

Abstract
The scope, chemoselectivity, and utility of the click-like tyrosine labeling reaction with 4-phenyl-3H-1,2,4-triazoline-3,5(4H)-diones (PTADs) is reported. To study the utility and chemoselectivity of PTAD derivatives in peptide and protein chemistry, we synthesized PTAD derivatives possessing azide, alkyne, and ketone groups and studied their reactions with amino acid derivatives and peptides of increasing complexity. With proteins we studied the compatibility of the tyrosine click reaction with cysteine and lysine-targeted labeling approaches and demonstrate that chemoselective tri-functionalization of proteins is readily achieved. In particular cases, we noted PTAD decomposition resulted in formation of a putative isocyanate by-product that was promiscuous in labeling. This side reaction product, however, was readily scavenged by the addition of a small amount of 2-amino-2-hydroxymethyl-propane-1,3-diol (Tris) to the reaction medium. To study the potential of the tyrosine click reaction to introduce poly(ethylene) glycol chains onto proteins (PEGylation), we demonstrate that this novel reagent provides for the selective PEGylation of chymotrypsinogen whereas traditional succinimide-based PEGylation targeting lysine residues provided a more diverse range of PEGylated products. Finally, we applied the tyrosine click reaction to create a novel antibody drug conjugate. For this purpose, we synthesized a PTAD derivative linked to the HIV entry inhibitor aplaviroc. Labeling of the antibody trastuzumab with this reagent provided a labeled antibody conjugate that demonstrated potent HIV-1 neutralization activity demonstrating the potential of this reaction in creating protein conjugates with small molecules. The tyrosine click linkage demonstrated stability to extremes of pH, temperature and exposure to human blood plasma indicating that this linkage is significantly more robust than maleimide-type linkages that are commonly employed in bioconjugations. These studies support the broad utility of this reaction in the chemoselective modification of small molecules, peptides, and proteins under mild aqueous conditions over a broad pH range using a wide variety of biologically acceptable buffers such as phosphate buffered saline (PBS) and 2-amino-2-hydroxymethyl-propane-1,3-diol (Tris) buffers as well as others and mixed buffered compositions.
INTRODUCTION
Bioconjugation reactions exploit either the intrinsic chemical reactivity of the biomolecule or introduces extrinsic functionalities that can then be subsequently reacted in a bio-orthogonal fashion. In recent years a wide-range of extrinsic bioorthogonal reactions have been developed (Figure 1a).(1–4) This two-step approach relies on the introduction of functionalities such as ketones, aldehydes, azides, alkenes, or alkynes into the target protein. In a second step, the introduced extrinsic functionalities are selectively reacted to form oximes/hydrazones,(1–12) Staudinger ligation products,(1–4, 13) triazole-Click products,(1–4, 14–18) or Diels-Alder,(1–4, 19–23) among others.(1–4, 24) In addition to chemical routes, extrinsic functional groups can be introduced into biomolecules via enzymatic modification,(1–5, 11, 12),(19–23) or genetic encoding.(1–4, 25–33) Thus extrinsic approaches involve a minimum of two modification steps of the biomolecule. In contrast, intrinsic approaches have the potential to be one-step (Figure 1b). Intrinsic approaches to bioconjugation are, however, limited to the development of chemistry compatible with the 20 naturally occurring amino acids and are challenged by the fact that any given amino acid is likely to occur more than once in a protein. Regardless of the approach, bioconjugation reactions ideally proceed rapidly, selectively, and in high yield under physiological conditions while preserving the target protein’s biological activity. (1–4, 29–31)
Figure 1.
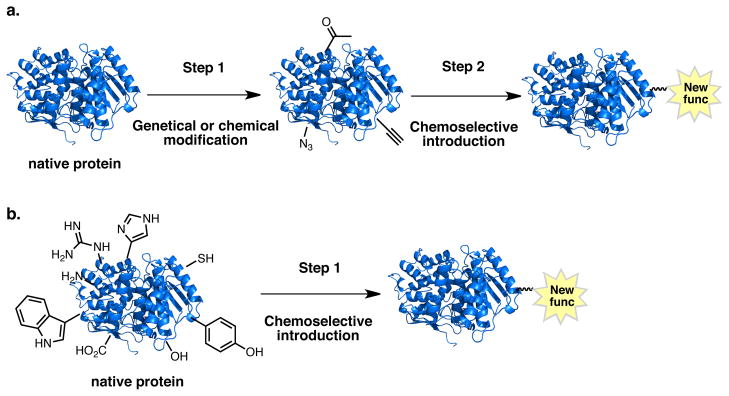
Bioconjugation reactions: a) two step extrinsic b) one step intrinsic approach.
