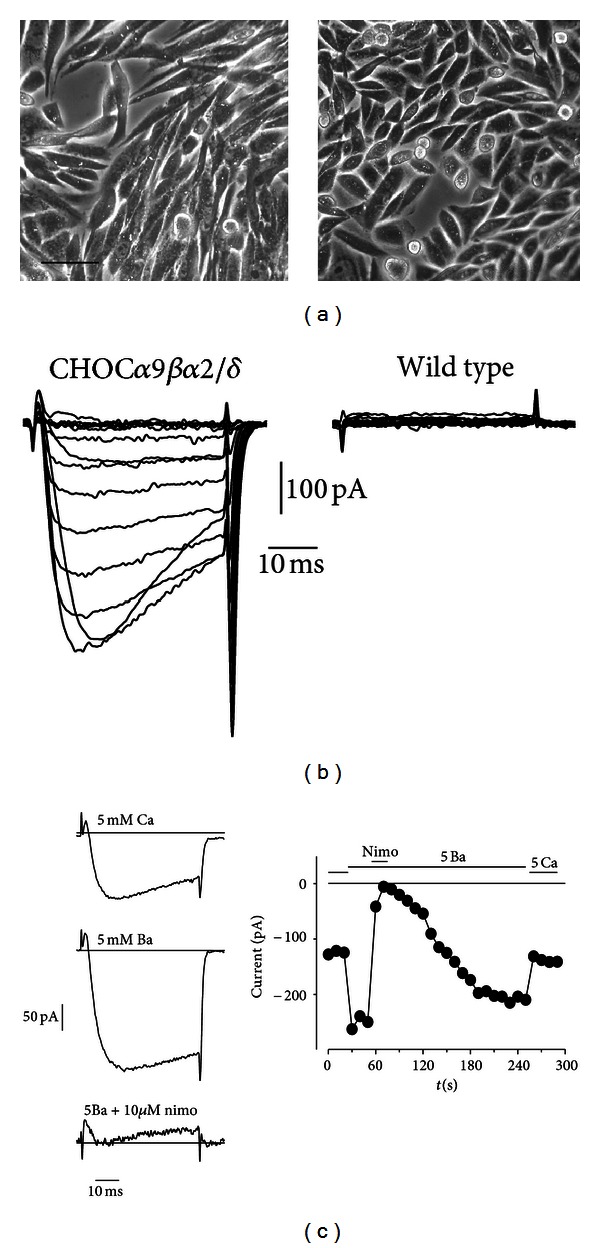
CHOCα cells express L-type VDCC. (a) Representative microphotographs of monolayer cell cultures of permanently transfected CHOCα cells (left) and wild-type CHO cells (right). Bar = 35 μM. (b) Representative current traces evoked by depolarizing voltage steps of 40 ms duration from −60 to +80 mV from a holding potential of −80 mV in a CHOCα cell (left) and a wild-type CHO cell (right). The external solution contained 5 mM CaCl2. This protocol evoked voltage-dependent calcium current in the permanently transfected cell, while no current was present in CHO cell of the wild-type. (c) Characterization of the Ca current in CHOCα cell. The current was increased by more than 50% when the external solution was changed from 5 mM CaCl2 to 5 mM BaCl2, and it was reversibly blocked by 10 μM nimodipine. Current traces evoked by 50 ms depolarizing steps from −80 mV (holding potential) to +10 mV in the three conditions are shown on the left. The graph on the right shows the time course of the experiment.
Both cell types (wild-type and CHOCα) were subjected to a “pulse treatment” with 50 or 100 μM Cd in serum-free culture medium and observed after 18–24 hour after wash. Cell suffering and mortality was negligible immediately after the treatment, but it was evident after at least 16 hours. As previously observed [21, 30, 31], Cd accumulates into the cell during treatment, but the toxic mechanism leading to cell death becomes effective at a later time. Figure 3 shows the appearance of the cell monolayer 24 hours after an incubation of 30 min with 100 μM Cd. Cells exposed to Cd were of smaller size than control cells and had an increased tendency to detach from their neighbors and from the substrate and float. In addition, CHOCα VDCC-expressing cells frequently acquired an irregular shape, with shredded edges (Figure 3). The number of viable cells was estimated from the number of adherent, trypan-blue excluding cells, and this number was normalized to that of adherent viable cells in control conditions, as in our previous works [21]. Despite some morphological features, the dose dependence of Cd sensitivity was very similar in the two cell types (Figure 3). Therefore, by this approach it is not possible to demonstrate a clear role of VDCC in mediating Cd-induced cell detachment.
