

Abstract
Motivation: Although chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) or tiling array hybridization (ChIP-chip) is increasingly used to map genome-wide–binding sites of transcription factors (TFs), it still remains difficult to generate a quality ChIPx (i.e. ChIP-seq or ChIP-chip) dataset because of the tremendous amount of effort required to develop effective antibodies and efficient protocols. Moreover, most laboratories are unable to easily obtain ChIPx data for one or more TF(s) in more than a handful of biological contexts. Thus, standard ChIPx analyses primarily focus on analyzing data from one experiment, and the discoveries are restricted to a specific biological context.
Results: We propose to enrich this existing data analysis paradigm by developing a novel approach, ChIP-PED, which superimposes ChIPx data on large amounts of publicly available human and mouse gene expression data containing a diverse collection of cell types, tissues and disease conditions to discover new biological contexts with potential TF regulatory activities. We demonstrate ChIP-PED using a number of examples, including a novel discovery that MYC, a human TF, plays an important functional role in pediatric Ewing sarcoma cell lines. These examples show that ChIP-PED increases the value of ChIPx data by allowing one to expand the scope of possible discoveries made from a ChIPx experiment.
Availability: http://www.biostat.jhsph.edu/∼gewu/ChIPPED/
Contact: hji@jhsph.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
ChIPx experiments, including ChIP-seq (Johnson et al., 2007) and ChIP-chip (Ren et al., 2000), have become a powerful tool used by individual investigators, as well as consortium projects, such as the ENCODE (Dunham et al., 2012) to study transcription factor-binding sites. Each individual ChIPx experiment is non-trivial to perform—extensive time and effort must be spent to acquire effective antibodies and design efficient protocols to generate high-quality ChIPx data—thus, it is important to develop methodology to help investigators to maximize the value of each individual ChIPx experiment.
One of the primary limitations of ChIPx is it may be difficult for individual laboratories to study TF regulation in a wide variety of biological contexts, which we define as the cell or tissue types and associated treatments or disease conditions (see definition details in Supplementary Method 1.1). This is largely because of the prohibitively high labor and time costs to perform each experiment. To resolve this limitation, we investigate whether publicly available gene expression data (PED) in the Gene Expression Omnibus (GEO; Barrett et al., 2009) can be used as a tool to increase the value of ChIPx experiments. Currently, >600 000 gene expression samples from a broad spectrum of biological contexts and species are deposited in the GEO and ArrayExpress (Parkinson et al., 2011). These data are freely available and contain rich information complementary to ChIPx, which may be extremely useful to help study TF regulation.
In this article, we demonstrate that this is indeed the case by proposing and evaluating a new approach, ChIP-PED. Given a TF regulatory pathway, i.e. a TF and the corresponding set of target genes defined using ChIPx and gene expression data in one or more biological contexts, ChIP-PED scans through a large collection of >20 000 human and mouse gene expression samples generated by hundreds of different laboratories by quickly surveying the TF and target gene activities across >2000 biological contexts to identify potentially new connections between the TF regulatory pathway and various cell types, tissues or diseases (Fig. 1). We will illustrate that the predictions from ChIP-PED are useful and can greatly expand the scope of discoveries one can make from ChIPx experiments. We also provide an R package for users to perform ChIP-PED analyses on their own ChIPx and TF perturbation data.
Fig. 1.

ChIP-PED overview. Gene expression profiles from TF perturbation experiments are intersected with ChIPx experiments to obtain a set of activated and repressed target genes. ChIP-PED then takes as input the TF and target genes and scans through a compendium of publicly available gene expression profiles to search for biological contexts in which the TF and target genes are enriched in activity. The final output is a ranked table of biological contexts enriched with a regulatory pattern of interest
ChIP-PED represents a novel conceptual approach to building computational tools for ChIPx data analysis. Most existing tools for analyzing ChIPx data, including those for detecting protein–DNA–binding sites (Laajala et al., 2009; Wilbanks and Facciotti, 2011), discovering DNA-binding motifs (Bailey et al., 2011; Liu et al., 2002), correlating ChIPx with gene expression data (Cheng et al., 2011; Ouyang et al., 2009) and so forth, focus on addressing analysis issues concerning a single or a few related ChIPx datasets. Their discoveries are also typically restricted to the biological context in which the ChIPx experiments are performed, and none of them systematically integrates information from PED. PED has been shown to be invaluable in other applications (Huang et al., 2010; Zilliox and Irizarry, 2007), but the possibility of using PED as a tool to boost the analysis of ChIPx data still remains largely unexplored. A number of methods do integrate large amounts of ChIPx and gene expression data to construct gene regulatory networks, but most are primarily used to study lower organisms (e.g. yeast; Faith et al., 2007; Zhu et al., 2008). The present study is different from those described works, as ChIP-PED focuses specifically on integrating ChIPx with large amounts of heterogeneous data in human and mouse to improve ChIPx analyses. Instead of attempting to construct a comprehensive gene regulatory network, the primary goal of ChIP-PED is to produce simple testable hypotheses, such as ‘TF A is functionally active in biological contexts X, Y and Z through target gene set S’.
