

Abstract
Ca2+ sparks are the elementary units of Ca2+ signaling in striated muscle fibers that appear as highly localized Ca2+ release events through ryanodine receptor (RyR) Ca2+ release channels in the sarcoplasmic reticulum (SR). While these events are commonly observed in resting cardiac myocytes, they are rarely seen in resting skeletal muscle fibers. Since Ca2+ spark analysis can provide extensive data on the Ca2+ handling characteritsics of normal and diseased striated muscle, there has been interest in developing methods for observing Ca2+ sparks in skeletal muscle. Previously, we discovered that stress generated by osmotic pressure changes induces a robust Ca2+ spark response confined in close spatial proximity to the sarcolemmal membrane in wild-type intact mammalian muscles. Our studies showed these peripheral Ca2+ sparks (PCS) were altered in dystrophic or aged skeletal muscles. Other methods to induce Ca2+ sparks include permea-bilization of the sarcolemmal membrane with detergents, such as saponin. In this chapter, we will discuss the methods for isolation of muscle fibers, the techniques for inducing Ca2+ sparks in these isolated fibers, and provide guidance on the analysis of data from these experiments.
Keywords: Calcium, Spark, Burst, Skeletal muscle, Permeabilization, Saponin, Osmotic shock, Osmotic stress, Myocyte, Muscle fiber, Ryanodine receptor
1. Introduction
The elementary units of Ca2+ release from sarcoplasmic reticulum (SR) in striated muscle fibers are discreet events known as Ca2+ sparks. Ca2+ sparks were first discovered in cardiac muscle as localized quantal Ca2+ release events originating from arrays of ryano-dine receptor (RyR) Ca2+ release channels inserted into the SR membrane. In cardiac muscle, they represent the elemental units of Ca2+-induced Ca2+ release (CICR) necessary for cardiac contractility (1–3). The discovery of Ca2+ sparks revolutionized our understanding of the physiology and pathophysiology of Ca2+ signaling in cardiac and smooth muscles (4–7). Ever since the discovery of Ca2+ sparks in cardiac muscle, investigators have had difficulty in detecting Ca2+ sparks in adult mammalian skeletal muscle where voltage-induced Ca2+ release (VICR) is the dominant mode of Ca2+ release from the SR (8).
Initial studies detecting Ca2+ sparks in skeletal muscle were performed with amphibian muscle (9, 10) or neonatal mammalian skeletal muscle (11) where they were attributed to the activity of the type 3 RyR (12), a RyR isoform present in mammalian skeletal muscle principally during development (13). While rare observations of Ca2+ sparks were made in resting intact mammalian muscle fibers (11, 14), until recently, significant numbers of events were only observed in skeletal fibers whose sarcolemmal integrity was disrupted by various physical or skinning methods (15, 16). Here we present one such method where saponin detergent is used to permeabilize the membrane of muscle fibers isolated from the extensor digitorum longus (EDL) muscle of rats. Because Ca2+ spark signaling rarely appears in intact mammalian muscle fibers, it was questioned if Ca2+ sparks appear in mammalian skeletal muscle at all, and if so, what sort of physiological role they would play in skeletal muscle function.
In 2003, we began a series of experiments that would reveal mammalian skeletal muscle display Ca2+ sparks at the periphery of the muscle fiber, directly under the sarcolemma. During these studies, we discovered that transient hypoosmotic stress (and non-physiological levels of hyperosmotic stress) induced peripheral Ca2+ sparks (PCS) adjacent to the sarcolemmal membrane in intact muscle fibers (17–19). Our studies suggested a physiological role for PCS in skeletal muscle by linking enhanced spark activity to exercise (17, 20). Further studies showed that this Ca2+ spark response was altered in different states, including muscular dystrophy (17) and aging muscle (21, 22). Additionally, we found osmotic stress-induced Ca2+ signaling is altered in a mouse model of amyotrophic lateral sclerosis (23), another disease state with compromised muscle function.
Several other groups have gone on to examine the phenomena of PCS in mammalian skeletal muscle using variations on this osmotic stress approach or simply through examination of such fibers without stimulation. Multiple investigators have used this technique to confirm and expand our findings of elevated PCS events in dystrophic muscle from mdx mice (24–26) and to link this altered response in dystrophic muscle to increased levels of oxidative stress (25). Additional studies show that PCS are modi-fied by the redox state of the cell, pointing to the physiological relevance of PCS (27). Further evidence of physiological regulation of PCS comes from a series of studies that show dihydropyri-dine receptor (DHPR) modulates the characteristics of PCS (28). Other investigators show that removal of inhibition by DHPR is essential for induction of PCS (29). These findings correspond nicely with other studies illustrating an essential role for DHPR in the repression of spontaneous Ca2+ sparks in differentiating skeletal myotubes (30) and in general regulation of RyR function in many cell types (31).
Here we present our protocols to measure Ca2+ sparks in per-meabilized EDL muscle fibers and intact flexor digitorum brevis (FDB) muscle fibers treated with hypoosmotic stress. These spe-cific methods detail how to isolate muscle fibers from each of these muscle types (Subheadings 3.1 and 3.2) and how to induce Ca2+ sparks in these fibers (Subheadings 3.3 and 3.4). Additionally, we provide a brief tutorial on recording and analyzing Ca2+ data while providing additional resources for further details on this extensive topic (Subheading 3.5).
