

Abstract
Reproducible and efficient affinity enrichment is increasingly viewed as an essential step in many investigations leading to the discovery of new biomarkers. In this work, we have evaluated the repeatability of lectin enrichment of glycoproteins from human blood serum through both qualitative and quantitative proteomic approaches. In a comprehensive evaluation of lectin binding, we have performed 30 separate microscale lectin affinity chromatography experiments, followed by a conventional sample purification, and LC-MS/MS analysis of the enriched glycoproteins. Two lectin affinity matrixes, both with Con A lectin immobilized to the same solid support but differing in the amount of immobilized lectin, were investigated to characterize their binding properties. Both qualitative and quantitative data indicate acceptable repeatability and binding efficiency for the lectin materials received from two different commercial sources.
Keywords: Glycoproteins, Lectin Affinity Chromatography, Quantitative Glycoproteomics, LC-MS/MS
1 Introduction
The glycosylation of proteins is one of the most common posttranslational modifications (PTM) observed in eukaryotic species. On average, over 50% of human serum and tissue proteins are likely to be glycosylated [1-3]. The glycosylation of proteins can be essential to their different functions and, consequently, it has been implicated in a multitude of cellular processes [4]. For example, the solubility and stability of a protein is dependent on glycosylation [5], while this type of modification directly influences intra- and extracellular trafficking of proteins, as well as mediating and modulating both cell signaling and cell adhesion [6-9]. Moreover, aberrant glycosylation of proteins has been recognized as an attribute of many diseases, including rheumatoid arthritis [10], prion diseases [11], congenital disorders of glycosylation [12], cardiovascular diseases [10], and cancer [5, 13-18].
Since glycoproteins, some of which may be potential disease biomarkers, are commonly present in only minute quantities in the analyzed tissue extracts and physiological fluids, their isolation and enrichment prior to analysis become mandatory. Consequently, numerous attempts have been made to develop methods aimed at the enrichment of glycoproteins present in complex biological samples [19]. Some of these enrichment methodologies depend on the use of immobilized lectins, which permit a more or less selective enrichment of the pools of glycoproteins for proteomic/glycomic studies [20-28].
Lectins are specialized proteins that have been isolated from various plants and animal sources. For a number of years, they have been employed to study interactions with glycoconjugates [29-32]. Lectins have also been widely used to isolate, purify and characterize glycoproteins and glycolipids in various modes of affinity chromatography [29, 30]. The lectin affinity separations in conventional columns have now been recognized as a powerful means for purification of glycoproteins prior to structural studies [33]. These techniques are based on a reversible, biospecific interaction of certain glycoproteins with the lectins immobilized to a solid support such as agarose and similar materials. Some of these materials have been routinely used to isolate glycoproteins on the basis of their different glycan structures. Commonly, a complex mixture of proteins is applied to an immobilized lectin bed, while the unbound proteins can be washed out with a binding buffer. The interacting glycoproteins are subsequently displaced by washing with an elution buffer, which usually contains a hapten sugar. Lectin affinity chromatography has been conducted in different formats, including tubes [23], packed columns [24], microfluidic channels [34], and unconventional solid surfaces, such as colloidal gold [35] and affinity membranes [36, 37]. Moreover, the capability of lectins to reversibly interact with a variety of glycoproteins has been extensively exploited in many proteomic investigations, where discovery of protein glycosylation has been a primary concern [2, 38].
Bioanalytical methods implementing a glycoprotein enrichment step through lectin affinity chromatography are becoming increasingly popular due to the diverse nature and ubiquity of glycosylation in mammalian systems. The importance of lectin-carbohydrate recognition has rapidly gained attention following the proven existence of aberrant glycosylation of proteins in the progression of malignant diseases [15-18], a characteristic which lectin enrichment methods can be uniquely tailored to recognize through the use of lectins with different binding specificity. However, no comprehensive studies demonstrating the repeatability of lectin enrichment have thus far been conducted. Previous glycoproteomic experiments that were concerned with the repeatability of lectin enrichment prior to nano LC-MS/MS have reported the results acquired from only two [25] or three analyses [39].
Although lectin affinity chromatography is employed routinely in numerous biochemical laboratories as a glycoprotein isolating technique, its analytical/quantitative use has been less common. This is expected to change with the increasing demand in contemporary proteomic investigations to perform differential measurements on a multitude of glycoproteins in complex mixtures within a wide concentration range. Additionally, the emphasis on analyzing small samples without the losses of trace analytes in the system distinctly favors miniaturization of all system's components, including the lectin preconcentration step. Although there have been promising attempts to miniaturize lectin affinity devices through surface-immobilized lectin arrays [40] or lectin microcolumns coupled to reversed-phase liquid chromatography [41, 42] and liquid chromatography/mass spectrometry [21, 22, 41-44], a comprehensive enrichment of glycoproteins from complex protein mixtures has not been systematically addressed. While the recent studies using single lectin or multi-lectin affinity chromatography [21, 25-28] have demonstrated the efficacy of glycoprotein enrichment through this technique, little effort has been aimed at addressing the repeatability of this technique and its quantitative limits.
Here, we address the repeatability of lectin affinity chromatography in quantitative proteomics, involving human blood serum, which was depleted from the six most abundant proteins prior to lectin-enrichment step and subsequent LC-MS/MS analysis. We also address the quantitative aspects and limits of this methodology. The lectin media acquired from two different vendors were employed separately to enrich glycoproteins from 30 blood serum samples. The amount of bound and unbound proteins was determined through both the Bradford assay and quantitative proteomics. The latter involves LC-MS/MS analysis and label-free quantification utilizing our recently developed ProteinQuant computer tool [45]. The data generated through this study support the repeatability of lectin affinity enrichment even when using media from different vendors.
2 Materials and Methods
2.1 Materials and chemicals
Concanavalin A (Con A) affinity media immobilized to agarose gel were purchased from Amersham Biosciences (Piscataway, NJ) and Vector Laboratories (Burlingame, CA), and were used as 50% (v/v) slurry in a binding buffer as recommended by each vendor. These lectin gels are referred to as “affinity matrix A” and “affinity matrix B,” respectively. Human blood serum sample pooled from different healthy female donors was obtained from Innovative Research, Inc. (Novi, MI), stored at -70°C, and thawed immediately prior to use. Dithiothreitol (DTT) and iodoacetamide (IAA) were acquired from Bio-Rad (Hercules, CA). Proteomic sequencing grade trypsin, chicken lysozyme, and all other chemicals were obtained from Sigma-Aldrich (St Louis, MO). All buffers were prepared by solubilizing the appropriate chemicals in deionized water (Millipore, Billerica, MA).