2 MATERIALS AND METHODS
2.1 Data collection
ChIP-PED relies on two large compendiums of gene expression profiles, consisting of 13 182 human gene expression samples generated from Affymetrix Human U133A (GPL96) and 9643 mouse samples generated from Affymetrix Mouse 430 2.0 (GPL1261) arrays (McCall et al., 2011). The gene expression profiles were downloaded from GEO (July 2010), pre-processed and normalized consistently using fRMA (McCall et al., 2010). fRMA is designed to normalize large amount of heterogeneous microarray samples to reduce the effect of batch on gene expression estimates. For each probeset, we standardized the fRMA values across all microarray samples from the same array platform to have zero mean and unit standard deviation. The biological context of each sample was recorded and manually verified based on the sample descriptions in GEO (see Supplementary Method 1.1 and Supplementary Fig. S1).
2.2 ChIP-PED
Given a TF and its activated and repressed target genes defined using ChIPx and gene expression data in one or more biological contexts, ChIP-PED searches for other contexts in which the TF is likely to be functionally active. Target genes (TG) are genes that are both TF-bound in the ChIPx experiments and differentially expressed in corresponding gene expression data in which the expression of the TF is perturbed. The latter are from TF perturbation experiments comparing wild-type with TF-knockout, control with TF-knockdown or control with TF-overexpression and so forth. Users will need to provide and analyze their own ChIPx and TF perturbation experiments to define the input target genes. Supplementary Method 1.2 discusses methods for generating target gene lists. To define target genes in a particular biological context, ideally one would like to have ChIPx and TF perturbation data from the same biological context. However, such data may not always be available, and it is not uncommon to have ChIPx and TF perturbation data collected from two different contexts. In that case, one can still intersect the data from different experiments to obtain a putative target gene set assumed to contain the shared targets.
ChIP-PED first measures the TF expression and TG activity in each microarray sample in our PED compendiums. TF expression, ETF, is defined as a simple average of the normalized probeset intensities, p:
| (1) |
where TF is the set of probesets that measure the expression of the TF, and nTF is the number of probesets for the TF. TG activity, ATG, is defined as:
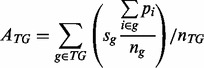 |
(2) |
Here, TG is the set of target genes of the TF, nTG is the number of target genes, ng is the number of probesets for a specific target gene, g, and sg is 1 or −1 depending on whether gene g is activated (positively regulated) or repressed (negatively regulated), respectively. sg is included to account for TFs that are capable of both activating and repressing different target genes. ATG is designed to describe the regulatory activity of a TF through its target genes, rather than measure the raw expression of the target genes. For example, if a TF acts mainly as a repressor in a biological context in which it is functionally active, we would observe low expression of its target genes, but high ATG because of the multiplier sg = −1 (Supplementary Method 1.3 and Supplementary Fig. S2A and B). Examples of the distributions of ETF and ATG for real TF ChIPx data are shown in Supplementary Figure S3.
After measuring TF expression and TG activity, users can choose cut-offs c1 ∼ c4 to define (i) high-TF expression (‘ETF > c1’), (ii) low-TF expression (‘ETF < c2’), (iii) high-TG activity (‘ATG > c3’) and (iv) low-TG activity (‘ATG < c4’), denoted by TF+, TF−, TG+ and TG−, respectively. By default, c1 ∼ c4 are chosen to be values corresponding to a one-sided P-value of 0.1 based on fitted normal distributions for ETF or ATG across all samples, with c1 and c3 taking values above the mean, and c2 and c4 taking values below the mean (Fig. 2A). ChIP-PED can then search for biological contexts associated with four regulatory patterns: (i) TF+TG+, (ii) TF+TG−, (iii) TF−TG+ and (iv) TF−TG−. The pattern TF+TG+ is of primary interest, as it focuses on discovering new contexts in which the TF is functionally active through its target genes (TF-active). This is because high-TF expression alone is not sufficient to imply the existence of functional TF protein because of possible post-transcriptional and translational regulation, but high-TG activity in addition to high-TF expression would strongly support the presence of active TF protein. Other regulatory patterns are discussed in more detail in the Supplementary Method 1.4.
Fig. 2.