Much of the chemistry developed to tap intrinsic amino acid reactivity is many decades old and is centered on the modification of lysine and cysteine side chains. The abundance of lysine residues on the protein surface, however, makes site-specific modification difficult. In contrast, cysteine is rare and most often found in disulfide-linked pairs in native proteins. For cysteine modification, pretreatment by reduction to cleave disulfide bonds is typically required; this is followed by reaction with a reagent like maleimide.(34) Recently, novel mono- and dibromomaleimides have expanded the potential of cysteine-targeted labeling.(35, 36) Less developed are methods for the selective modification of the aromatic amino acid side chains of tryptophan(37, 38) and tyrosine,(39–43) but a number of promising approaches have been recently reported. Among nucleophilic amino acids, tyrosine has unique reactivity due to the acidic proton of the phenol ring.(44) The alkyation or acylation reaction of tyrosine with under basic conditions proceeds at the oxygen. Under acidic conditions, an ene-like reaction occurs at a carbon atom on the aromatic ring. Bioconjugation at carbon in mild, biocompatible, metal-free conditions was reported using Mannich-type additions to imines or cerium (IV) ammonium nitrate (CAN) as an oxidation reagent.(39, 43) However, the former, the modification of tyrosine with imines formed in situ is limited by the requirement of an excess of highly reactive formaldehyde. Tryptophan and amine containing side chains and reduced disulfides form formaldehyde adducts under these reaction conditions. The latter, oxidative coupling with electron-rich aniline derivatives, results in the coupling of both tyrosine and tryptophan. An alternative tyrosine modification with cyclic imines solves this problem but often reacts in an uncontrolled way with either one or two equivalents of cyclic imine. Recently, we discovered that we could exploit the reactivity of certain diazodicarboxylate-related molecules and the intrinsic reactivity of tyrosine to create an rapid aqueous ene-type reaction, the tyrosine-click reaction.(45) As a background for the development of this reaction, it should be noted that substituted phenols react with highly reactive electrophiles such as diazodicarboxylates in organic solvents in the presence of activating protic or Lewis acid additives.(46–54) However, highly reactive diazodicarboxylate reagents decompose rapidly in aqueous media and stabile diazodicarboxyamide reagents do not react efficiently with phenols in aqueous media; thus most diazodicarboxyamides are not suitable as bioconjugation reagents.(55) Although cyclic diazodicarboxylate reagents can be activated by interaction with cationic species such as protons or metals, cyclic diazodicarboxyamides like 4-phenyl-3H-1,2,4-triazoline-3,5(4H)-dione (PTAD) are not activated by protic or Lewis acid additives.(55) Our initial study involved a survey of the reactivities and stabilities of diazodicarboxylate and diazodicarboxyamide; these studies led us to develop the tyrosine-click reaction of PTAD with the phenolic side chain of tyrosine (Scheme 1).
Scheme 1.
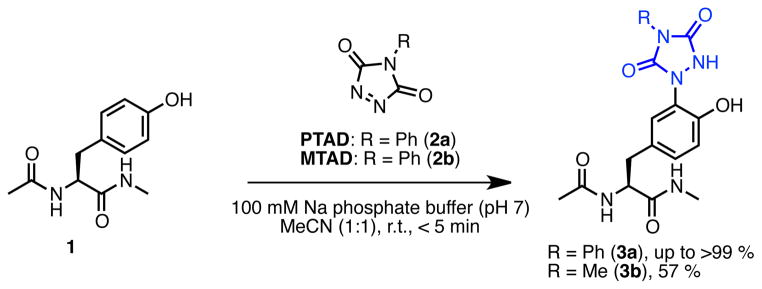
Model tyrosine click reaction
The tyrosine click reaction is illustrated in Scheme 1 involving the reaction in this case of N-acyl tyrosine methyl amide 1 with PTAD 2a in mixed organic solvent/aqueous media. In sodium phosphate buffer (pH 7)/acetonitrile (1:1), 1 reacted rapidly (reaction was complete in less than 5 min) with 1.1 equivalents of PTAD 2a to provide 3a in quantitative yield. PTAD labeling was more efficient than 4-methyl-3H-1,2,4-triazoline-3,5(4H)-dione (MTAD) labeling;(56) reaction of MTAD 2b with amide 1 gave 3b in 57% isolated yield as electronic modulation of the triazolinedione system is key to controlling this reaction.(45) Here we present a comprehensive study concerning the synthesis and characteristics of versatile triazolinedione-based labeling reagents, study of the scope, stability, and chemoselectivity of these reagents, and examine their applications in bioconjugation reactions with small molecules, peptides, and proteins.