Figure 3.
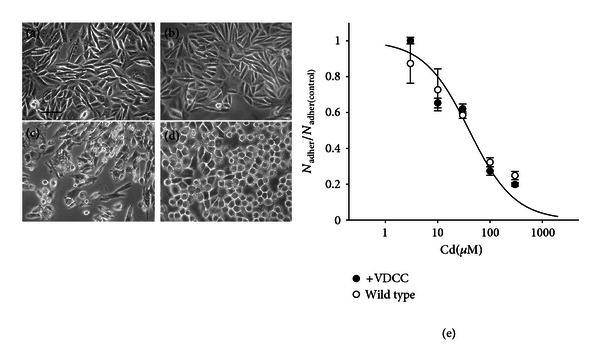
Effect of pulse treatment with Cd in CHOCα VDCC-expressing and wild-type CHO cells. Microphotographs of control (a, b) and Cd-treated (c, d) cells: (a, c) VDCC-expressing CHOCα cells and (b, d) wild-type CHO cells. Both cell types were incubated in 100 μM Cd for 60 minutes in the presence of 30 mM KCl. Pictures were taken 24 h after wash of the metal. Bar = 35 μM. The graph (e) shows the effect of a 30 min pulse treatment with Cd on cell adhesion, as a function of concentration. Trypan-blue excluding adherent cells were counted 24 h after wash of the metal. Points are average ± sem of 3 experiments in the same condition in CHOCα (filled circles) and wild-type CHO (empty circles) and were best fitted to the function. Nadher/Nadher(control) = 1/(1 + ([Cd]/ED50)), where Nadher/Nadher(control) is the number of adherent cells after Cd treatment normalized to the number of adherent cells in control culture; [Cd] is the concentration of Cd and ED50 is the concentration of Cd that causes detachment from the substrate of 50% of cells. The best fit yielded ED50 = 40 μM for CHOCα and 43 μM for wild-type CHO. The two curves are overlapped. In contrast with the different appearance, the two cell types were similarly affected by Cd treatment.
In subsequent experiments, we challenged the cells with Cd in the presence of 1 μM DHP modifiers. When VDCC-expressing CHOCα cells were exposed to Cd in the presence of the antagonist (nimodipina) or agonist (BayK 8644), the effect of the metal was clearly modified with respect to cells exposed to Cd alone (Figure 4), with a sizeable increase of mortality in the presence of BayK and protection in the presence of nimodipine. DHP modifiers were largely ineffective in wild-type CHO cells exposed to Cd (not shown). Therefore, it seems that the actual contribution of L-type VDCC to Cd uptake is clearly underscored only in the presence of these modifiers.
Figure 4.
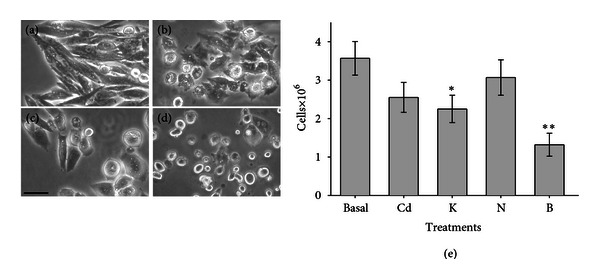
Effect of dihydropyridines on Cd cytotoxicity in VDCC-expressing CHOCα cells. Cells were treated for 30 min in (a) control medium containing 30 mM KCl, (b) 100 μM Cd + 30 mM KCl, (c) 100 μM Cd + 30 mM KCl + 1 μM nimodipine, (d) 100 μM Cd + 30 mM KCl + 1 μM BayK8644. Note the partial recovery in shape of the cells treated in the presence of nimodipine and definitive lost of adhesion when the treatment was performed in the presence of BayK. Bar = 25 μM. The graph (e) represents counts of viable adherent cells in the same experiment (average ± sem in 3 samples) in control (basal), following for a 30 min treatment with 100 μM Cd (Cd), Cd + 30 mM KCl (K), Cd + 30 mM KCl and 1 μM nimodipine (N), and Cd + 30 mM KCl and 1 μM BayK (B). * indicates significantly different from control with P < 0.05, and ** with P < 0.0001.