ChIP-PED plots show strong correlation between TF expression, ETF (x-axis) and TG activity, ATG (y-axis), for mouse Oct4 (A) and Gata1 (C) in 9643 Affymetrix Mouse 430 2.0 array samples and human MYC (B) and STAT1 (D) in 13 182 Affymetrix Human HGU133a samples. Number of TGs in each plot is shown in the parentheses. Solid lines correspond to TF+ (c1) and TF− (c2) ETF cut-offs and TG+ (c3) and TG− (c4) ATG cut-offs. Samples from a few biological contexts with enriched TF+TG+ (in A–D), TF−TG+ (in B) and TF+TG− (in C) functional activity are shown in color. Also plotted in color are ‘Diff ESC, EBs’ (purple) to show the separation between differentiated and undifferentiated ESCs in (A), ‘A673’ (purple) and ‘Ewing tumor’ (blue), both of which are Ewing tumor samples in (B), and ‘PBMC-normal’ (orange), which fall outside of the TF+TG+ region in contrast to infected PBMCs in (D). All other samples are plotted in gray. ‘Cor’: Pearson correlation coefficient between ETF and ATG. ‘Pval’: P-values that are empirically calculated from ETF and ATG correlations of randomly drawn pseudo-TG sets of the same size 10 000 times. For comparison, an example plot of a random sample of pseudo-TGs is shown for each TF (bottom-right)
Then given a compendium of N gene expression profiles, ChIP-PED searches among all biological contexts with at least three samples, for contexts that are associated with the regulatory pattern of interest (e.g. TF+TG+). For each context c, it counts (i) K, the total number of samples in the compendium that exhibit the pattern, (ii) nC, the total number of samples in context c, and (iii) kC, the number of samples in context c that exhibit the pattern. Fisher’s exact test is then applied to the quadruplet (nC, N, kC and K) to test the association between c and the regulatory pattern of interest (i.e. whether kC is significantly larger than random expectation). To account for testing multiple contexts, the P-values are adjusted using the Bonferroni correction. The final output of ChIP-PED is a ranked table of statistically significant biological contexts at a default Bonferroni corrected P-value cut-off of 0.05 (Supplementary Method 1.5).
After the initial ChIP-PED analysis, ChIP-PED can perform the following analyses to further explore each predicted context: (i) search for related contexts in the compendium based on user-specified keyword(s), (ii) extract the ETF and ATG values for the set of contexts found, (iii) calculate, sort and plot the mean and standard deviation of the ETF and ATG values for each context and (iv) perform t-tests between all pairwise combinations of the contexts for significant differences in mean ETF or ATG. See Supplementary Methods 1.6–1.7 for details and Section 3.3 for an example analysis.
2.3 ChIP-PED evaluation
We evaluated ChIP-PED by applying it to multiple TFs—Oct4, Gata1 and Jarid2 in mice and MYC, STAT1 and ESR1 in human—using the datasets listed in Supplementary Table S1. The TF target genes were constructed by intersecting TF-bound genes predicted from ChIPx data with differentially expressed genes [false discovery rate (FDR) ≤ 10%] in TF perturbation data. TF-bound genes were defined as genes with a significant peak (FDR ≤ 10%) overlapping with the −10- to +5-kb region around the transcription start site of the gene. Details are provided in Supplementary Method 1.2, and full target gene lists can be found in Supplementary Tables S2–S7.
Predictions were verified by a thorough search of existing literature to identify whether each prediction was functionally validated or suggested in previous experiments. ‘Functional’ validations required previous experimental data from the predicted biological context demonstrating observable changes in phenotype when the expression of the TF is perturbed or TF binding coupled with transcriptional responses to TF binding of target genes. ‘Suggested’ predictions must be supported by other lines of indirect evidence, such as experimentally observed high-TF protein levels in the predicted context. All supporting references are recorded in Supplementary Tables S2–S7. We also experimentally validated a novel ChIP-PED functional connection between MYC and Ewing sarcoma (Supplementary Method 1.8).
3 RESULTS
3.1 PED are capable of measuring TF regulatory activities in spite of data heterogeneity
We first investigated whether it was appropriate to compare gene expression across thousands of heterogeneous microarray samples generated by different laboratories. To this end, we asked whether laboratory and batch effects were a significant and detrimental source of variation (Leek et al., 2010). Previous efforts have been made using our gene expression compendiums to demonstrate that similar tissue types do cluster together (Zilliox and Irizarry, 2007), and that it is possible to accurately predict tissue types from a single gene expression profile in spite of the laboratory or batch effects (McCall et al., 2011). We reaffirmed these findings by observing that samples from the same tissues from different laboratories were more similar in expression compared with samples from different tissues from the same laboratory (Supplementary Fig. S4 and Supplementary Method 1.9).
We then examined the correlation between TF expression (ETF) and TG activity (ATG) for multiple TFs, including mouse Oct4 and Gata1 and human MYC and STAT1. We reasoned that if there were strong laboratory or batch effects that overwhelmed the biological signal, we would observe weak to zero correlation between ETF and ATG across the heterogeneous samples. Instead, we found significant correlation between TF expression and TG activity; the Pearson correlation coefficients between ETF and ATG for Oct4, MYC, Gata1 and STAT1 were 0.679, 0.418, 0.303 and 0.699, respectively (P < 0.02; Fig. 2). As this observation holds for multiple mouse and human TFs from different microarray platforms (GPL1261 and GPL96), our results suggest that biological variability in the publicly available Affymetrix microarray data is stronger than the laboratory or batch effects. This is consistent with earlier observations made by Lukk et al. (2010).