2.2 Immunoaffinity depletion
Human blood serum samples were depleted of the six most abundant proteins (albumin, IgA, IgG, haptoglobin, anti-trypsin, transferrin) using the Multiple Affinity Removal System (MARS) column (4.6 mm × 100 mm, Agilent Technologies, Santa Clara, CA). This column has a 30-μL loading capacity. Accordingly, 60 30-μL aliquots of serum sample were depleted according to the protocol recommended by the manufacturer prior to a buffer exchange into the lectin-binding buffer (10 mM TRIS.HCl, pH 7.5, 500 mM NaCl, 1 mM MnCl2, 1 mM CaCl2, 0.08% NaN3) using 5 kDa MWCO spin concentrators (Agilent). Prior to lectin binding, all depleted human blood serum aliquots were pooled together and the total protein content was determined by Bradford assay [46]. The total volume of the pooled sample after buffer exchange was 7.5 ml which was evenly divided into 60 aliquots (125 μl each). Each aliquot was subjected to lectin affinity chromatography as described next.
2.3. Lectin affinity chromatographic enrichment
Due to the difference in the amounts of Con A immobilized on both affinity matrixes used here (see Table 1), it was necessary to adjust the volumes of gel slurries accordingly, so that the same amount of lectin was utilized in all binding experiments. Accordingly, a 125-μL aliquot of depleted blood serum (ca. 213 μg of proteins) solubilized in the lectin binding buffer was added to an appropriate volume of lectin gel (Table 1), vortexed and shaken overnight at 4°C. After washing the lectin gels 3-times with 100 μL of the binding buffer, glycoproteins were displaced from the lectin media through washing with a 100- μL aliquot of the recommended elution buffer twice (Table 1). In order to reliably evaluate the repeatability of the method, 30 lectin-binding experiments were performed in parallel, with the two gel-based media obtained from different sources. Collected Con A bound fractions were then filtered and dialyzed overnight on a 96-well plate dialyzer (Harvard Apparatus, Holliston, MA) against 50 mM ammonium bicarbonate. Next, dialyzed samples were dried and resuspended in a 250-μL aliquot of 50 mM ammonium bicarbonate buffer prior to the enzymatic digestion. A Bradford assay was used for the determination of a total protein content remaining in all lectin unbound and bound fractions.
Table 1.
Experimental conditions used for the small-scale enrichment of human blood serum glycoproteins on two different lectin affinity gels.
| Lectin gel A | Lectin gel B | |
|---|---|---|
| Average amount of immobilized lectin [mg/mL] | 13 | 6 |
| Volume of gel used [μL] | 16 | 35 |
| Binding buffer | 10 mM TRIS.HCl (pH 7.5), 500 mM NaCl, 1 mM MnCl2, 1 mM CaCl2, 0.08% NaN3 | |
| Elution buffer | 200 mM α-D-methylmannoside in binding buffer | 200 mM α-D-methylmannoside, 200 mM α-D-methylglucoside in binding buffer |
2.4 Trypsin digestion of Con A-bound fractions
The lectin-bound fractions were denatured by heating at 95°C for 10 min and reduced with a 2.5-μL aliquot of 200 mM DTT followed by incubation at 60°C for 30 min. After alkylation with 10 μL of 200 mM IAA at room temperature for 30 min in the dark, and subsequent quenching of the reaction with 2.5 μL of 200 mM DTT, the glycoproteins were digested with trypsin (enzyme:substrate ratio of 1:50 w/w) at 37°C for 18 hours. Enzymatic digestion was then stopped through the addition of 2 μL neat formic acid. In order to monitor the run-to-run consistency of LC-MS/MS experiments, the digested samples were spiked with a tryptic digest of chicken lysozyme, which was separately prepared under the same conditions.
2.5 Nano ESI-LC-MS/MS, data processing and high-throughput quantification
A 4-μL aliquot of each digested lectin-bound fraction, spiked with 1 pmol of chicken lysozyme tryptic digest, was subjected to nano-LC-MS/MS analysis performed on an 1100 LC system (Agilent Technologies, Santa Clara, CA) connected to an LC/MSD Trap XCT Ultra mass spectrometer (Agilent Technologies) fitted with nano ESI source. Peptides were on-line desalted on a C18 PepMap300 trapping cartridge (5 μm, 300 Å, 300 μm id × 5 mm; Dionex, Sunnyvale, CA) and separated on a Zorbax C18 capillary column (3.5 μm, 75 μm i.d. × 150 mm; Agilent Technologies) over 55 mins with a linear gradient from 3-55% aqueous acetonitrile mobile-phase containing 0.1% formic acid. The LC system was controlled by ChemStation software (Agilent Technologies), while the data generated by the mass spectrometer were acquired in EsquireControl software (Bruker Daltonics, Billerica, MA). The mass spectrometer capillary voltage was kept at 1700 V, while the desolvation temperature was maintained at 300°C. The ion charge control value (ICC) of the mass spectrometer was set to 200,000 with a maximum ion accumulation time of 200 ms. An MS/MS fragmentation of the five most intense precursor ions in the spectra was performed automatically with an exclusion window of 0.5 min.
Data acquired with the mass spectrometer were processed with Data Analysis Software (Bruker Daltonics) and subjected to Mascot database searching. The data were then submitted to another search against a reversed version of the same protein database in order to estimate the false positive rate [47, 48]. This estimation was then used to set a MOWSE-based peptide filtering criteria, where the ion score threshold represented a false positive rate of less than 5%. In addition, the peptide hits found with the same query numbers in both regular and reversed databases were excluded from the results, since they are most likely false positives. Identified proteins and peptides were then processed and quantified using our in-house developed ProteinQuant Suite software [45].