Another indicator of apoptosis is cell shrinkage, which can be quantified by cell capacitance measurements. Both wild-type and CHOCα cells showed a decrease in cell capacitance 18–24 hours after Cd treatment. In wild-type, cell capacitance was 24 ± 2 pF (n = 15) in control and 15 ± 3 pF (n = 6) after incubation with 100 μM Cd, significantly different from control with P < 0.05. In CHOCα cells, cell capacitance was 35 ± 3 pF (n = 17) in control and 22 ± 2 pF (n = 39) after incubation with 100 μM Cd, significantly different from control with P < 0.001. In CHOCα cells, the effect of Cd treatment was partially prevented by nimodipine: the capacitance was 35 ± 3 pF (n = 10) and the difference between this condition and control condition was not significant (Figure 5). This indicates again that the contribution of L-type VDCC in Cd-induced toxicity is evident only in the presence of DHPs.
Figure 5.
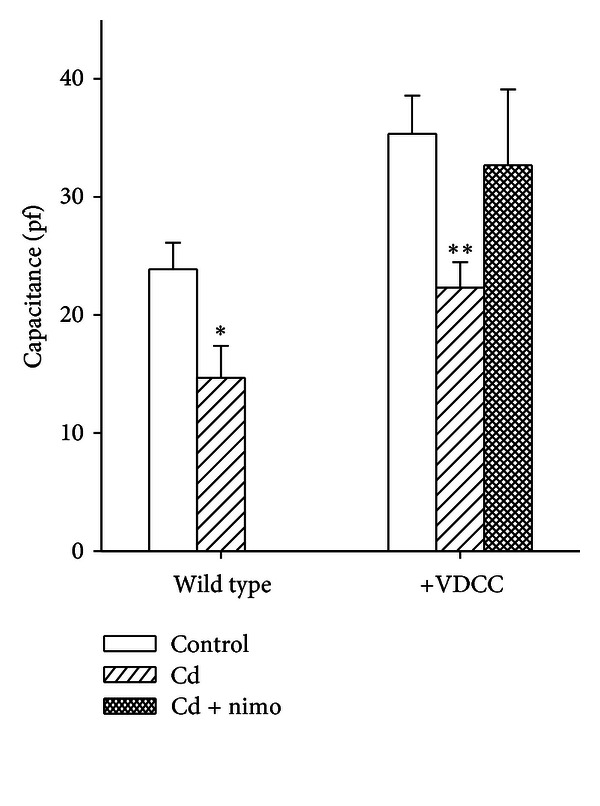
Evidence of Cd-induced apoptosis: change in cell capacitance. Cells were incubated with 100 μM Cd for 60 minutes, and electrical measurements were performed 24 h after wash. Membrane capacitance was estimated from transient compensation (see Section 2.4). Bars represent mean ± sem. Treatment with Cd caused shrinkage of all cell, as revealed by reduction of the cell capacitance in both wild-type (n = 15 in control and n = 6 with Cd treatment) and CHOCα cells (n = 17 in control and n = 39 with Cd treatment). Nimodipine protected CHOCα cells from Cd-induced shrinkage (n = 10). * indicates significantly different from control with P < 0.05, and ** with P < 0.001.
Finally, we performed measurements of total Cd accumulation during 60 min incubation time by atomic absorption spectroscopy. For these experiments, cells were harvested immediately after the treatment, and the total amount of Cd was normalized to the cell volume, estimated from the single-cell capacitance. Assuming that both cell types are approximately spherical and have a specific membrane capacitance of 1 μF/cm2, 106 wild-type CHO cells comprise a volume of 11 μL, while 106 CHOCα cells a volume of 19.5 μL. This difference is not negligible, and with this method, the results were considerably more reproducible than using a standard normalization to the total protein content. Data shown in Figure 6 indicate that the difference in Cd accumulation is barely or not significant when both cell types are incubated in 100 μM Cd with or without elevated KCl. On the other hand, BayK 8644 significantly enhanced and nimodipine significantly decreased metal accumulation in depolarized CHOCα cells treated with 50 or 100 μM Cd, but not in wild-type CHO cells.