3.2 ChIP-PED predicts known TF-active contexts
After verifying that it is meaningful to compare ETF and ATG across heterogeneous samples, we asked whether the samples observed with high-TF expression and high-TG activity (TF+TG+) and the biological contexts enriched with a TF+TG+ regulatory pattern were biologically meaningful. In this regard, we performed and evaluated ChIP-PED analyses of six TFs: mouse Oct4, Gata1 and Jarid2 and human MYC, STAT1 and ESR1.
Oct4 is a master regulator in mouse embryonic stem cells (ESCs). We obtained 519 activated and 337 repressed Oct4 target genes by combining ChIP-seq data from mouse ESCs with gene expression data from ESCs in which Oct4 was knocked down via siRNA (Supplementary Tables S1 and S2). Using these target genes as input, Oct4 target gene activity was plotted against Oct4 expression after excluding the PED samples used to construct the target genes (Fig. 2A). We found that undifferentiated ESCs clustered together with high-TF expression and high-TG activity. In contrast, differentiated ESCs or embryoid bodies (EBs) had lower TF expression and TG activity. This is consistent with the self-renewal and pluripotency role of Oct4 in ESCs and its decrease in expression when ESCs differentiate (Chen et al., 2008; Loh et al., 2006).
Of the 9643 mouse samples in the compendium, 480 were labeled as TF+TG+ using the default cut-offs. Among them, 69.2% (332/480) were known Oct4-expressing (+Oct4) biological contexts—most commonly, undifferentiated ESCs (Niwa et al., 2000), primordial germ cells (Kehler et al., 2004), induced pluripotent stem cells (Wernig et al., 2007) and embryonic carcinomas (Wang and Schultz, 1996)—covering 96.0% (332/346) of all +Oct4 samples in the compendium. In all, 18.1% (87/480) of the TF+TG+ samples were differentiating ESCs or EBs, and the remaining 12.7% (61/480) were other contexts, such as embryos, mouse embryonic fibroblasts (MEFs) and so forth. The observation that a large proportion (30.8% = 18.1% + 12.7%) of TF+TG+ samples were biological contexts not known to express Oct4 (-Oct4) shows the noisy nature of PED. This makes it challenging to correctly predict whether an individual sample truly exhibits functional TF activity. However, the primary goal of ChIP-PED is not to correctly identify TF-active samples, but to identify TF-active biological contexts. Thus, ChIP-PED takes advantage of the fact that each biological context has multiple samples in the PED compendium to predict TF-active biological contexts by reporting the contexts with a statistically significant proportion of TF+TG+ samples.
In total, ChIP-PED predicted 28 biological contexts were enriched with TF+TG+ activity at a Bonferroni-corrected P-value cut-off of 0.05 (Supplementary Table S2). Among these, 89.3% (25/28) were different +Oct4 contexts, and 10.7% (3/28) were -Oct4 contexts related to differentiating ESCs and EBs. The 28 statistically enriched contexts covered 47.9% (230/480) of the TF+TG+ samples. These samples were from multiple laboratories (e.g. normal undifferentiated ESCs: 11 experiments), confirming that the observed enrichment in ESCs was unlikely to be caused by experimental artifacts or laboratory or batch effects. More importantly, ChIP-PED filtered out most -Oct4 biological contexts: 30.8% (148/480) TF+TG+ samples were from -Oct4 contexts, whereas only 10.7% (3/28) of the TF+TG+-enriched contexts were from -Oct4 contexts, and among the samples of the 28 TF+TG+-enriched contexts, only 8.7% (20/230) were -Oct4 samples. Therefore, by integrating information from multiple samples, predictions made at the context level are more accurate than at the sample level.
Next, we analyzed human MYC, a TF known to be involved in multiple tumors (Zeller et al., 2003). We identified 1716 activated and 617 repressed target genes from a compilation of eight TF perturbation datasets along with 12 ChIPx datasets (Supplementary Tables S1 and S3). The target genes were required to be differentially expressed in the same direction in ≥50% of the TF perturbation datasets and MYC-bound in ≥50% of the ChIPx datasets. The aim was to identify the core MYC regulatory target genes that were cell-type independent across multiple MYC-active contexts. As expected, MYC regulatory activity was significantly enriched in numerous tumor types (Figs 2B and 4A and Supplementary Table S3): 74.7% of the 521 TF+TG+ samples were tumors, which was significantly higher than the background percentage of 46.0% for all 13 182 samples in the human PED compendium (one-sided P < 0.001, binomial test). Among these samples, ChIP-PED predicted 33 TF+TG+-enriched biological contexts. Many of the predictions were found to be correct, such as B-cell lymphomas, which have been shown to have functionally active MYC protein (Zeller et al., 2003).