Experimental Procedures
General
1H NMR and(44) C NMR spectra were recorded on Bruker DRX-600 (600 MHz) or Varian MER-300 (300 MHz) spectrometers in the stated solvents using tetramethylsilane as an internal standard. Chemical shifts were reported in parts per million (ppm) on the δ scale from an internal standard (NMR descriptions: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad). Coupling constants, J, are reported in Hertz. Mass spectroscopy was performed by The Scripps Research Institute Mass Spectrometer Center. Analytical thin-layer chromatography and flash column chromatography were performed on Merck Kieselgel 60 F254 silica gel plates and Silica Gel ZEOprep 60 ECO 40–63 Micron, respectively. Visualization was accomplished with anisaldehyde or KMnO4. High performance liquid chromatography (HPLC) was performed on SHIMADZU GC-8A using VYDAC 218TP C18 GRACE Column (22 mm × 250 mm) for purification and Hewlett Packard series 1100 using MCI GEL C18 Mitubishi Chemical Column (4.6 mm × 250 mm) for analysis. Unless otherwise noted, all the materials were obtained from commercial suppliers, and were used without further purification. All solvents were commercially available grade. All reactions were carried out under argon atmosphere unless otherwise mentioned. Amide starting materials, tyrosine, histidine, tryptophan, serine, cysteine, lysine, and (Ile3)-pressionoic acid, were commercially available compounds or prepared according to published procedures1). A peptide, H2N-VWSQKRHFGY-CO2H, was custom-synthesized by Abgent, Inc. Chymotrypsinogen A (MW 25kDa) (ImmunO) was used as a model protein for PEGylation.
Synthesis of 1,2,4-triazolidine-3,5-diones 8
Method A: To a 0.2 M solution of ethyl hydrazinecarboxylate 4 (1.0 eq.) in THF was added 1,1′-carbonyldiimidazole (CDI, 1.0 eq.) at room temperature. The resulting solution was stirred at room temperature. After 2 h, aniline 5 (1.0 eq.) and Et3N (2.0 eq.) were added at room temperature and stirred overnight. Then, EtOAc and 10% HCl were added. The organic layer was separated and washed once with 10% HCl and water. The resulting aqueous layer was extracted once with EtOAc. The combined organic layer was dried over MgSO4, and concentrated in vacuo. The obtained crude solid was washed with EtOAc, dried and dissolved in MeOH (0.2M solution) followed by addition of K2CO3 (3.0 eq.). The calculation was done based on the crude material. The suspension was stirred under reflux for 3 h. The reaction mixture was acidified with 12N HCl to pH 2 and then concentrated in vacuo. The generated white solids were washed with water and EtOAc to give 8. 4-(4-Propargyloxyphenyl)-1,2,4-triazolidine-3,5-dione (8a). The title compound 8a was obtained as white solid (2 steps, 28%). 1H NMR (300 MHz, DMSO-d6): δ 10.6 (br, 2H), 7.60-7.57 (m, 2H), 7.54-7.50 (m, 2H), 4.27 (s, 1H). 13C NMR (75 MHz, DMSO-d6): δ158.83, 133.36, 133.07, 126.67, 121.59, 83.81, 82.41. HRMS: calcd for C10H8N3O2 (MH+) 202.0611, found 202.0619. 4-(4-(2-Azidoethoxy)phenyl)-1,2,4-triazolidine-3,5-dione (8b). The title compound 18b was obtained as white solid (2 steps, 28%). 1H NMR (300 MHz, DMSO-d6): δ 10.4 (br, 2H), 7.36-7.33 (m, 2H), 7.07-7.03 (m, 2H), 4.21 (t, J = 6 Hz, 2H), 3.66 (t, J = 6 Hz, 2H). 13C NMR (75 MHz, DMSO-d6): δ 158.16, 154.63, 128.67, 125.85, 115.63, 68.03, 50.44. HRMS: calcd for C10H11N6O3 (MH+) 263.0887, found 263.0889. 