Figure 6.
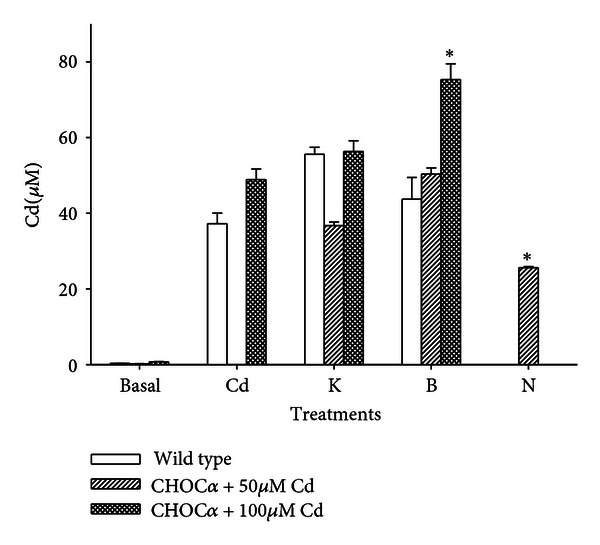
Total Cd accumulation measured by FAAS in CHOCα and wild-type CHO cells. Both cell types were incubated for 1 hour in medium containing 0, 50, or 100 μM Cd and other modifiers. Cd determination was normalized to the cell volume (see text). Treatments were as follows: control 0 Cd (basal), 60 min Cd (Cd), Cd + 30 mM KCl (K), Cd + 30 mM KCl, and 1 μM BayK (B), Cd + 30 mM KCl and 1 μM nimodipine (N). Depolarization (treatment K) did not increase significantly the intracellular concentration of Cd (P > 0.05). In CHOCα cells, but not in wild-type CHO, BayK significantly enhanced and nimodipine significantly reduced Cd accumulation, both with P < 0.001 (*), with respect to the same treatments in depolarization. Bars represent mean ± sem in at least 10 experiments in each group.
Toxicity of both Cd and Pb is mainly due to their ability to permeate not simply mammalian cell membranes, but specifically tight membrane barriers, such as the epithelial lining in the gastrointestinal tract, luminal membrane of proximal tubules in the kidney, and the blood-brain barrier. In early work, the influx of Pb and Cd into mammalian cells was frequently ascribed to permeation through calcium channels, and in particular L-type DHP-sensitive VDCC [16, 18, 20, 32]. More recently other types of VDCC, different from the classical DHP-sensitive L-type, have been implicated both in Cd [19, 33] and Pb [23] uptake. Moreover, Pb has been shown to use different pathways in several systems [34–37], and it is largely independent of VDCC even in neurons [23, 24].
Here I have shown that expression of L-type VDCC is not sufficient to modify Cd uptake to a toxicological significant extent, at least in the case of a brief, intense exposure, as those used in this study. Cd accumulation and sensitivity were comparable in two cell lines whose only difference was the presence of L-type VDCC in one of the two. Indeed, wild-type CHO cells, which do not possess VDCC, are vulnerable to Cd poisoning and take up Cd during relatively brief exposures to an extent sufficient to trigger cell death by apoptosis or necrosis similarly to VDCC expressing CHOCα cells. Although VDCCs play an important role in Cd uptake in CHOCα cells, as indicated by the sizeable effect of L-type channel DHP modifiers, wild-type CHO cells use other transport proteins with similar efficiency and outcome.
It is thus clear that VDCCs are not the main mechanism responsible for toxic metal permeation through cell membranes and tight barriers. Rapid passive transport of Pb, independent of VDCC, was reported at the brain endothelium [34, 36, 37], and even protein-independent transport of lead has been described [35]. In astroglia cell models, the uptake of Pb was shown to be driven by store-depletion-activated channels [38] and by two distinct pH-sensitive transport mechanisms [39]. VDCC-independent uptake of Pb by cerebellar granule neurons was inhibited by La [24], and this observation can provide some clue as for the mechanism of permeation. The involvement of a La-sensitive store-depletion activated channel, as proposed in other systems [36, 37, 40], is unlikely because Pb influx did not require drainage of the stores. Another possibility is that Pb2+ enters the neuron via reverse operation of an exchanger similar (or identical) to the Na–Ca exchanger, which is also very sensitive to La block [41] and whose role in Zn permeation was demonstrated in cortical neurons [42]. Also this possibility was discarded because Na–Ca exchanger does not play a significant role when the Na concentrations are close to the physiological value, as in Esposito et al. [24]. It is more probable that Pb is taken up through a La-sensitive ion carrier similar to the Zn transporter found in brain neurons [43], whose identity and features still wait full definition.