2. Materials
Water reaching “ultrapure” requirements (18 MΩ per cm, TOC < 10 ppb) was used for all solutions prepared for these studies.
2.1. FDB Muscle Fiber Isolation
To prepare the dissection chamber, add 8 mL liquid Sylgard (DOW CORNING, Sylgard® 184 Silicone Elastomer Kit) into an 100-mm cell culture dish and wait 48 h to let the Sylgard become solid. Store at room temperature.
Isotonic Tyrode Solution: 140 mM NaCl, 5 mM KCl, 10 mM HEPES (free acid), 5.5 mM D-glucose, 2.5 mM CaCl2, 2 mM MgCl2. Adjust pH to 7.2 with NaOH. Filter sterilize. Osmolality of this solution should be measured at 290 ± 5 mOsm. Store at 4°C for up to 2 months. Solution should be warmed to room temperature before use.
Minimal Ca2+ Tyrode Solution: 140 mM NaCl, 5 mM KCl, 10 mM HEPES (free acid), 5.5 D-glucose, 2 mM MgCl2. Adjust pH to 7.2 with NaOH. Filter sterilize. Osmolatity of this solution should be measured at 280 ± 5 mOsm. Store at 4°C for up to 2 months. Solution should be warmed to room temperature before use.
Digestion Solution I: Minimal Ca2+ Tyrode Solution supplemented with 2 mg/mL collagenase type I (#4196 from Worthington). Solution should be prepared in advance and 0.75 mL aliquots should be stored in 1.5 mL snap-cap tubes at −20°C for up to a month. Solution should be warmed to 37°C before use.
Dissecting Tools: Heavy Mayo dissecting scissors, curved iris dissection scissors, Dumont 46 blunt forceps, Noyes spring scissors, dissecting pins.
2.2. EDL Muscle Fiber Isolation
Modified Krebs Solution: 136 mM NaCl, 5 mM KCl, 10 mM HEPES, 10 mM glucose, 2.6 mM CaCl2, 1 mM MgCl2. Adjust pH to 7.0 with NaOH. Osmolatity of this solution should be measured at 310 ± 5 mOsm. Filter sterilize. Store at 4°C for up to 2 months. Solution should be warmed to room temperature before use.
Relaxing Solution: 150 mM K-glutamate, 10 mM HEPES, 2 mM MgCl2, 1 mM EGTA. Adjust pH to 7.0 with NaOH. Filter sterilize. Store at 4°C for up to 2 months. Solution should be warmed to room temperature before use.
Digestion Solution II (in mM): 136 mM NaCl, 5 mM KCl, 10 mM HEPES, 10 mM glucose, 10% fetal bovine serum (FBS), 2 mg/mL collagenase type I (#4196 from Worthington). Adjust pH to 7.0 with NaOH. Solution should be made fresh daily.
Dissection tools: Moria spring scissors, Moria ultra fine-tipped forceps, and dissecting pins.
2.3. Ca2+ Spark Imaging of Intact FDB Fibers
35 mm Delta TPG dishes.
Fluorescent Ca2+ imaging dyes (fluo-4AM, cell permeant, or fluo-3AM, cell permeant) are prepared as 1 mM stocks in DMSO and individual tubes are prepared with 10 μL of stock per tube. Individual tubes are stored desiccated in the dark at −20°C for up to 3 months.
Hypotonic Tyrode Solution: 70 mM NaCl, 5 mM KCl, 10 mM HEPES (free acid), 2.5 mM CaCl2, 2 mM MgCl2. Adjust pH to 7.2 with NaOH. Filter sterilize. Osmolatity of this solution should be measured at 170 ± 2 mOsm. Store at 4°C for up to 2 months. Solution should be warmed to room temperature before use.
Conventional laser scanning confocal microscope with ≥40× objective lens with a minimum of a 1.2-NA. The confocal microscope must be configured for an excitation at wavelength of 488 nm and an emission range at 510–580 nm for recording the fluorescence signal from fluo-3 or fluo-4.
Perfusion system with at least two independent perfusion channels capable of perfusing >1 mL/min of solution through a single perfusion tip of >0.2 mm in diameter.
Three-axis micromanipulator capable of accommodating the perfusion system.
2.4. Ca2+ Spark Imaging of Permeabilized EDL Fibers
Custom made 100 μL Lucite chamber with a glass bottom (16, 32, 33). Commercially available glass bottom dishes can also be used. Some possibilities include Delta TPG dishes, MatTek glass bottom dishes, or confocal imaging chambers.
Fluorescent Ca2+ imaging dyes (fluo-4, pentapotassium salt, or fluo-3, pentapotassium salt) are prepared as 10 mM stocks in millipure water and individual tubes are prepared with 10 μL of stock per tube. Individual tubes are stored desiccated in the dark at −20°C for up to 3 months.