3 Results and Discussions
This study is aimed at evaluating the repeatability of glycoprotein enrichment using the lectin affinity principle. As shown in Figure 1, which outlines the experimental workflow, we have used human blood serum sample depleted of the six most abundant proteins, thus representing, in terms of complexity, the majority of real samples typically subjected to quantitative glycoproteomic analyses. The repeatability of lectin binding was examined through analyzing large sets of samples (n=30) prepared through mixing the aliquots of depleted serum with Con A immobilized on agarose gel. The choice of Concanavalin A was predominantly due to its facile availability, low cost and a relatively broad specificity toward N-glycans [29]. Since the interaction of glycoproteins with a specific lectin covalently attached to the agarose gel can also be influenced by the nature of coupling chemistry and the amount of loaded lectin, the binding performance of two different Con A-agarose affinity media has been compared. In order to avoid other sources of variation, each sample aliquot was always added to the appropriate volume of these gels, so that the amount of Con A used for the binding was the same regardless of the source. The interpretation of binding repeatability was based on the statistical evaluation of the results acquired from 30 affinity chromatography experiments conducted in parallel for both Con A-agarose media.
Figure 1.
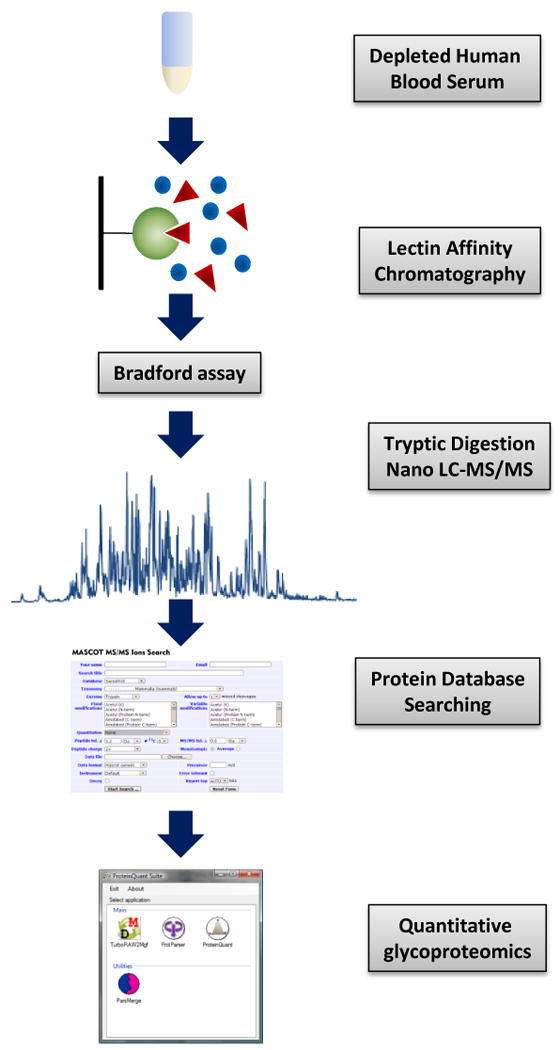
Experimental workflow chart.
3.1 Efficiency of lectin binding and protein recovery
The lectin-enriched glycoproteins of depleted human blood serum were eluted, desalted, enzymatically digested, and subjected to nano-LC-MS/MS analysis. Acquired data were then evaluated through both a qualitative approach based on reliable protein identification, and a quantitative approach, which relies on a software-assisted data evaluation. While it is apparent that there is a certain level of variation introduced through the experimental workflow, a miniaturized lectin affinity enrichment will likely have the most significant influence on the overall repeatability. However, this is expected to be minimal. Additionally, the use of microscale lectin affinity enrichment was dictated by the fact that many real samples, such as body fluids or tissue extracts, may be available to proteomic studies only in small quantities, thus making it necessary to perform lectin enrichment in small volumes. Despite continuous improvements, affinity enrichment, in comparison with some other chromatographic enrichment methods (reversed-phase, ion-exchange, etc.), still suffers from relatively low recoveries, caused mainly by the irreversible non-specific binding of analyzed components to the surface of a chromatographic support. The extent of non-specific interactions depends predominantly on the type of matrix, surface activation, and nature of coupling chemistry [38].
Recognizing the existence of such non-specific interactions and their possible impact on sample losses experienced in microscale glycoproteomic experiment [21], we have characterized both lectin affinity media in terms of their binding efficiency as well as the total protein recovery. The average percentage of protein content determined in all bound and unbound fractions by the Bradford assay is shown in Figure 2. The data suggest that both Con A affinity media (matrix A and B) exhibited very similar binding properties. Moreover, statistical evaluation based on 30 separate Con A affinity experiments for each matrix suggests that both lectin media have very comparable binding efficiency as reflected by the measured protein content, where 33% (69 μg) of the loaded protein content was bound to matrix A, and 30% (64 μg) was bound to matrix B. These values seem realistic, since about 50% of all proteins originating from mammalian systems are believed to be glycosylated [1-3]. This high number accounts for all different types of glycans attached to the studied proteins, some of which exhibit a weak affinity toward Concanavalin A lectin. As for the total protein recovery calculated from the protein content retrieved from both bound and unbound fractions and the amount of proteins initially loaded on a gel, 69% (147 μg) of proteins were recovered from matrix A, whereas 77% (164 μg) was recovered from matrix B. Although both Con A-agarose gels under comparison showed somewhat similar non-specific binding - a finding that is in rough agreement with what is commonly observed in affinity chromatography [5-26] - a lower run-to-run variation was exhibited by matrix A (Figure 2).
Figure 2.

Box graphs of the Bradford assay results for the Con A lectin affinity chromatography of the two different affinity matrixes (A and B). The boxes represent an interquartile range measuring the statistical distribution, the horizontal line outlines the median, and the vertical line shows the spread(N=30).
The relatively low protein recoveries observed here were mainly attributed to both sample dialysis and strong binding of some glycoproteins to lectin media, as suggested by Bradford protein assay (data not shown). A very limited loss was due to the latter (less than 5%). Although the elution of residual glycoproteins that are bound strongly to the media could be achieved using acidic elution buffer, the elution step utilized in this study was performed according to the manufacturer's recommended protocol which is routinely employed by many laboratories.
3.2 Qualitative proteomic evaluation
The statistical evaluation of lectin binding repeatability, which relies solely on a comparison of the total protein content, helps to characterize the overall trapping efficiency as well as the major sample losses caused by non-specific interactions. However, this measurement does not indicate whether the lectin covalently attached to a chromatographic support repeatably binds a group of glycoproteins. The importance of addressing this issue is unquestionable, since reproducible identification is vital to any subsequent quantification of potential glycosylated biomarkers. As mentioned above, only a limited number of studies have thus far addressed the repeatability of lectin enrichment, analyzing a very small number of samples (n=2 or 3) [25, 39]. These studies showed the identification of proteins with more than a 70% common occurrence in all runs. This is similar to what we observe here.