Fig. 4.

MYC analysis and validation. (A) MYC TF+TG+ biological contexts (similar contexts are grouped) and the number of MYC+TG+ samples (orange) and number of non-MYC+TG+ samples (blue) are shown. The majority of TF+TG+ samples are tumor types. (B) Decrease in proliferation of TC71 cells on knockdown of MYC. Control and shMyc TC71 cells were evaluated for changes in proliferation rates by using a cell viability reagent, CCK-8. Two thousand cells were initially plated into individual 96 wells and assessed daily for changes in growth and proliferation. (C) Decreased tumorigenicity as assessed by soft-agar assay for shMyc TC71 cells. Control TC71 cells developed significant soft-agar colonies within 2–3 weeks, whereas the shMyc cells formed only a few miniscule colonies over the same time. (D) Graphic display of differences in tumor weight comparing control and shMyc tumors. On average, the shMyc tumors weighed only 20% of control tumors. Vertical error bars indicate the standard deviation of the tumor volume. The P-value is obtained from a two-sided t-test. (E) Western blot analysis for MYC protein, c-Myc, in control and shMyc cells at 0 (Pre) and 6 weeks (Post). Actin is provided as a loading control. Blot displays decrease in MYC protein levels on stable expression of shMyc in TC71 cells
Successful predictions were also made when analyzing mouse Gata1 target genes from erythroid contexts, mouse Jarid2 target genes from ESCs, human STAT1 target genes from HeLaS3 cells and human ESR1 target genes from estrogen-treated MCF7 cells (Supplementary Tables S1, S4–S7). ChIP-PED found enriched Gata1 expression and target gene activity in expected biological contexts related to erythrocyte and megakaryocyte development, such as in fetal liver, common myeloid progenitor cells and murine erythroleukemia cells (Fig. 2C; Iwasaki et al., 2003). ChIP-PED also predicted STAT1 functional activity in peripheral blood mononuclear cells (PBMC) infected with hepatitis C and malaria, consistent with current knowledge of STAT1 regulatory functions (Fig. 2D; Kim et al., 2008; Taylor et al., 2007). Jarid2 activity, a known repressor with an essential role in embryonic development, was enriched in expected cell types, such as undifferentiated ESCs and induced pluripotent stem cells (Supplementary Fig. S2A; Landiera and Fisher, 2011). Finally, ChIP-PED correctly predicted ESR1 functional activity in breast cancer-related cell types, such as MCF7 cells (Supplementary Fig. S2C; Frasor et al., 2009).
For the six TFs analyzed, ChIP-PED made 178 TF+TG+ predictions listed in Supplementary Tables S2–S7 (Oct4: 28, MYC: 33, Gata1: 37, STAT1: 12, Jarid2: 41 and ESR1: 27). To systematically evaluate ChIP-PED prediction accuracy, we examined all predictions through a survey of existing literature. We found that 90 of 178 (50.6%) biological contexts predicted to be enriched with TF+TG+ activity were functionally validated in previous experiments (see Section 2). For example in the Oct4 analysis, 20 of the 28 predictions were functionally validated, even though 25 of the 28 predictions were known to express Oct4 RNA (i.e. +Oct4 as described earlier). This is because functional experiments demonstrating changes in phenotype after perturbing Oct4 or showing TF binding with associated transcriptional response of target genes could only be found for 20 predictions; therefore, we only counted those 20 predictions as functionally validated. The 50.6% accuracy rate is a conservative estimate, as the remaining predictions may not necessarily be false positives, but instead may represent unknown/novel functional relationships. Altogether, these results demonstrate that given the target genes of a TF defined from ChIPx and TF perturbation data from one or a few biological contexts, ChIP-PED is capable of discovering TF-active contexts from a broad spectrum of PED samples.
Searching through PED only for TF+ samples or only for TG+ samples, rather than TF+ and TG+ samples, may result in substantially decreased ChIP-PED prediction accuracy and number of functionally validated predictions. For instance, when we modified ChIP-PED to search only for TF+ samples, we found that only 40.0% (62/155) of the predicted TF+ contexts were functionally validated in previous experiments compared with 50.6% (90/178) when using ChIP-PED to search for TF+ and TG+ samples (Supplementary Tables S2–S7). Conversely, searching for only TG+ samples resulted in only 34.0% (67/197) functionally validated TG+ predictions (Supplementary Tables S2–S7). Thus, it is useful to check both TF expression and target gene activity of each context to identify TF+ and TG+ samples when predicting TF-active biological contexts.