Saponin Solution: 150 mM K-Glutamate, 10 mM HEPES, 2 mM MgCl2, 1 mM EGTA. Adjust pH to 7.0 with NaOH. Add 0.002% saponin, 4% dextran, and 50 μM fluo-4 (or fluo-3) pentapotassium salt (see Note 1). Store at 4°C for up to 2 weeks. Solution should be warmed to room temperature before use.
Internal Solution: 97 mM K2SO4, 10 mM trizma maleate, 10 mM Na2PC (phosphocreatine), 5 mM Na2ATP, 5 mM glucose, 1 mM EGTA, 0.32 mM CaCl2 (200 nM free Ca2+), 10.7 mM MgCl2 (2 mM free Mg2+), 8% dextran, pH 7.0. Make 10 mL stock solution and individual tubes are prepared with 1 mL of stock per tube. Store at −20°C for up to 1 year. Before the experiment, add 100 μM fluo-4 (or fluo-3) into 1 mL internal stock solution (see Notes 2 and 3).
Conventional laser scanning confocal microscope with ≥40× objective lens with a minimum of a 1.2-NA. The confocal microscope must be configured for an excitation at wavelength of 488 nm and an emission range at 510–580 nm for recording the fluorescence signal from fluo-3 or fluo-4.
3. Methods
All manipulations of the experimental preparations were conducted at room temperature unless otherwise noted in the procedure. Adherence to the recommended temperatures for digestion of anatomical muscles with collagenase and for storage of muscle fibers is important for the success of the protocol as a whole.
3.1. FDB Muscle Fiber Isolation
Euthanize the mouse by CO2 inhalation followed by cervical dislocation. Remove the foot from the carcass using a pair of heavy dissecting scissors to cut through the leg slightly above the ankle joint.
Fill a Sylgard dissection chamber with a sufficient quantity of Minimal Ca2+ Tyrode Solution to fully submerge the mouse foot. The foot is then pinned, bottom side facing up, to the dissection chamber using dissection pins. Remove the FDB from the foot using a dissection scope if necessary (see Note 4).
Use forceps to transfer the FDB muscle into a thawed aliquot of Digestion Solution I warmed to 37°C. Always handle muscles by the tendons to avoid additional damage.
Incubate the tube containing the FDB upright at 37°C with orbital shaking at 160 rpm for 60–90 min (see Note 5).
Wash digested FDB muscle by using blunt forceps to transfer the muscle through three dishes containing at least 1 mL of Minimal Ca2+ Tyrode Solution.
Transfer digested FDB muscle into a 1.5 mL snap-cap tube containing 700 μL of Isotonic Tyrode Solution using blunt forceps.
Cut the tip of a standard 20 μL plastic micropipette tip with a razor blade so that the diameter of the tip is just large enough to allow the muscle to be drawn through the tip.
Slowly draw the tendon end of the digested FDB muscle through the pipette tip 5–6 times until it passes through with no resistance. It should be possible to observe portions of the muscle coming loose from the muscle bundle. Allow these released pieces to settle at the bottom of the tube and do not disturb with subsequent pipetting (until step 12 if necessary). This process is known to some as “tituration.”
Cut another plastic micropipette tip to a new diameter that matches the current width of the FDB muscle.
Repeat steps 8 and 9 until the muscle is completely broken into smaller pieces. Let sit for 5 min at room temperature (see Note 6).
Flick the bottom of the tube with a finger to suspend the pieces of the muscle in the Tyrode Solution. Draw out 35 μL with a cut 200 μL plastic micropipette tip and add to the center of a 35-mm Delta TPG dish containing 1 mL of Isotonic Tyrode Solution. The tip should be placed against the glass bottom and the muscle fibers added slowly so that they do not diffuse throughout the dish.
Examine the muscle fibers using a dissection microscope. This will allow determination if the muscle fibers have undergone sufficient tituration and if these can be used for experimentation (see Note 7). Additionally, the number of useful fibers can be determined for preparation of additional dishes (see Note 8).
Additional aliquots of the FDB muscle fibers can be added until there are at least three useful fibers on the dish. The presence of three fibers on the dishes makes it likely that at least one will survive loading with Ca2+ indicator dye.
FDB fibers can be added to more dishes using the volume added to the initial dish as a guide to plate at least three useful fibers. Usually three dishes are prepared at a time for Ca2+ imaging. The tube of muscle fibers can be stored at 4°C for plating fibers into additional dishes later in the day.
3.2. EDL Muscle Fiber Isolation
Euthanize the rat (Sprague–Dawley) with CO2 inhalation. Separate the EDL muscle at the tendon and dissect it out from the leg (see Note 9). Pin down the muscle to the Sylgard dissection chamber filled with Digestion Solution II at room temperature. The muscle should be stretched to the length that it was in the body of the animal.
Place the Sylgard dissection chamber in an orbital shaker (10 rpm) at 37°C for 60 min. The EDL muscle will appear only partially digested with ragged edges but no loss of portions of the muscle into the Digestion Solution.
Wash out the Digestion Solution from the chamber by changing to Krebs Solution 3 times.
Let the digested muscle rest in Krebs Solution plus 10% FBS for at least 2 h at 4°C.