Generally, addressing the uncertainty of protein identification and quantification in high-throughput proteomic investigations requires statistical evaluation of a large number of replicates. Therefore, we have extended the scope of this study to demonstrate the variation in lectin affinity enrichment of certain glycoproteins, in addition to deducing the repeatability of lectin binding from the total protein content determined by the Bradford assay. The qualitative proteomic results are summarized in Table 2; they were statistically evaluated after the removal of outliers recognized from their substantially lower number of identified peptides. More than 50 proteins (from over 300 peptides) were identified on average for each enrichment experiment (Table 2). The same numbers (53 proteins on average) were observed in the bound fractions for both affinity matrixes. However, protein identification was based on 334 and 316 peptides in case of matrix A and B, respectively.
Table 2.
Qualitative proteomic comparison of LC-MS/MS analyses of 30 bound fractions collected from lectin enrichment using two different affinity media.
| Affinity matrix A | Affinity matrix B | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Run | # of proteins | # of peptides | Proteins out of >1 peptide [%] |
Common proteins [%] |
Run | # of proteins | # of peptides | Proteins out of >1 peptide [%] |
Common proteins [%] |
| 1 | 52 | 289 | 69 | 60 | 1 | 53 | 326 | 74 | 57 |
| 2 | 53 | 320 | 72 | 58 | 2 | 56 | 332 | 70 | 54 |
| 3 | 52 | 312 | 71 | 60 | 3 | 56 | 327 | 66 | 54 |
| 4 | 47 | 296 | 74 | 66 | 4 | 53 | 327 | 72 | 57 |
| 5 * | 43 | 259 | 72 | - | 5 * | 37 | 187 | 70 | - |
| 6 | 55 | 354 | 71 | 56 | 6 | 56 | 340 | 68 | 54 |
| 7 | 55 | 358 | 75 | 56 | 7 | 54 | 308 | 67 | 56 |
| 8 * | 40 | 257 | 65 | - | 8 | 56 | 351 | 77 | 54 |
| 9 | 57 | 358 | 77 | 54 | 9 | 53 | 351 | 77 | 57 |
| 10 | 54 | 324 | 65 | 57 | 10 | 52 | 324 | 75 | 58 |
| 11 | 57 | 325 | 63 | 54 | 11 | 55 | 321 | 71 | 55 |
| 12 | 49 | 328 | 71 | 63 | 12 | 54 | 307 | 67 | 56 |
| 13 | 56 | 334 | 73 | 55 | 13 | 53 | 303 | 68 | 57 |
| 14 | 56 | 351 | 70 | 55 | 14 | 56 | 291 | 63 | 54 |
| 15 | 54 | 334 | 70 | 57 | 15 | 51 | 310 | 73 | 59 |
| 16 | 56 | 350 | 71 | 55 | 16 | 51 | 305 | 73 | 59 |
| 17 | 50 | 353 | 84 | 62 | 17 | 48 | 289 | 77 | 63 |
| 18 | 57 | 352 | 68 | 54 | 18 | 54 | 314 | 67 | 56 |
| 19 | 50 | 349 | 78 | 62 | 19 | 48 | 285 | 71 | 63 |
| 20 | 57 | 351 | 68 | 54 | 20 | 50 | 299 | 70 | 60 |
| 21 | 48 | 300 | 77 | 65 | 21 | 51 | 299 | 71 | 59 |
| 22 | 52 | 323 | 73 | 60 | 22 | 51 | 297 | 67 | 59 |
| 23 | 49 | 323 | 80 | 63 | 23 | 51 | 318 | 75 | 59 |
| 24 | 52 | 302 | 63 | 60 | 24 | 51 | 275 | 73 | 59 |
| 25 | 52 | 335 | 81 | 60 | 25 | 52 | 321 | 75 | 58 |
| 26 | 55 | 341 | 73 | 56 | 26 | 53 | 329 | 72 | 57 |
| 27 | 54 | 349 | 76 | 57 | 27 | 53 | 305 | 68 | 57 |
| 28 | 50 | 326 | 74 | 62 | 28 | 55 | 322 | 69 | 55 |
| 29 | 56 | 341 | 68 | 55 | 29 | 53 | 335 | 75 | 57 |
| 30 | 53 | 361 | 77 | 58 | 30 | 54 | 344 | 70 | 56 |
|
| |||||||||
| CI (95%) | 53 ± 1 | 334 ± 8 | 73 ± 2 | 59 ± 2 | CI (95%) | 53 ± 1 | 316 ± 7 | 71 ± 2 | 57 ± 1 |
| RSD [%] | 6 | 6 | 7 | 6 | RSD [%] | 4 | 6 | 5 | 4 |
- these runs were removed as outliers, CI - confidence interval
These numbers also include proteins identified from one peptide only, which are not commonly considered as reliable assignments. This is due to the fact that some proteins may contain segments of identical amino acid sequence, thus eliminating the confident assignment of these proteins. The content of proteins identified with one peptide in each fraction was generally about 30%, which may have contributed to significant run-to-run variations in the reported protein hits. Therefore, we only considered those proteins which were commonly observed in all experiments. After filtering the data sets in this way, the number of identified proteins was 31 and 30 for matrixes A and B, respectively. None of these commonly observed proteins were identified from only one peptide, and all of them were positively confirmed as glycoproteins, according to Expasy proteomic server at http://ca.expasy.org/.
In general, the extent of affinity interactions toward certain glycoproteins also depends on the amount of lectin immobilized on the surface of a matrix. Due to the current availability of many affinity gels loaded with different amounts of lectin attached through various coupling reactions, all experiments involved in this study were conducted simultaneously employing two gels with different amounts of Con A loaded per unit volume of agarose. To ensure that the same quantity of Con A participated in binding (see Materials and Methods), a larger volume of lectin matrix B had to be used for the affinity experiments, resulting in lower concentration of a lectin in the reaction mixture. Since a higher lectin load facilitates the accessibility of certain glycoproteins to the binding site, more lectin immobilized on a gel might induce stronger affinity interactions. Thus, more glycosylated components could be theoretically found in the bound fraction. The comparison between the numbers of glycoproteins commonly observed in the Con A-bound fractions from all experiments performed using the two lectin matrixes is depicted in the Venn diagrams shown in Figure 3. There are 26 glycoproteins which were commonly observed in both lectin matrixes. Only 5 unique glycoproteins were identified using matrix A, while 4 unique proteins were identified using matrix B. This suggests a relatively consistent lectin binding to both affinity matrixes used here, with a small variation which might be partially attributed to the difference in the concentration of Con A bound to each matrix.