Involvement of different transport proteins in Cd transport has been described. At the epithelial lining of the gastrointestinal barrier, DMT1, the main transporter that absorbs Fe in the brush border membrane of the mammalian intestine has been shown to carry also different toxic metals [44–46]. The role of this transporter in Cd absorption is underscored by the fact that iron deficiency creates a significant risk for increased cadmium exposure by increasing gastrointestinal absorption from 5% to as much as 20%. In general, a low level of essential metals favors uptake of nonessential (toxic) elements that compete for their site, and this stresses the importance of evaluating metal balance, in contrast with metal concentration. In addition, other metal-specific transporters have been identified at the cDNA level [47] and studied in expression systems [48].
4. Conclusion
Ionic mimicry is a useful framework to study the mechanisms of metal toxicity. Uptake of toxic metals, such as Cd and Pb, by mammalian cells of different tissues occurs through many different pathways, of which VDCCs have frequently been regarded as prominent. Because of the abundance of VDCC, excitable cells may be more susceptible to accumulate Cd and Pb than nonexcitable cells. In this chapter I have shown that although Cd and Pb can permeate through VDCC, this is not the most relevant mechanism of passive uptake of Pb, while the mere presence of VDCC is not sufficient to make cells significantly more vulnerable to Cd injury. Other factors, including different transport systems and expression of detoxification proteins, may be more relevant in the accumulation process of these toxic metals.
Acknowledgments
The author thanks Ilaria Zanardi and Irena Baranowska-Bosiacka (PUM, Szczecin, Poland) for help in Cd treatment experiments and Elisabetta Morelli for FAAS measurements. The author is also grateful to Franz Hofmann and Norbert Klugbauer (Institut für Pharmakologie und Toxikologie, Münich, Germany) for their permission to use the permanently transfected CHO cells and to Maurizio Taglialatela and Mauro Cataldi (Università Federico II, Naples, Italy) for sending him the cells.
References
- 1.Castellani RJ, Moreira PI, Liu G, et al. Iron: the redox-active center of oxidative stress in Alzheimer disease. Neurochemical Research. 2007;32(10):1640–1645. doi: 10.1007/s11064-007-9360-7. [DOI] [PubMed] [Google Scholar]
- 2.Smith DG, Cappai R, Barnham KJ. The redox chemistry of the Alzheimer’s disease amyloid βpeptide. Biochimica et Biophysica Acta. 2007;1768(8):1976–1990. doi: 10.1016/j.bbamem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Xu Y, Feng L, Jeffrey PD, Shi Y, Morel FMM. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature. 2008;452(7183):56–61. doi: 10.1038/nature06636. [DOI] [PubMed] [Google Scholar]
- 4.Clarkson TW. Molecular and ionic mimicry of toxic metals. Annual Review of Pharmacology and Toxicology. 1993;33:545–571. doi: 10.1146/annurev.pa.33.040193.002553. [DOI] [PubMed] [Google Scholar]
- 5.Bridges CC, Zalups RK. Molecular and ionic mimicry and the transport of toxic metals. Toxicology and Applied Pharmacology. 2005;204(3):274–308. doi: 10.1016/j.taap.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouyang H, Vogel HJ. Metal ion binding to calmodulin: NMR and fluorescence studies. BioMetals. 1998;11(3):213–222. doi: 10.1023/a:1009226215543. [DOI] [PubMed] [Google Scholar]