We used a custom-built glass bottom chamber for these experiments. If a custom chamber is not available, commercial glass bottom dishes can also be used. To prepare the glass bottom for imaging, apply ~1 μL of grease (DOW CORNING high vacuum grease) at both ends of the glass bottom ~7 mm apart. Place a small piece of double-side tape (1.5 × 3 mm) on the top of each spot of grease and then add a similar amount of the grease on the top of the tape. Then fill the glass bottom chamber/dish with Relaxing Solution.
Replace the Krebs Solution in the Sylgard dissection chamber with Relaxing Solution. Use Moria spring scissors to make a small cut at one end of the EDL muscle. From the location of the small cut, use Moria ultra fine-tipped forceps to separate a small muscle bundle (containing 3–5 muscle fibers) about 1 cm in length from the whole EDL muscle (do not cut it off from the EDL). Then, use two Moria ultra fine-tipped forceps to peel one muscle fiber from this small bundle. The digestion should be sufficient to loosen the connective tissue around the muscle bundles. So it should not be difficult to separate a single muscle fiber from the bundle. Otherwise, it is necessary to optimize the digestion conditions by increasing the digestion time. Finally, cut out a single fiber segment (about 1 cm). Use a glass Pasteur pipette to transfer the fiber segment to the imaging chamber filled with the Relaxing Solution (see Note 10).
Position either end of the EDL fiber on one of the pieces of tape covered with grease. Then fix the fiber in place against the glass bottom by placing another small piece of double-side tape (1.5 × 3 mm) at each end of the fiber.
3.3. Ca2+ Spark Imaging of Intact FDB Fibers
Transfer 500 μL of Isotonic Tyrode Solution from dishes with plated FDB fibers to a tube of fluo-4 in DMSO. Mix 3 times by pipetting and add back to the dish drop-wise.
Incubate for 60 min at room temperature in the dark.
Load two channels of the perfusion system, one with Isotonic Tyrode Solution and other with Hypotonic Tyrode Solution.
Wash the fibers by gently removing 500 μL of Isotonic Tyrode Solution from the dish and replacing with fresh Isotonic Tyrode Solution. Repeat this process a total of 4 times.
The dish can now be transferred to the confocal microscope. Select a target fiber for experimentation (see Note 8). The tip of the perfusion system should be positioned directly adjacent to the target muscle fiber using the micromanipulator controls.
Begin the flow of Isotonic Tyrode Solution from the perfusion system onto the muscle fiber to test if the FDB fiber remains in place.
Start acquisition of fluo-4 fluorescent signal using the confocal microscope. Individual manufacturers will have significantly different software to control the microscope. Thus, this should be done according to manufacturer’s directions. General guidelines on the collection of data can be found in Subheading 3.5.
After collecting at least 60 s of baseline recordings, switch the perfusion solution to Hypotonic Tyrode Solution for 60 s. The muscle fiber should swell in volume in the presence of the Hypotonic Tyrode Solution.
After 60 s of exposure to Hypotonic Tyrode Solution, switch the perfusion solution back to the Isotonic Tyrode Solution. As the muscle fiber shrinks back to the original volume, there will be a robust Ca2+ spark response directly under the sarcolemma of the muscle fiber (Fig. 1a). Other osmotic stress methods can be used to induce Ca2+ sparks as well (see Note 11).
Record and analyze Ca2+ spark data as described in Subheading 3.5.
Fig. 1.

(a) A xy field scan image of an intact FDB muscle fibers loaded with Fluo-4-AM. When perfused with isotonic Tyrode solution (left), the fiber only displays minimal Ca2+ sparks. Perfusion of hypotonic Tyrode solution causes the fiber to swell (center) and a return to perfusion of isotonic Tyrode solution induces robust Ca2+ sparks in the periphery of the muscle fiber (right). (b) A xt line scan image along the dotted line in (a) shows the varying kinetics that occur in individual Ca2+ sparks following osmotic stress.
3.4. Ca2+ Spark Imaging of Permeabilized EDL fibers
Mount the imaging chamber on the stage of a confocal microscope.
Replace the Relaxing Solution with the Saponin Solution, and monitor the appearance of the fluo-4 fluorescence inside the fiber in the confocal microscope system. Immediately after the appearance of fluo-4 inside the fiber (usually about 2–3 min after the addition of the Saponin Solution), wash the muscle fiber 3 times with the Relaxing Solution plus 4% dextran.
Replace the Relaxing Solution with the Internal Solution (see Note 12).
Record Ca2+ sparks using a confocal scanning microscope. Recording conditions and analysis of these recordings are discussed in Subheading 3.5.
3.5. Analysis of Ca2+ Sparks Imaging from Isolated Muscle Fibers
Ca2+ sparks can be recorded under xy confocal scan mode to evalu- ate the distribution and frequency of Ca2+ sparks (15, 30). Figure 2a shows a representative two-dimensional (xy) field scan image obtained from a permeabilized EDL fiber with a conventional confocal scanning microscope. Note that Ca2+ sparks inside the muscle fiber.
Fig. 2.