Figure 3.

The number of glycoproteins (A) and their corresponding peptides (B) identified in all LC-MS/MS runs of bound fractions collected separately from all lectin enrichments using two different affinity matrixes.
3.3 Quantitative proteomic evaluation
Generally, a specific disease progression could be either attributed to the aberrant glycosylation of certain glycoproteins or any significant changes in the overall expression of particular glycoproteins. For example, the diagnosis of prostate cancer has recently been based on monitoring the abundance of prostate-specific antigen (PSA) and the isoforms of apolipoproteins in urine [49]. Although changes in the glycosylation of these biomarkers were observed as a result of cancer progression [50], alterations in the concentrations of these proteins seem to be more significant. Therefore, glycoproteomic studies employing lectins for a selective enrichment of glycoproteins should combine both qualitative and quantitative approaches to determine whether there is a complete loss or just a change in the affinity of certain glycoproteins toward specific lectins. While the qualitative approach helps to recognize substantial changes in glycosylation, quantitative studies permit the characterization of any alterations in the abundances of respective glycoproteins in the analyzed samples. The issue of whether or not lectins bind identified glycoproteins quantitatively and reproducibly is thus important.
It should be stressed that the overall quantitative variation of lectin binding, in microscale affinity enrichment and proteomics, is dependent not only on the quality of sample preparation but also on the consistent performance of a mass spectrometer. Thus, the quantitative evaluation of the lectin binding repeatability discussed in this section covers all major errors introduced in a typical glycoproteomic investigation. The variations in LC-MS/MS analyses over a long period of time, using a reliable mass spectrometer, are deemed to be less than 10% [45]. We ensured fairly quantitative results through spiking each sample with a constant amount of chicken lysozyme tryptic digest prior to LC-MS/MS analyses. The intensities of peaks corresponding to extracted ion chromatograms constructed from the lysozyme peptides were compared among all injections. The recorded data did not indicate any significant run-to-run variations which might be attributed to a change in the performance of our mass spectrometer (data not shown). In addition, the run-to-run variations in signal consistency were further lowered through the implementation of the global normalization, where the area of each peptide peak was normalized to the sum of peak areas calculated for all peptides present in a sample [45].
The quantitative repeatability of lectin binding was evaluated for both matrixes utilizing label-free quantitative glycoproteomics facilitated by ProteinQuant Suite software package [45]. As mentioned above, 31 glycoproteins were commonly observed in lectin matrix A bound fractions, while 30 glycoproteins were commonly observed in lectin matrix B bound fractions. The amounts of identified glycoproteins were expressed as a total glycoprotein area defined as the sum of peak areas corresponding to every peptide identified for that glycoprotein. Since all quantified glycoproteins were reliably identified in all analyzed fractions, it is very likely that, under the assumption of repeatable enrichment, those proteins should generate similar numbers of peptides after enzymatic digestion. Some of these peptides may not, however, be identified in certain fractions due to the overall sample complexity and the duty cycle limitations of a mass spectrometer. The chromatographic peaks for such peptides are not used in the calculation of total protein areas, thus potentially resulting in an additional variation in the results. Therefore, rather than quantifying only specific peptides identified in the individual samples, all peptides identified across all bound fractions for each lectin affinity matrix were compiled together into a master file which was then used for quantification [45, 51, 52].
All identified glycoproteins and their total areas are listed in Table 3. When we compare the average normalized areas calculated for the same glycoproteins common in all samples, it can be concluded that the binding of these glycoproteins to Con A is similar regardless of the type of lectin matrix used. Normalized areas of some glycoproteins listed in Table 3 also correlate with their natural abundances in human blood serum. For example, alpha-2-macroglobulin (A2MG_HUMAN), complement C3 (CO3_HUMAN), and hemopexin (HEMO_HUMAN) which are among the most abundant glycoproteins in serum sample, were experimentally determined as such (Table 3). The area ratios of these glycoproteins are in agreement with their natural abundance ratios. Nevertheless, this observation cannot be generalized, since there was no agreement between the natural abundance of other glycoproteins and their total area determined experimentally. In fact, the quantification of some proteins with similar natural abundances resulted in quite different normalized areas. Hence, these discrepancies are believed to be partially due to the extent of glycosylation. Glycoproteins with more glycosylation sites generate more proteolytic glycopeptides which are not readily identified in MS2 (due to poor fragmentation of their peptide backbone) and, therefore, are not quantified. When the attention is aimed at the variation of the protein areas calculated from all carried experiments, it can be seen that the relative standard deviations (RSD) fall within 5 to 30% for the majority of listed proteins, and in some rare cases, exceed 50 %. The high deviations in RSDs were observed only in those glycoproteins, which were present at lower quantities in the analyzed samples, as suggested by their low number of identified peptides. This indicates inefficient enrichment, or the presence of multiple glycosylation sites. While, overall, the results still demonstrate binding with a generally acceptable repeatability, these observations warrant further investigations.
Table 3.