- 7.Kern M, Wisniewski M, Cabell L, Audesirk G. Inorganic lead and calcium interact positively in activation of calmodulin. NeuroToxicology. 2000;21(3):353–364. [PubMed] [Google Scholar]
- 8.Markovac J, Goldstein GW. Picomolar concentrations of lead stimulate brain protein kinase C. Nature. 1988;334(6177):71–73. doi: 10.1038/334071a0. [DOI] [PubMed] [Google Scholar]
- 9.Sun X, Tian X, Tomsig JL, Suszkiw JB. Analysis of differential effects of Pb2+ on protein kinase C isozymes. Toxicology and Applied Pharmacology. 1999;156(1):40–45. doi: 10.1006/taap.1999.8622. [DOI] [PubMed] [Google Scholar]
- 10.Tomsig JL, Suszkiw JB. Multisite interactions between Pb2+ and protein kinase C and its role in norepinephrine release from bovine adrenal chromaffin cells. Journal of Neurochemistry. 1995;64(6):2667–2673. doi: 10.1046/j.1471-4159.1995.64062667.x. [DOI] [PubMed] [Google Scholar]
- 11.Bouton CM, Frelin LP, Forde CE, Godwin HA, Pevsner J. Synaptotagmin I is a molecular target for lead. Journal of Neurochemistry. 2001;76(6):1724–1735. doi: 10.1046/j.1471-4159.2001.00168.x. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Ellinor PT, Sather WA, Zhang JF, Tsien RW. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature. 1993;366(6451):158–161. doi: 10.1038/366158a0. [DOI] [PubMed] [Google Scholar]
- 13.Wakamori M, Strobeck M, Niidome T, Teramoto T, Imoto K, Mori Y. Functional characterization of ion permeation pathway in the N-type Ca2+ channel. Journal of Neurophysiology. 1998;79(2):622–634. doi: 10.1152/jn.1998.79.2.622. [DOI] [PubMed] [Google Scholar]
- 14.Peng S, Hajela RK, Atchison WD. Characteristics of block by Pb2+ of function of human neuronal L-, N-, and R-type Ca2+ channels transiently expressed in human embryonic kidney 293 cells. Molecular Pharmacology. 2002;62(6):1418–1430. doi: 10.1124/mol.62.6.1418. [DOI] [PubMed] [Google Scholar]
- 15.Usai C, Barberis A, Moccagatta L, Marchetti C. Pathways of cadmium uptake in excitable mammalian cells: a microspectrofluorimetric study. European Journal of Histochemistry. 1997;41:189–190. [PubMed] [Google Scholar]
- 16.Hinkle PM, Kinsella PA, Osterhoudt KC. Cadmium uptake and toxicity via voltage-sensitive calcium channels. Journal of Biological Chemistry. 1987;262(34):16333–16337. [PubMed] [Google Scholar]
- 17.Hinkle PM, Shanshala ED, Nelson EJ. Measurements of intracellular cadmium with fluorescent dyes—further evidence for the role of calcium channels in cadmium uptake. The Journal of Biological Chemistry. 1992;267:25553–25559. [PubMed] [Google Scholar]
- 18.Hinkle PM, Osborne ME. Cadmium toxicity in rat pheochromocytoma cells: studies on the mechanism of uptake. Toxicology and Applied Pharmacology. 1994;124(1):91–98. doi: 10.1006/taap.1994.1012. [DOI] [PubMed] [Google Scholar]
- 19.Usai C, Barberis A, Moccagatta L, Marchetti C. Pathways of cadmium influx in mammalian neurons. Journal of Neurochemistry. 1999;72(5):2154–2161. doi: 10.1046/j.1471-4159.1999.0722154.x. [DOI] [PubMed] [Google Scholar]
- 20.Souza V, Bucio L, Gutiérrez-Ruiz MC. Cadmium uptake by a human hepatic cell line (WRL-68 cells) Toxicology. 1997;120(3):215–220. doi: 10.1016/s0300-483x(97)00057-7. [DOI] [PubMed] [Google Scholar]
- 21.Gavazzo P, Morelli E, Marchetti C. Susceptibility of insulinoma cells to cadmium and modulation by L-type calcium channels. BioMetals. 2005;18(2):131–142. doi: 10.1007/s10534-004-5789-1. [DOI] [PubMed] [Google Scholar]