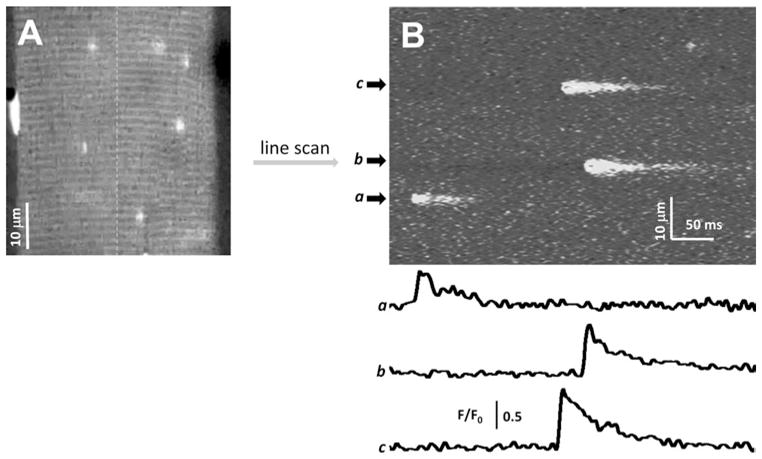
(a) A xy field scan image of a permeabilized EDL muscle fiber loaded with the internal solution containing 100 μM fluo-4, 200 nM free Ca2+, and 2 mM free Mg2+. (b) A xt line scan image of the same muscle fiber obtained through repeatedly scanning the fiber along the dashed line. Traces a, b, and c are the quantitative measurement of Ca2+ sparks by evalu-2+ sparks.
For more precise analysis of the kinetics and morphology, individual Ca2+ sparks images can be collected using a confocal line scan (xt) mode. Figure 2b shows a representative xt line scan image obtained by repeatedly scanning the permeabilized EDL fiber (shown in Fig. 2a) along the dashed line for 512 times at a time interval of 1.25 ms. Traces a, b, and c are quantitative indication of the changes of fluorescence intensity (F/F0) along the area of Ca2+ sparks.
By collecting a large number of such traces, the morphology of individual Ca2+ sparks can be evaluated, i.e., the amplitude, rise time, duration, time to the peak, full width of half magnitude ating the changes of fluo-4 fluorescence intensity (F/F0) along the area occupied by Ca (FWHM), and full duration of half magnitude (FDHM) of the recorded sparks (16, 32, 33). Cheng et al. (1999) developed an automated algorithm to detect and measure sparks in line scan images without human intervention (34). The algorithm was coded in the image-processing language IDL (Research System, Boulder, CO). This method was further refined by González et al. (2000) for analyzing sparks recorded from amphibian skeletal muscle (35). Further refinement led to an optimized computer routine to characterize Ca2+ sparks in permeabilized rat EDL muscle fibers (16). In the case of Ca2+ sparks induced in FDB fibers by hypoosmotic stress, the unique kinetics of Ca2+ release (19, 20) (Fig. 1b) are better assessed using another semi-automated IDL routine, sparkfit, which provides flexibility to select certain events not recognized by automatic detection programs (17).
Fig. 3.

(a) Clumped muscle fibers indicate that the FDB muscle is not sufficiently digested or requires additional disruption with a pipette. (b) Hypercontracted muscle fibers result from excessive pipetting of muscle fibers. (c) Damaged FDB fibers (arrow) are generally wider than health fibers (center) and lack the smooth membrane surface of healthy fibers. (d) Curled ends of FDB fibers (arrow) should exclude such fibers from use in Ca2+ spark experiments. (e) Wrinkled sarcolemmal membranes (arrow) also should exclude FDB fibers from use in experiments. (f) Wide ends of FDB fibers (arrow) indicate a fiber that has not properly resealed at the end and should be excluded from experimentation. (g) Excessively long or bent FDB fibers so are not optimal for use in Ca2+ spark experiments. (h) Slight bending or twisting of an FDB fiber usually does not exclude a fiber from use in experiments. (i) An example of an ideal FDB fiber for experimentation. The long, rod-like structure, smooth membrane, and average size are all characteristics that indicate a fiber useful for experimentation.
Acknowledgments
This work was supported by NIH grants to Drs. Weisleder (AR54793), Zhou (AR57404) and Ma (AG28614, HL69000 and AG28856), and MDA funding to Dr. Zhou (MDA4351). The authors thank Ms. Andoria Tjondrokoesoemo for helpful comments during the preparation of the final manuscript.
Footnotes
Before beginning this step, prepare 1% saponin stock solution (1 mL) and Relaxing Solution plus 4% dextran (referred to as relaxing + dextran, 20 mL) and store them at 4°C. Before an experiment, add 2 μL 1% saponin and 50 μM fluo-4 to 1 mL of this relaxing + dextran stock. Because it can be difficult to suspend saponin evenly inside a solution, vigorously vortex the saponin stock solution before use and also vigorously vortex the final saponin solution before adding it to the chamber.
Ca2+ sparks in permeabilized mammalian skeletal muscle fibers were first recorded by Kirsch et al. (2001) using an internal solution containing glutamate as the major anion (15). We found that muscle fibers immersed in such internal solution with glutamate produced events transiently at a very low frequency, and some fibers did not show sparks at all. Substitution of glutamate with SO42− as the major anion caused an increase in spark frequency for any given fibers (16).