The quantitative proteomic evaluation of LC-MS/MS analyses of 30 bound fractions collected from lectin enrichment experiments using two different affinity media. All areas were calculated using ProteinQuant, a quantitative proteomic computer tool [45].
| Index | Protein | Description | Mass | Lectin matrix A | Lectin matrix B | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Normalized area, confidence interval (95%) (×10-4) | RSD [%] |
Normalized area, confidence interval (95%) (×10-4) | RSD [%] |
||||
| 1 | A1AG1_HUMAN | (P02763) Alpha-1-acid glycoprotein 1 | 23725 | 56 ± 4 | 37 | 68 ± 6 | 23 |
| 2 | A1BG_HUMAN | (P04217) Alpha-1B-glycoprotein | 54809 | 192 ± 5 | 7 | 160 ± 9 | 16 |
| 3 | A2MG_HUMAN | (P01023) Alpha-2-macroglobulin | 164600 | 2449 ± 25 | 3 | 2761 ± 45 | 5 |
| 4 | AACT_HUMAN | (P01011) Alpha-1-antichymotrypsin | 47792 | 69 ± 4 | 17 | 20 ± 2 | 22 |
| 5 | AFAM_HUMAN | (P43652) Afamin | 70963 | 26 ± 2 | 17 | - | - |
| 6 | AMBP_HUMAN | (P02760) AMBP protein | 39886 | - | - | 33 ± 3 | 26 |
| 7 | ANGT_HUMAN | (P01019) Angiotensinogen | 53406 | 53 ± 2 | 12 | 36 ± 3 | 20 |
| 8 | ANT3_HUMAN | (P01008) Antithrombin-III | 53025 | 61 ± 3 | 12 | 33 ± 3 | 21 |
| 9 | APOA1_HUMAN | (P02647) Apolipoprotein A-I | 30759 | 110 ± 6 | 14 | 108 ± 11 | 30 |
| 10 | C1R_HUMAN | (P00736) Complement C1r subcomponent | 81661 | - | - | 43 ± 2 | 16 |
| 11 | C4BP_HUMAN | (P04003) C4b-binding protein alpha chain | 69042 | 123 ± 4 | 10 | 152 ± 8 | 14 |
| 12 | CERU_HUMAN | (P00450) Ceruloplasmin (EC 1.16.3.1) | 122983 | 441 ± 8 | 7 | 433 ± 12 | 8 |
| 13 | CFAB_HUMAN | (P00751) Complement factor B | 86847 | 159 ± 4 | 7 | 124 ± 7 | 16 |
| 14 | CFAH_HUMAN | (P08603) Complement factor H | 143654 | 244 ± 5 | 5 | 293 ± 14 | 13 |
| 15 | CLUS_HUMAN | (P10909) Clusterin | 53031 | 20 ± 1 | 12 | - | - |
| 16 | CO3_HUMAN | (P01024) Complement C3 | 188585 | 1878 ± 33 | 5 | 1948 ± 54 | 8 |
| 17 | CO5_HUMAN | (P01031) Complement C5 | 189923 | 18 ± 1 | 13 | - | - |
| 18 | CO6_HUMAN | (P13671) Complement component C6 | 108425 | 16 ± 1 | 17 | 25 ± 16 | 183 |
| 19 | CPN2_HUMAN | (P22792) Carboxypeptidase N subunit 2 | 61431 | 25 ± 1 | 16 | 28 ± 3 | 32 |
| 20 | HEMO_HUMAN | (P02790) Hemopexin | 52385 | 728 ± 18 | 7 | 611 ± 37 | 17 |
| 21 | HEP2_HUMAN | (P05546) Heparin cofactor 2 | 57205 | 31 ± 3 | 26 | - | - |
| 22 | HPT_HUMAN | (P00738) Haptoglobin | 45861 | 221 ± 9 | 12 | 285 ± 21 | 20 |
| 23 | HRG_HUMAN | (P04196) Histidine-rich glycoprotein | 60510 | - | - | 31 ± 2 | 22 |
| 24 | IC1_HUMAN | (P05155) Plasma protease C1 inhibitor | 55347 | 139 ± 9 | 18 | 130 ± 12 | 27 |
| 25 | IGHA1_HUMAN | (P01876) Ig alpha-1 chain C region | 38486 | 35 ± 2 | 15 | 57 ± 2 | 11 |
| 26 | IGJ_HUMAN | (P01591) Immunoglobulin J chain | 16041 | 2 ± 0.3 | 52 | 2 ± 0.4 | 55 |
| 27 | ITIH1_HUMAN | (P19827) Inter-alpha-trypsin inhibitor heavy chain H1 | 101782 | 143 ± 4 | 8 | 183 ± 7 | 11 |
| 28 | ITIH2_HUMAN | (P19823) Inter-alpha-trypsin inhibitor heavy chain H2 | 106826 | 95 ± 4 | 11 | 140 ± 7 | 14 |
| 29 | ITIH4_HUMAN | (Q14624) Inter-alpha-trypsin inhibitor heavy chain H4 | 103522 | 113 ± 3 | 8 | 106 ± 8 | 21 |
| 30 | KAC_HUMAN | (P01834) Ig kappa chain C region | 11773 | 55 ± 3 | 13 | 61 ± 9 | 39 |
| 31 | KNG1_HUMAN | (P01042) Kininogen-1 | 72984 | - | - | 47 ± 3 | 18 |
| 32 | LAC_HUMAN | (P01842) Ig lambda chain C regions | 11401 | 20 ± 1 | 17 | - | - |
| 33 | MUC_HUMAN | (P01871) Ig mu chain C region | 50210 | 84 ± 3 | 12 | 136 ± 6 | 11 |
| 34 | SAMP_HUMAN | (P02743) Serum amyloid P-component | 25485 | 63 ± 3 | 12 | 76 ± 8 | 31 |
| 35 | VTNC_HUMAN | (P04004) Vitronectin | 55069 | 85 ± 3 | 10 | 41 ± 2 | 16 |
Although our results demonstrate repeatable binding, it may not necessarily mean that all the glycoproteins associated with the analyzed serum sample are completely enriched on the lectin media. The efficiency of enrichement process is dependent on the specificity of lectin and protein glycosylation. Some glycoproteins will always be present in both the unbound and bound fractions. For example, Con A strongly recognizes high mannose and complex glycan structures but has a relatively weaker affinity to acidic glycans. Therefore, glycoprotein possessing acidic glycans will always be detected in both bound and unbound Con A lectin affinity fractions.
The experiment-to-experiment repeatability is also expressed as the scatter plots which are shown in Figure 4. These plots represent a comparison between the normalized protein areas measured for the glycoproteins identified after the first run and the normalized areas calculated for the same glycoproteins after injection 10, 20, and 30. Figure 4A depicts the results obtained after the analysis of Con A bound fractions collected on affinity matrix A, whereas Figure 4B illustrates the same comparison for the experiments performed with affinity matrix B. Ideally, if the areas reflecting the abundances of glycoproteins commonly observed in two different experiments are identical, the slope of a trend line constructed for all acquired points will have a value of 1. As can be deduced from the slopes calculated for each plot displayed in Figure 4, the experiment-to-experiment comparison showed only minimal variations, and thus seems analytically acceptable.
Figure 4.