TF target genes can vary from one cell type to another. If two known TF-active contexts do not share any target genes, then ChIP-PED will not be able to predict either context using target genes constructed from the other context. To test whether ChIP-PED can still be effective when only a minority of the target genes are shared, we used ChIP-PED to analyze Stat3 target genes constructed from mouse CD4+ T cells and Th17 cells, which are both contexts in which Stat3 plays an important regulatory role (Durant et al., 2010; Kwon et al., 2009). We found that ChIP-PED was able to successfully recover both CD4+ T cells and Th17 cells when analyzing target genes defined from the other context, even though <30% of the target genes were in common (Supplementary Method 1.10, Supplementary Table S8 and Supplementary Fig. S5).
3.3 ChIP-PED can expand the scope of possible functional discoveries
ChIP-PED would not be useful if the predicted biological contexts were always closely related to the context in which the experimental data were generated. Our results indicate otherwise: among the 90 of 178 (50.6%) predictions supported by previous functional experiments, 40 (44.4%) are in contexts unrelated to the context(s) in which the experimental data used to construct the TF target genes were obtained (Supplementary Tables S2–S7).
Furthermore, ChIP-PED can provide additional biological insights that otherwise could not be made using standard ChIPx analyses. For example, after the initial STAT1 ChIP-PED analysis described in Section 3.2, we found many hepatitis C-infected PBMCs predictions from experiment GSE7123 (Supplementary Table S5 and Supplementary Method 1.11). To examine STAT1 functional activity in hepatitis C-infected PBMCs in more detail, we searched for all contexts in GSE7123 and also found healthy PBMCs along with the predicted hepatitis C-infected PBMCs. We then used ChIP-PED to compare TF expression and TG activity in each context and found that ETF and ATG values were significantly different between healthy and hepatitis C-infected PBMCs, with a gradual decrease in ETF and ATG values as patients recovered from infection (Supplementary Table S5, Fig. 3 and Supplementary Fig. S6). When reviewing both of the original publications, the STAT1 ChIPx study (Robertson et al., 2007) and the study that generated the gene expression profiles from hepatitis C-infected PBMCs (Taylor et al., 2007), we found that neither study had reported this finding. To verify whether this observation was correct, we searched through existing literature and found an entirely independent experiment that showed in a series of overexpression and siRNA-mediated knock-down experiments of STAT1 in hepatitis C virus-infected PBMCs that STAT1 protein was indispensable for the control of hepatitis C virus expression (Lin et al., 2005).
Fig. 3.

Series of ChIP-PED plots depicting the gradual decrease in STAT1 expression and target gene (TG) activity when blood samples are successively drawn from hepatitis C-infected patients as they recover after treatment with interferon and ribavirin from Day 1, 2, 7, 14 to 28 of recovery (GSE7123). Gray points are all samples in the GPL96 compendium, and colored points are the samples from the infected PBMCs in GSE7123. The x-axis is STAT1 expression (ETF) and the y-axis is TG activity (ATG). The mean ETF and ATG of each group of PBMCs are indicted at the top of each plot. Normal PBMCs (bottom right in black) in GSE7123 fall almost entirely out of the TF+TG+ cut-offs (the dashed lines), which suggests that only when infected with hepatitis C is STAT1 functionally active in PBMCs
3.4 ChIP-PED can discover novel TF-active contexts
Besides verifying that ChIP-PED is able to correctly predict known TF-active biological contexts, we also experimentally investigated whether the predictions that were not functionally validated could possibly represent unknown TF-active biological contexts. As a proof-of-concept, we used our ChIP-PED analysis of human MYC to illustrate the discovery of a novel MYC-active context. Among the enriched TF+TG+ predictions from the MYC ChIP-PED analysis, 18 of 33 (54.5%) biological contexts were not supported by functional experiments that demonstrated MYC functional activity (Supplementary Table S3). One of the non-functionally validated contexts was A673 cells (Fig. 4A), which were established from a patient with Ewing sarcoma (Martínez-Ramírez et al., 2003). Although Ewing tumor has been previously shown to exhibit high-MYC expression (Dauphinot et al., 2001), the functional role of MYC protein in Ewing tumor currently remains uncharacterized. To verify the novel prediction that MYC protein plays a functional role in Ewing tumor, we assessed the phenotype changes of independent Ewing sarcoma cell lines on MYC knockdown. Knocking down of MYC using shMYC in TC71 and MHH-ES Ewing sarcoma cell lines resulted in a substantially slower proliferation rate and tumorigenicity when compared with control cells (Fig. 4B and C and Supplementary Figs S7 and S8). Furthermore, xenograft of control and shMYC TC71 Ewing sarcoma cells into immunodeficient mice (NOD/SCID/IL-2γ null) resulted in a significant decrease in volume and weight for the MYC knockdown tumors after 6 weeks of growth (Fig. 4D). Subsequent isolation of the tumors confirmed the decrease in MYC protein by western blot analysis (Fig. 4E). These results strongly support the novel prediction that the MYC protein plays a key functional role in Ewing tumor.