The concentration of free Ca2+ and Mg2+ in an internal solution is critical for inducing Ca2+ sparks in permeabilized muscle fibers. We found that 200–300 nM free Ca2+ and 2 mM free Mg2+ produced maximal frequency of Ca2+ sparks in mammalian muscle fibers (32). The concentrations of free Ca2+ and Mg2+ were calculated using public domain program WinMaxC 2.10 (http://www.stanford.edu/~cpatton/maxc.html). Kd values of all Ca2+ and Mg2+, buffers (SO42− ATP, PC, EGTA) were from Martel and Smith (36) and extrapolated within WinMaxC.
While the dissection of the mouse foot can be done with the naked eye, some find it easier to use a dissection scope during the removal of the FDB. Effective dissection can best be done by removing the skin with a pair of curved dissection scissors. The FDB is the most superficial muscle of the foot and appears in the center on the foot once the skin is removed. It is attached to the medial tubercle of the calcaneum by a thick tendon that then runs through the mass of the muscle and branches into four thinner tendon strands that eventually insert into the middle phalanx of the four lateral toes. Once the thicker portion of the tendon near the ankle joint has been identified, it can be grasped with a pair of blunt forceps just above the union between the tendon and the mass of the FDB muscle. The tendon can then be cut near the ankle using curved dissection scissors while maintaining grip on the tendon using the blunt forceps. Once the tendon is cut, the forceps can be used to pull at the muscle and begin to peel it off of the layer of tendon on which is rests. The FDB is held in place by connective tissue fascia that will provide resistance as you begin to pull the muscle free from the foot. This connective tissue can be removed using a pair of spring scissors to make short, sweeping cuts along the edges of the FDB with the points of the scissors always running out and away from the FBD proper. Continue this approach until you reach the point when the FDB connects with the tendon layer that runs beneath the FDB. The FDB can then be removed from the animal using a single cross-cut of the spring scissors as far down as possible towards the point where the FDB joins this lower tendon.
Determination of the appropriate time of digestion for each lot of collagenase used is a critical step for the success of FDB fiber isolation. In general, 60 min is sufficient. The best indication that the FDB has been properly digested is that the edges of the muscle itself begin to appear ragged. If the muscle begins to fall apart in the tube of collagenase or upon transfer into Tyrode Solution before titration, then the digestion has gone far too long.
The FDB fibers used for experiments are fragments of longer muscle fibers that have been broken apart by tituration of the fiber. The ends of these fibers must reseal to allow the fiber to remain intact. This membrane resealing takes a few minutes. So allow the fibers to sit before plating in Isotonic Tyrode Solution.
If there has not been sufficient tituration of the muscle, many of the muscle fibers will appear in clumps when examined under the microscope (Fig. 3a). To remedy this situation, a 10-μL plastic micropipette tip can be used to conduct a series of 4–5 additional gentle tituration strokes that pull through the FDB fragments in the bottom of the snap-cap tube. If this does not reduce the number of muscle fibers clumps, then it is likely the FDB was not digested with collagenase for a sufficient amount of time.
It is important to determine how many useful fibers are on a dish before beginning to load Ca2+ indicator dye into the fibers. To be useful for the measurement of Ca2+ sparks, a fiber must be firmly attached to the bottom of the dish and display intact morphology. Intact morphology can be assessed by the following characteristics: (1) a straight, rod-like appearance (2) a length between 70 and 100 μm, (3) a width between 8 and 18 μm, (4) a clear, uniform striation pattern, and (5) a smooth sarcolemmal membrane with no patches of “wrinkled” membrane. The size of the fiber may be altered in aged mice, or in transgenic and disease models such as dystrophic animals. Some curvature of the fiber may be acceptable as long as the membrane is not wrinkled at the site where the fiber bends. See Fig. 3, for example, of isolated FDB fibers with proper morphology and common types of defects.
The origin tendon of the EDL is found at the lateral condyle of the tibia, while the insertion tendon passes under the extensor retinaculum ligament. Generally, it is easier to identify the insertion tendon, grasp the tendon with a pair of blunt forceps, and then cut the tendon while holding the forceps to keep tension of the EDL. Then you can run a separate pair of forceps under the length of the muscle to remove it from the weak layer of fascia surrounding the muscle. The origin tendon can be removed from the body using a pair of spring scissors.
After dissecting out a single fiber from the EDL muscle, the Relaxing Solution in the dissection chamber should be immediately replaced with Krebs Solution plus 10% FBS, and the muscle should be returned to a refrigerator (4°C) immediately. The digested EDL muscle kept in Krebs Solution plus 10% FBS at 4°C can be used repeatedly in 24 h.