Scatter plots showing quantitative run-to-run reproducibility of LC-MS/MS analyses of Con A bound fractions acquired through the enrichment on affinity matrix A (a,b,c) and matrix B (d,e,f). Normalized protein areas were calculated with the ProteinQuant Suite software tool.
We have also compared the quantification results acquired from all experiments performed on both Con A agarose gels. The data listed in Table 3 did not suggest preferential binding of identified glycoproteins to either one of the used affinity matrixes. This was further supported by the scatter plot shown in Figure 5. The y-axis represents normalized glycoprotein areas calculated from the analysis of data generated using matrix B, while the x-axis depicts normalized areas for the same proteins calculated from the analysis of data generated using matrix A. The normalized areas calculated for the same proteins observed in two different lectin gels did not significantly differ. The slope of the linear regression trend line is 1.078, clearly suggesting very similar binding properties of both affinity matrixes. Furthermore, a high linearity coefficient (R2 > 0.99) shows good correlation of the amounts of individual glycoproteins bound to the two lectin media under comparison, which also indicates that both media provide repeatable enrichment of the commonly observed glycoproteins with a minimum bias.
Figure 5.

A scatter plot describing similar trend in binding of glycoproteins identified in all lectin-bound fractions collected from two different affinity matrixes.
4 Concluding Remarks
Using state-of-the-art quantitative proteomics and the Bradford assay, we have demonstrated the repeatability and efficiency of lectin binding. Our data were recorded in 30 separate analyses of the bound fractions collected from two lectin matrixes acquired from different sources, showing an overall recovery of approximately 70%. Approximately 30% of all proteins in the sample were bound to Con A (RSD < 11%), as suggested by the Bradford assay. More than half of the identified proteins were commonly observed in all analyzed fractions. The repeatability of lectin enrichment was demonstrated through a label-free quantitative proteomics procedure that was aided by our recently developed ProteinQuant suite, which allows quantification of all components identified in the analyzed samples. Our data show that the abundances of glycoproteins found in all lectin-bound fractions, when represented as normalized peak areas for all the identified peptides, varied with the relative standard deviations of less than 30%. Data were comparable for both lectin matrixes investigated in this study.
Additionally, our results indicate that lectin affinity enrichment is a suitable technique for a repeatable enrichment of specific glycoproteins from complex biological samples, in both qualitative and quantitative terms. However, some variations observed in the abundances of enriched glycoproteins across all analyzed samples (RSD < 30%) have to be taken into account, when the described approach is to be implemented in a general lectin-carbohydrate recognition study or, more specifically, in a biomarker discovery investigation.
Acknowledgments
This work was supported by grant No. GM24349 from the National Institute of General Medical Sciences, U.S. Department of Health and Human Services, and grant No. RR018942 from the National Center for Research Resources, a component of the National Institute of Health (NIH-NCRR), for the National Center for Glycomics and Glycoproteomics (NCGG) at Indiana University.
References
- 1.Anderson LN, Anderson NG. Mol Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 2.Ping PP, Vondriska TM, Creighton CJ, Gandhi TKB, Yang ZP, Menon R, Kwon MS, Cho SY, Drwal G, Kellmann M, Peri S, Suresh S, Gronborg M, Molina H, Chaerkady R, Rekha B, Shet AS, Gerszten RE, Wu HF, Raftery M, Wasinger V, Schulz-Knappe P, Hanash SM, Paik YK, Hancock WS, States DJ, Omenn GS, Pandey A. Proteomics. 2005;5:3506–3519. doi: 10.1002/pmic.200500140. [DOI] [PubMed] [Google Scholar]
- 3.Spiro RG. Glycobiology. 2002;12:43r–56r. doi: 10.1093/glycob/12.4.43r. [DOI] [PubMed] [Google Scholar]
- 4.Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dwek RA. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 6.Clark GF, Dell A, Morris HR, Patankar M, et al. Mol Hum Reprod. 1997;3:5–13. doi: 10.1093/molehr/3.1.5. [DOI] [PubMed] [Google Scholar]
- 7.Pochec E, Litynska A, Amoresano A, Casbarra A. Biochim Biophys Acta. 2003;1643:113–123. doi: 10.1016/j.bbamcr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Science. 2001;291:2370–2376. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- 9.Zachara NE, Hart GW. Chem Rev. 2002;102:431–438. doi: 10.1021/cr000406u. [DOI] [PubMed] [Google Scholar]
- 10.Carroll L, Hannawi S, Marwick T, Thomas R. Wien Med Wochenschr. 2006;156:42–52. doi: 10.1007/s10354-005-0242-9. [DOI] [PubMed] [Google Scholar]
- 11.Durand G, Seta N. Clin Chem. 2000;46:795–805. [PubMed] [Google Scholar]
- 12.Freeze HH. Nature Rev Gene. 2006;7:537–551. doi: 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- 13.Dennis JW, Granovsky M, Warren CE. Bioassays. 1999;21:412–421. doi: 10.1002/(SICI)1521-1878(199905)21:5<412::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Lowe JB, Marth JD. Ann Rev Biochem. 2003;72:643–691. doi: 10.1146/annurev.biochem.72.121801.161809. [DOI] [PubMed] [Google Scholar]