- 22.Kirberger M, Yang JJ. Structural differences between Pb2+- and Ca2+-binding sites in proteins: implications with respect to toxicity. Journal of Inorganic Biochemistry. 2008;102(10):1901–1909. doi: 10.1016/j.jinorgbio.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazzolini M, Traverso S, Marchetti C. Multiple pathways of Pb2+ permeation in rat cerebellar granule neurones. Journal of Neurochemistry. 2001;79(2):407–416. doi: 10.1046/j.1471-4159.2001.00557.x. [DOI] [PubMed] [Google Scholar]
- 24.Esposito A, Robello M, Pellistri F, Marchetti C. Two-photon analysis of lead accumulation in rat cerebellar granule neurons. Neurochemical Research. 2005;30(8):949–954. doi: 10.1007/s11064-005-5980-y. [DOI] [PubMed] [Google Scholar]
- 25.Marchetti C, Amico C, Usai C. Functional characterization of the effect of nimodipine on the calcium current in rat cerebellar granule cells. Journal of Neurophysiology. 1995;73(3):1169–1180. doi: 10.1152/jn.1995.73.3.1169. [DOI] [PubMed] [Google Scholar]
- 26.Welling A, Bosse E, Cavalie A, et al. Stable co-expression of calcium channel α1,β and α2/δ subunits in a somatic cell line. Journal of Physiology. 1993;471:749–765. doi: 10.1113/jphysiol.1993.sp019926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diaspro A, Annunziata S, Raimondo M, Ramoino P, Robello M. A single-pinhole confocal laser scanning microscope for 3-D imaging of biostructures. IEEE Engineering in Medicine and Biology Magazine. 1999;18(4):106–110. doi: 10.1109/51.775496. [DOI] [PubMed] [Google Scholar]
- 28.Pellistri F, Cupello A, Esposito A, Marchetti C, Robello M. Two-photon imaging of calcium accumulation in rat cerebellar granule cells. NeuroReport. 2004;15(1):83–87. doi: 10.1097/00001756-200401190-00017. [DOI] [PubMed] [Google Scholar]
- 29.Cataldi M, Secondo A, D’Alessio A, et al. Studies on maitotoxin-induced intracellular Ca2+ elevation in chinese hamster ovary cells stably transfected with cDNAs encoding for L-type Ca2+ channel subunits. Journal of Pharmacology and Experimental Therapeutics. 1999;290(2):725–730. [PubMed] [Google Scholar]
- 30.Galán A, Troyano A, Vilaboa NE, Fernández C, De Blas E, Aller P. Modulation of the stress response during apoptosis and necrosis induction in cadmium-treated U-937 human promonocytic cells. Biochimica et Biophysica Acta. 2001;1538(1):38–46. doi: 10.1016/s0167-4889(00)00134-8. [DOI] [PubMed] [Google Scholar]
- 31.Poliandri AHB, Cabilla JP, Velardez MO, Bodo CCA, Duvilanski BH. Cadmium induces apoptosis in anterior pituitary cells that can be reversed by treatment with antioxidants. Toxicology and Applied Pharmacology. 2003;190(1):17–24. doi: 10.1016/s0041-008x(03)00191-1. [DOI] [PubMed] [Google Scholar]
- 32.Tomsig JL, Suszkiw JB. Permeation of Pb2+ through calcium channels: fura-2 measurements of voltage- and dihydropyridine-sensitive Pb2+ entry in isolated bovine chromaffin cells. Biochimica et Biophysica Acta. 1991;1069(2):197–200. doi: 10.1016/0005-2736(91)90124-q. [DOI] [PubMed] [Google Scholar]
- 33.Leslie EM, Liu J, Klaassen CD, Waalkes MP. Acquired cadmium resistance in metallothionein-I/II(−/−) knockout cells: role of the T-type calcium channel Cacnα1G in cadmium uptake. Molecular Pharmacology. 2006;69(2):629–639. doi: 10.1124/mol.105.014241. [DOI] [PubMed] [Google Scholar]
- 34.Deane R, Bradbury MWB. Transport of lead-203 at the blood-brain barrier during short cerebrovascular perfusion with saline in the rat. Journal of Neurochemistry. 1990;54(3):905–914. doi: 10.1111/j.1471-4159.1990.tb02337.x. [DOI] [PubMed] [Google Scholar]