Other investigators have used a number of different solutions of varying composition to induce Ca2+ sparks. Frequently, these are hyperosmotic solutions that can induce Ca2+ sparks immediately following perfusion of the solution and shrinking of the fiber. We have previously used a high Ca2+ solution to induce hyperosmotic conditions and trigger Ca2+ sparks (17). Others have perfused solutions using sucrose to elevate osmolality and observed similar effects (29). Hyperosmotic approaches using buffers with an osmolality >420 mOsm are effective and valid, but in some cases can result in more damage to the fiber than the hypoosmotic approach detailed here. Some of the solutions used, particularly the high Ca2+ solution, also go beyond the physiological range that may be experienced by cells in vivo. These effects can elevate the intracellular Ca2+ level of the muscle fiber and complicate imaging and analysis of Ca2+ spark signaling.
The concentration of free Ca2+ and Mg2+ in an internal solution is critical for inducing Ca2+ sparks in permeabilized muscle fibers. We found that 200–300 nM free Ca2+ and 2 mM free Mg2+ usually gave the maximal frequency of Ca2+ sparks in mammalian muscle fibers (32).
References
- 1.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 2.Wang SQ, Stern MD, Rios E, Cheng H. The quantal nature of Ca2+ sparks and in situ operation of the ryanodine receptor array in cardiac cells. Proc Natl Acad Sci USA. 2004;101:3979–3984. doi: 10.1073/pnas.0306157101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wier WG, ter Keurs HE, Marban E, Gao WD, Balke CW. Ca2+ ‘sparks’ and waves in intact ventricular muscle resolved by confocal imaging. Circ Res. 1997;81:462–469. doi: 10.1161/01.res.81.4.462. [DOI] [PubMed] [Google Scholar]
- 4.Kamishima T, Quayle JM. Ca2+ -induced Ca2+ release in cardiac and smooth muscle cells. Biochem Soc Trans. 2003;31:943–946. doi: 10.1042/bst0310943. [DOI] [PubMed] [Google Scholar]
- 5.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 6.Zhuge R, Fogarty KE, Baker SP, McCarron JG, Tuft RA, Lifshitz LM, Walsh JV., Jr Ca(2+) spark sites in smooth muscle cells are numerous and differ in number of ryanodine receptors, large-conductance K(+) channels, and coupling ratio between them. Am J Physiol Cell Physiol. 2004;287:C1577–1588. doi: 10.1152/ajpcell.00153.2004. [DOI] [PubMed] [Google Scholar]
- 7.Zhuge R, Fogarty KE, Tuft RA, Lifshitz LM, Sayar K, Walsh JV., Jr Dynamics of signaling between Ca(2+) sparks and Ca(2+)- activated K(+) channels studied with a novel image-based method for direct intracellular measurement of ryanodine receptor Ca(2+) current. J Gen Physiol. 2000;116:845–864. doi: 10.1085/jgp.116.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rossi AE, Dirksen RT. Sarcoplasmic reticulum: the dynamic calcium governor of muscle. Muscle Nerve. 2006;33:715–731. doi: 10.1002/mus.20512. [DOI] [PubMed] [Google Scholar]
- 9.Klein MG, Cheng H, Santana LF, Jiang YH, Lederer WJ, Schneider MF. Two mechanisms of quantized calcium release in skeletal muscle. Nature. 1996;379:455–458. doi: 10.1038/379455a0. [DOI] [PubMed] [Google Scholar]
- 10.Tsugorka A, Rios E, Blatter LA. Imaging elementary events of calcium release in skeletal muscle cells. Science. 1995;269:1723–1726. doi: 10.1126/science.7569901. [DOI] [PubMed] [Google Scholar]
- 11.Shirokova N, Garcia J, Rios E. Local calcium release in mammalian skeletal muscle. J Physiol. 1998;512:377–384. doi: 10.1111/j.1469-7793.1998.377be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ward CW, Schneider MF, Castillo D, Protasi F, Wang Y, Chen SR, Allen PD. Expression of ryanodine receptor RyR3 produces Ca2+ sparks in dyspedic myotubes. J Physiol. 2000;525:91–103. doi: 10.1111/j.1469-7793.2000.t01-2-00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutko JL, Airey JA, Murakami K, Takeda M, Beck C, Deerinck T, Ellisman MH. Foot protein isoforms are expressed at different times during embryonic chick skeletal muscle development. J Cell Biol. 1991;113:793–803. doi: 10.1083/jcb.113.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conklin MW, Barone V, Sorrentino V, Coronado R. Contribution of ryanodine receptor type 3 to Ca(2+) sparks in embryonic mouse skeletal muscle. Biophys J. 1999;77:1394–1403. doi: 10.1016/S0006-3495(99)76988-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirsch WG, Uttenweiler D, Fink RH. Spark- and ember-like elementary Ca2+ release events in skinned fibres of adult mammalian skeletal muscle. J Physiol. 2001;537:379–389. doi: 10.1111/j.1469-7793.2001.00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou J, Brum G, Gonzalez A, Launikonis BS, Stern MD, Rios E. Ca2+ sparks and embers of mammalian muscle. Properties of the sources. J Gen Physiol. 2003;122:95–114. doi: 10.1085/jgp.200308796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Weisleder N, Collet C, Zhou J, Chu Y, Hirata Y, Zhao X, Pan Z, Brotto M, Cheng H, Ma J. Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat Cell Biol. 2005;7:525–530. doi: 10.1038/ncb1254. [DOI] [PubMed] [Google Scholar]