- 15.Block TM, Comunale MA, Lowman M, Steel LF, Romano PR, Fimmel C, Tennant BC, London WT, Evans AA, Blumberg BS, Dwerk RA, Mattu TS, Mehta AS. Proc Nat Acad Sci USA. 2005;102:779–784. doi: 10.1073/pnas.0408928102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dabelsteen E, Clausen H, Mandel U. J Oral Pathol Med. 1991;20:361–368. doi: 10.1111/j.1600-0714.1991.tb00945.x. [DOI] [PubMed] [Google Scholar]
- 17.Turner GA. Clin Chim Acta. 1992;208:149–171. doi: 10.1016/0009-8981(92)90073-y. [DOI] [PubMed] [Google Scholar]
- 18.Hakomori S. Proc Natl Acad Sci USA. 2002;99:10231–10233. doi: 10.1073/pnas.172380699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hjorth R, Vretblad . In: Lect Chem Soc Int Symp. Epton R, editor. Ellis Horwood Ltd.; Uppsala, Sweden: 1976. pp. 330–336. [Google Scholar]
- 20.Hage DS. Clin Chem. 1999;45:593–615. [PubMed] [Google Scholar]
- 21.Madera M, Mechref Y, Klouckova I, Novotny MV. J Proteome Res. 2006;5:2348–2363. doi: 10.1021/pr060169x. [DOI] [PubMed] [Google Scholar]
- 22.Madera M, Mechref Y, Klouckova I, Novotny MV. J Chromatogr B. 2007;845:121–137. doi: 10.1016/j.jchromb.2006.07.067. [DOI] [PubMed] [Google Scholar]
- 23.Nawarak J, Phutrakul S, Chen ST. J Proteome Res. 2004;3:383–392. doi: 10.1021/pr034052+. [DOI] [PubMed] [Google Scholar]
- 24.Xiong L, Andrews D, Regnier F. J Proteome Res. 2003;2:618–625. doi: 10.1021/pr0340274. [DOI] [PubMed] [Google Scholar]
- 25.Yang Z, Hancock WS. J Chromatogr A. 2004;1053:79–88. [PubMed] [Google Scholar]
- 26.Yang Z, Hancock WS. J Chromatogr A. 2005;1070:57–64. doi: 10.1016/j.chroma.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z, Hancock WS, Chew TR, Bonilla L. Proteomics. 2005;5:3353–3366. doi: 10.1002/pmic.200401190. [DOI] [PubMed] [Google Scholar]
- 28.Yang Z, Harris LE, Palmer-Toy DE, Hancock WS. Clin Chem. 2006;52:1897–1905. doi: 10.1373/clinchem.2005.065862. [DOI] [PubMed] [Google Scholar]
- 29.Cummings RD. Meth Enzymol. 1994;230:66–86. doi: 10.1016/0076-6879(94)30008-9. [DOI] [PubMed] [Google Scholar]
- 30.Cummings RD, Kornfeld S. J Biol Chem. 1982;257:11235–11240. [PubMed] [Google Scholar]
- 31.Gravel P, Golaz O. The Protein Protocols Handbook. Humana Press Inc.; Totowa, NJ: 1996. [Google Scholar]
- 32.Gravel P, Walzer C, Aubry C, Balant LP, Yersin B, Hochstrasser DF, Guimon J. Biochem Biophys Res Commun. 1996;220:78–85. doi: 10.1006/bbrc.1996.0360. [DOI] [PubMed] [Google Scholar]
- 33.Carlsson SR. Glycobiology: A Practical Approach. Oxford University Press; Oxford, UK: 1993. [Google Scholar]
- 34.Mao X, Luo Y, Dai Z, Wang K, Du Y, Lin B. Anal Chem. 2004;76:6941–6947. doi: 10.1021/ac049270g. [DOI] [PubMed] [Google Scholar]
- 35.Hermo L, Winikoff R, Kan FWK. Histochemistry. 1992;98:93–103. doi: 10.1007/BF00717000. [DOI] [PubMed] [Google Scholar]
- 36.Bundy J, Fenselau C. Anal Chem. 1999;71:1460–1463. doi: 10.1021/ac981119h. [DOI] [PubMed] [Google Scholar]
- 37.Bundy JL, Fenselau C. Anal Chem. 2001;73:751–757. doi: 10.1021/ac0011639. [DOI] [PubMed] [Google Scholar]
- 38.Mechref Y, Madera M, Novotny MV. In: Lectins - Analytical Technologies. Nilsson CL, editor. Elsevier; Amsterdam: 2007. p. 442. [Google Scholar]
- 39.Plavina T, Wakshull E, Hancock WS, Hincapie M. J Proteome Res. 2007;6:662–671. doi: 10.1021/pr060413k. [DOI] [PubMed] [Google Scholar]
- 40.Pilobello KT, Krishnamoorthy L, Slawek D, Mahal LK. ChemBiochem. 2005;6:985–989. doi: 10.1002/cbic.200400403. [DOI] [PubMed] [Google Scholar]
- 41.Bedair M, Rassi ZE. J Chromatogr A. 2004;1044:177–186. doi: 10.1016/j.chroma.2004.03.080. [DOI] [PubMed] [Google Scholar]
- 42.Bedair M, Rassi ZE. J Chromatogr A. 2005;1079:236–245. doi: 10.1016/j.chroma.2005.02.084. [DOI] [PubMed] [Google Scholar]
- 43.Madera M, Mechref Y, Novotny MV. Anal Chem. 2005;77:4081–4090. doi: 10.1021/ac050222l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang B, Palcic MM, Schriemer DC, Alvarez-Manilla G, Pierce M, Hindsgaul O. Anal Biochem. 2001;299:173–182. doi: 10.1006/abio.2001.5417. [DOI] [PubMed] [Google Scholar]
- 45.Mann B, Madera M, Mechref Y, Sheng QH, Tang H, Novotny MV. Rapid Comm Mass Spec. 2008 doi: 10.1002/rcm.3781. in press. [DOI] [PubMed] [Google Scholar]
- 46.Bradford MM. Anal Biochem. 1972;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 47.Choi H, Nesvizhskii AI. J Proteome Res. 2008;7:47–50. doi: 10.1021/pr700747q. [DOI] [PubMed] [Google Scholar]
- 48.Wang Lh, Li DQ, Fu Y, Wang HP, Zhang JF, Yuan ZF, Sun RX, Zeng R, He SM, Gao W. Rapid Commun Mass Spectrom. 2007;21:2985–2991. doi: 10.1002/rcm.3173. [DOI] [PubMed] [Google Scholar]
- 49.Drake RR, Schwegler EE, Malik G, Diaz J, Block T, Mehta A, Semmes OJ. Mol Cell Proteomics. 2006;5:1957–1967. doi: 10.1074/mcp.M600176-MCP200. [DOI] [PubMed] [Google Scholar]
- 50.Janković MM, Kosanović MM. Clin Biochem. 2005;38:58–65. doi: 10.1016/j.clinbiochem.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 51.Higgs RE, Knierman MD, Gelfanova V, Butler JP, Hale JE. J Proteome Res. 2005;4:1442–1450. doi: 10.1021/pr050109b. [DOI] [PubMed] [Google Scholar]
- 52.Andreev VP, Li L, Cao L, Gu Y, Rejtar T, Wu SL, Karger BL. J Proteome Res. 2007;6:2186–2194. doi: 10.1021/pr0606880. [DOI] [PMC free article] [PubMed] [Google Scholar]