When studying the 88 functionally unverified predictions across the six TFs analyzed, we found that 51 of the 88 (58.0%) predictions were supported by other lines of indirect evidence in existing literature, such as experimentally observed high-TF protein level in the predicted context (Supplementary Tables S2–S7). Thus, these predictions are likely to represent previously unknown functional predictions between each TF regulatory pathway and context, which further demonstrates that ChIP-PED can discover known and unknown TF-active biological contexts. In total, 141 of 178 (79.2%) TF+TG+ predictions for the six TFs analyzed were either directly supported by functional evidence (90 of 178) or indirectly supported in existing literature (51 of 178).
3.5 Effect of modifications to the ChIP-PED analysis
Many TFs regulate a subset of their target genes through distal enhancers. Recent tools, such as GREAT (McLean et al., 2010), have shown that by properly accounting for distal regulatory sites, one can improve the functional analysis of TF-binding sites. In our ChIP-PED analyses, we assigned peaks to genes if the peak overlapped with the −10- to +5-kb region around each gene transcription start site, which may miss distal TF regulatory activity. This in turn may affect ChIP-PED prediction accuracy. To investigate, we generated ChIP-PED predictions for ESR1 using target genes in estrogen-treated MCF7 cells derived from chromatin interaction analysis by paired-end tag sequencing (ChIA-PET), a method better able to link distal regulatory sites to TF-binding targets (Fullwood et al., 2009), and compared them with predictions made using target genes defined by ChIP-seq using the −10- to +5-kb window. We found that ChIA-PET-based predictions were similar to ChIP-seq–based predictions, and the former had slightly higher functional prediction accuracy of 43.5% compared with 40.7% (Supplementary Method 1.12 and Supplementary Table S7). We also analyzed all six TFs by using multiple annotation window sizes to annotate ChIPx peaks. Different window sizes produced comparable prediction accuracies at the default significance cut-off. However, the −10- to +5-kb window size produced the largest number (i.e. highest power) of functionally validated and/or indirectly supported predictions (Supplementary Method 1.12 and Supplementary Tables S9 and S10). Thus, our results suggest that the −10- to +5-kb window represents a reasonable choice as a default annotation region.
We also compared how well a median, rather than mean, target gene activity measure would perform and found the predictions and prediction accuracy to be almost the same; across all six TFs, 171 predictions were identical between the two measures accounting for 98.8% (171/173) of the median-based predictions and 96.1% (171/178) of the mean-based predictions (Supplementary Method 1.13 and Supplementary Fig. S9). In addition, we checked whether predicted biological contexts with more samples in the compendium were more or less accurate than predicted contexts with fewer samples. Our results were unable to find a clear monotone relationship between sample count for a given biological context and prediction accuracy (Supplementary Method 1.14 and Supplementary Table S11).
4 DISCUSSION
We have shown that ChIP-PED can improve the analysis of ChIPx data by integrating publicly available gene expression data. Given a TF and its target genes, ChIP-PED examines the expression of the TF and the activity of its target genes across an assortment of diverse biological contexts to search for contexts with enriched regulatory activity of the TF. This process may lead to the discovery of novel functional connections between TF regulatory pathways and diseases, thus providing a cost effective way to expand knowledge from one ChIPx study to other research areas.
We view ChIP-PED as an exploratory tool for fast and cost-effective hypothesis generation and screening. In this respect, the default cut-offs that define high- or low-TF expression or TG activity should be primarily used for initial exploration or first-pass automatic hypothesis screening, rather than as strict optimal cut-offs that apply to all TFs. Based on our real data analysis experience, we found it difficult to set a single consistent cut-off that was optimal across all TFs, as TFs can vary greatly in terms of regulatory behavior (Fig. 2). We, therefore, provide users with the flexibility to choose their own cut-offs, which can be adjusted to decrease or increase the number of predicted contexts (Supplementary Method 1.15).
ChIP-PED acts primarily as a guide to highlight biological contexts that would be good leads for experimental investigation. As such, we do not expect all ChIP-PED predictions to be correct nor for ChIP-PED to recover all TF-active biological contexts. This, however, does not prevent ChIP-PED from being a useful and unique tool: our analyses have shown that it can predict many known and new TF-active contexts with reasonable accuracy, and there currently exists no other computational method for analyzing ChIPx data that performs a similar task.
Although we have shown that ChIP-PED is able to capture pertinent biological information in PED, better statistical models are still needed to address technical biases and variations because of laboratory and batch effects. A natural extension of ChIP-PED would be to analyze multiple TFs and their TGs together to better connect cooperative TF regulatory pathways to cell types and diseases. Similarly, more work is also needed to understand how homologous TFs or other TFs with similar regulatory functions impact the regulatory activity of the TF of interest in different contexts.