- 35.Diaz RS, Monreal J. Protein-independent lead permeation through myelin lipid liposomes. Molecular Pharmacology. 1995;47(4):766–771. [PubMed] [Google Scholar]
- 36.Kerper LE, Hinkle PM. Cellular uptake of lead is activated by depletion of intracellular calcium stores. Journal of Biological Chemistry. 1997;272(13):8346–8352. doi: 10.1074/jbc.272.13.8346. [DOI] [PubMed] [Google Scholar]
- 37.Kerper LE, Hinkle PM. Lead uptake in brain capillary endothelial cells: activation by calcium store depletion. Toxicology and Applied Pharmacology. 1997;146(1):127–133. doi: 10.1006/taap.1997.8234. [DOI] [PubMed] [Google Scholar]
- 38.Legare ME, Barhoumi R, Hebert E, Bratton GR, Burghardt RC, Tiffany- Castiglioni E. Analysis of Pb2+ entry into cultured astroglia. Toxicological Sciences. 1998;46(1):90–100. doi: 10.1006/toxs.1998.2492. [DOI] [PubMed] [Google Scholar]
- 39.Cheong JH, Bannon D, Olivi L, Kim Y, Bressler J. Different mechanisms mediate uptake of lead in a rat astroglial cell line. Toxicological Sciences. 2004;77(2):334–340. doi: 10.1093/toxsci/kfh024. [DOI] [PubMed] [Google Scholar]
- 40.Evans TJ, James-Kracke MR, Kleiboeker SB, Casteel SW. Lead enters Rcho-1 trophoblastic cells by calcium transport mechanisms and complexes with cytosolic calcium-binding proteins. Toxicology and Applied Pharmacology. 2003;186(2):77–89. doi: 10.1016/s0041-008x(02)00030-3. [DOI] [PubMed] [Google Scholar]
- 41.Kaczorowski GJ, Slaughter RS, King VF, Garcia ML. Inhibitors of sodium-calcium exchange: identification and development of probes of transport activity. Biochimica et Biophysica Acta. 1989;988(2):287–302. doi: 10.1016/0304-4157(89)90022-1. [DOI] [PubMed] [Google Scholar]
- 42.Sensi SL, Canzoniero LMT, Yu SP, et al. Measurement of intracellular free zinc in living cortical neurons: routes of entry. Journal of Neuroscience. 1997;17(24):9554–9564. doi: 10.1523/JNEUROSCI.17-24-09554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colvin RA. Characterization of a plasma membrane zinc transporter in rat brain. Neuroscience Letters. 1998;247(2-3):147–150. doi: 10.1016/s0304-3940(98)00302-4. [DOI] [PubMed] [Google Scholar]
- 44.Picard V, Govoni G, Jabado N, Gross P. Nramp 2 (DCT1/DMT1) expressed at the plasma membrane transports iron and other divalent cations into a calcein-accessible cytoplasmic pool. Journal of Biological Chemistry. 2000;275(46):35738–35745. doi: 10.1074/jbc.M005387200. [DOI] [PubMed] [Google Scholar]
- 45.Okubo M, Yamada K, Hosoyamada M, Shibasaki T, Endou H. Cadmium transport by human Nramp 2 expressed in Xenopus laevis oocytes. Toxicology and Applied Pharmacology. 2003;187(3):162–167. doi: 10.1016/s0041-008x(02)00078-9. [DOI] [PubMed] [Google Scholar]
- 46.Illing AC, Shawki A, Cunningham CL, Mackenzie B. Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. The Journal of Biological Chemistry. 2012;287:30485–30496. doi: 10.1074/jbc.M112.364208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bressler JP, Olivi L, Cheong JH, Kim Y, Maerten A, Bannon D. Metal transporters in intestine and brain: their involvement in metal-associated neurotoxicities. Human and Experimental Toxicology. 2007;26(3):221–229. doi: 10.1177/0960327107070573. [DOI] [PubMed] [Google Scholar]
- 48.Liu Z, Li H, Soleimani M, et al. Cd2+ versus Zn2+ uptake by the ZIP8 HCO3—dependent symporter: kinetics, electrogenicity and trafficking. Biochemical and Biophysical Research Communications. 2008;365(4):814–820. doi: 10.1016/j.bbrc.2007.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]