- 18.Ward CW, Lederer WJ. Ghost sparks. Nat Cell Biol. 2005;7:457–459. doi: 10.1038/ncb0505-457. [DOI] [PubMed] [Google Scholar]
- 19.Weisleder N, Ferrante C, Hirata Y, Collet C, Chu Y, Cheng H, Takeshima H, Ma J. Systemic ablation of RyR3 alters Ca2+ spark signaling in adult skeletal muscle. Cell Calcium. 2007;42:548–555. doi: 10.1016/j.ceca.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weisleder N, Ma J. Ca2+ sparks as a plastic signal for skeletal muscle health, aging, and dystrophy. Acta Pharmacol Sin. 2006;27:791–798. doi: 10.1111/j.1745-7254.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- 21.Weisleder N, Brotto M, Komazaki S, Pan Z, Zhao X, Nosek T, Parness J, Takeshima H, Ma J. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J Cell Biol. 2006;174:639–645. doi: 10.1083/jcb.200604166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weisleder N, Ma J. Altered Ca2+ sparks in aging skeletal and cardiac muscle. Ageing Res Rev. 2008;7:177–188. doi: 10.1016/j.arr.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou J, Yi J, Fu R, Liu E, Siddique T, Rios E, Deng HX. Hyperactive intracellular calcium signaling associated with localized mito-chondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J Biol Chem. 2010;285:705–712. doi: 10.1074/jbc.M109.041319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lovering RM, Michaelson L, Ward CW. Malformed mdx myofibers have normal cytoskeletal architecture yet altered EC coupling and stress-induced Ca2+ signaling. Am J Physiol Cell Physiol. 2009;297:C571–580. doi: 10.1152/ajpcell.00087.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shkryl VM, Martins AS, Ullrich ND, Nowycky MC, Niggli E, Shirokova N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 2009;458:915–928. doi: 10.1007/s00424-009-0670-2. [DOI] [PubMed] [Google Scholar]
- 26.Teichmann MD, Wegner FV, Fink RH, Chamberlain JS, Launikonis BS, Martinac B, Friedrich O. Inhibitory control over Ca(2+) sparks via mechanosensitive channels is disrupted in dystrophin deficient muscle but restored by mini-dystrophin expression. PLoS One. 2008;3:e3644. doi: 10.1371/journal.pone.0003644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martins AS, Shkryl VM, Nowyck MC, Shirokova N. Reactive oxygen species contribute to Ca2+ signals produced by osmotic stress in mouse skeletal muscle fibres. J Physiol. 2008;586:197–210. doi: 10.1113/jphysiol.2007.146571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Apostol S, Ursu D, Lehmann-Horn F, Melzer W. Local calcium signals induced by hyper-osmotic stress in mammalian skeletal muscle cells. J Muscle Res Cell Motil. 2009;30:97–109. doi: 10.1007/s10974-009-9179-8. [DOI] [PubMed] [Google Scholar]
- 29.Pickering JD, White E, Duke AM, Steele DS. DHPR activation underlies SR Ca2+ release induced by osmotic stress in isolated rat skeletal muscle fibers. J Gen Physiol. 2009;133:511–524. doi: 10.1085/jgp.200910191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou J, Yi J, Royer L, Launikonis BS, Gonzalez A, Garcia J, Rios E. A probable role of dihydropyridine receptors in repression of Ca2+ sparks demonstrated in cultured mammalian muscle. Am J Physiol Cell Physiol. 2006;290:C539–553. doi: 10.1152/ajpcell.00592.2004. [DOI] [PubMed] [Google Scholar]
- 31.Dirksen RT. Bi-directional coupling between dihydropyridine receptors and ryanodine receptors. Front Biosci. 2002;7:d659–670. doi: 10.2741/A802. [DOI] [PubMed] [Google Scholar]
- 32.Zhou J, Launikonis BS, Rios E, Brum G. Regulation of Ca2+ sparks by Ca2+ and Mg2+ in mammalian and amphibian muscle. An RyR isoform-specific role in excitation-contraction coupling? J Gen Physiol. 2004;124:409–428. doi: 10.1085/jgp.200409105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou J, Brum G, Gonzalez A, Launikonis BS, Stern MD, Rios E. Concerted vs. sequential. Two activation patterns of vast arrays of intracellular Ca2+ channels in muscle. J Gen Physiol. 2005;126:301–309. doi: 10.1085/jgp.200509353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng H, Song LS, Shirokova N, Gonzalez A, Lakatta EG, Rios E, Stern MD. Amplitude distribution of calcium sparks in confocal images: theory and studies with an automatic detection method. Biophys J. 1999;76:606–617. doi: 10.1016/S0006-3495(99)77229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonzalez A, Kirsch WG, Shirokova N, Pizarro G, Stern MD, Rios E. The spark and its ember: separately gated local components of Ca(2+) release in skeletal muscle. J Gen Physiol. 2000;115:139–158. doi: 10.1085/jgp.115.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martell AE, Smith RM. Critical Stability Constants. Plenum Press; New York: 1974. [Google Scholar]