Our study is not necessarily the best or only way to integrate ChIPx and PED; however, to the best of our knowledge, this is the first systematic study of using PED to enhance ChIPx analyses in human and mouse. We hope that ChIP-PED will inspire new computational approaches that continue to maximize the value of ChIP-seq and ChIP-chip experiments.
Funding: National Institutes of Health (R01HG005220 and R01HG006282 to R.A.I and H.J.); National Institutes of Health training (T32GM074906 to G.W.); St. Baldrick’s Career Development Award (to J.T.Y.).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Cheng C, et al. A statistical framework for modeling gene expression using chromatin features and application to modENCODE datasets. Genome Biol. 2011;12:R15. doi: 10.1186/gb-2011-12-2-r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauphinot L, et al. Analysis of the expression of cell cycle regulators in Ewing cell lines: EWS-FLI-1 modulates p57KIP2 and c-Myc expression. Oncogene. 2001;20:3258–3265. doi: 10.1038/sj.onc.1204437. [DOI] [PubMed] [Google Scholar]
- Dunham I, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–615. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith J, et al. Large-scale mapping and validation of Escherichia coli transcription regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasor J, et al. Positive cross-talk between estrogen receptor and NF-kB in breast cancer. Cancer Res. 2009;69:8918–8925. doi: 10.1158/0008-5472.CAN-09-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood MJ, et al. An oestrogen-recepter-α-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, et al. Bayesian approach to transforming public gene expression repositories into disease diagnosis databases. Proc. Natl Acad. Sci. USA. 2010;107:6823–6828. doi: 10.1073/pnas.0912043107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H, et al. GATA-1 converts lymphoid and myelomonocytic progenitors into megakaryocyte/erythrocyte lineages. Immunity. 2003;19:451–462. doi: 10.1016/s1074-7613(03)00242-5. [DOI] [PubMed] [Google Scholar]
- Johnson DS, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Kehler J, et al. Oct4 is required for primordial germ cell survival. EMBO Rep. 2004;5:1078–1083. doi: 10.1038/sj.embor.7400279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, et al. Experimental malaria infection triggers early expansion of natural killer cells. Infect. Immun. 2008;76:5873–5882. doi: 10.1128/IAI.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H, et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity. 2009;18:941–952. doi: 10.1016/j.immuni.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laajala TD, et al. A practical comparison of methods for detecting transcription factor binding sites in ChIP-seq experiments. BMC Genomics. 2009;10:618. doi: 10.1186/1471-2164-10-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landiera D, Fisher AG. Inactive yet indispensable: the tale of Jarid2. Trends Cell Biol. 2011;21:74–80. doi: 10.1016/j.tcb.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010;11:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, et al. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology. 2005;128:1034–1041. doi: 10.1053/j.gastro.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Liu XS, et al. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat. Biotechnol. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- Loh Y, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Lukk M, et al. A global map of human gene expression. Nat. Biotechnol. 2010;28:322–324. doi: 10.1038/nbt0410-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Ramírez A, et al. Characterization of the A673 cell line (Ewing tumor) by molecular cytogenetic techniques. Cancer Genet. Cytogenet. 2003;141:138–142. doi: 10.1016/s0165-4608(02)00670-2. [DOI] [PubMed] [Google Scholar]
- McCall MN, et al. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11:242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MN, et al. The gene expression barcode: leveraging public data repositories to begin cataloging the human and marine transcriptomes. Nucleic Acids Res. 2011;39:D1011–D1015. doi: 10.1093/nar/gkq1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, et al. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- Ouyang Z, et al. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc. Natl Acad. Sci. USA. 2009;106:21521–21526. doi: 10.1073/pnas.0904863106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson H, et al. ArrayExpress update—an archive of microarray and high-throughput sequencing-based functional genomics experiments. Nucleic Acids Res. 2011;39:D1002–D1004. doi: 10.1093/nar/gkq1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- Taylor M, et al. Changes in gene expression during pegylated interferon and ribavirin therapy of chronic hepatitis C virus distinguish responders from nonresponders to antiviral therapy. J. Virol. 2007;81:3391–3401. doi: 10.1128/JVI.02640-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Schultz GA. Expression of Oct-4 during differentiation of murine F9 cells. Biochem. Cell Biol. 1996;74:579–584. doi: 10.1139/o96-062. [DOI] [PubMed] [Google Scholar]
- Wernig M, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- Wilbanks E, Facciotti M. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS One. 2011;5:e11471. doi: 10.1371/journal.pone.0011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller K, et al. An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008;40:854–861. doi: 10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilliox M, Irizarry R. A gene expression bar code for microarray data. Nat. Methods. 2007;4:911–913. doi: 10.1038/nmeth1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.