4-(4-(2-Oxopropoxy)phenyl)-1,2,4-triazolidine-3,5-dione (8c). The title compound 8c was obtained as white solid (2 steps, 12%). This compound was purified by short column chromatography (CHCl3/MeOH). 1H NMR (300 MHz, DMSO-d6): δ 10.4 (br, 2H), 7.31-7.27 (m, 2H), 7.02-6.95 (m, 2H), 4.87 (s, 2H), 2.16 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ204.81, 157.96, 154.67, 128.57, 125.75, 115.57, 73.13, 27.16. HRMS: calcd for C11H12N3O4 (MH+) 250.0822, found 250.0826. 4-(4-Azidophenyl)-1,2,4-triazolidine-3,5-dione (8d). The title compound 8d was prepared from 4-azidoaniline hydrochloride, and was obtained as white solid (2 steps, 35%). 1H NMR (300 MHz, DMSO-d6): δ 10.5 (br, 2H), 7.50 (d, J = 9.0 Hz, 2H), 7.23 (d, J = 9.0 Hz, 2H). 13C NMR (75 MHz, DMSO-d6): δ154.25, 139.59, 129.76, 128.53, 120.47. HRMS: calcd for C8H7N6O2 (MH+) 219.0625, found 219.0617. General procedure B: To a 0.5 M solution of compound 6 (1.0 eq.) and Et3N (1.8 eq.) in THF (5 mL) was added 4-nitrophenyl chloroformate (1.8 eq.) at 0 °C. The resulting solution was stirred at room temperature overnight. Ethyl hydrazinecarboxylate 4 (2.6 eq.) and Et3N (2.6 eq.) were added at room temperature and stirred at 40 °C for 4 h. Then, EtOAc and water were added. The organic layer was separated and washed once with water. The resulting aqueous layer was combined and extracted twice with EtOAc. The combined organic layer was dried over MgSO4, and concentrated in vacuo. The obtained crude solid was washed with EtOAc, dried and dissolved in MeOH (0.2M solution) followed by addition of K2CO3 (3.0 eq.). The calculation was done based on the crude material. The suspension was stirred under reflux for 3 h. The reaction mixture was acidified with 12N HCl to pH 2 and then concentrated in vacuo. The generated white solids were washed with water and Et2O to give 8. 4-(4-Ethynylphenyl)-1,2,4-triazolidine-3,5-dione (8e). The title compound 8e was prepared from 4-ethynylaniline hydrochloride, and was obtained as white solid (2 steps, 38%). 1H NMR (300 MHz, DMSO-d6): δ 10.6 (br, 2H), 7.60-7.57 (m, 2H), 7.54-7.50 (m, 2H), 4.27 (s, 1H). 13C NMR (75 MHz, DMSO-d6): δ153.83, 133.36, 133.07, 126.67, 121.59, 83.81, 82.41. HRMS: calcd for C10H8N3O2 (MH+) 202.0611, found 202.0619. 4-(4-Acetylphenyl)-1,2,4-triazolidine-3,5-dione (8f). The title compound 8f was prepared from 4′-aminoacetophenone, and was obtained as white solid (2 steps, 15%). 1H NMR (300 MHz, DMSO-d6): δ 10.7 (br, 2H), 8.09-8.06 (m, 2H), 7.71-7.68 (m, 2H), 2.62 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ198.18, 153.67, 137.09, 136.26, 129.74, 126.17, 27.76. HRMS: calcd for C10H10N3O3 (MH+) 220.0717, found 220.0713.
Oxidation of 3H-1,2,4-triazole-3,5(4H)-diones
To a 0.05 M solution of compound 8 (1.0 eq.) in CH2Cl2 was added 1,3-dibromo-5,5-dimethylhydantoin 10 (1.0 eq.) at room temperature. The resulting solution was stirred at room temperature. After 2 h, silica sulfuric acid(57) (SiO2-OSO3H, 4 times weight to starting material) was added at room temperature and stirred at room temperature. After 30 min, the silica sulfuric acid was removed by filtration. Then, volatile materials were evaporated in vacuo to give 9. The obtained material was relatively unstable against light and humidity in solution at room temperature. Therefore, it was used for next reaction without additional purification after confirmation of purity by 1H-NMR (see SI). 4-(4-(Propargyloxy)phenyl)-3H-1,2,4-triazole-3,5(4H)-dione (9a). The title compound 9a was prepared from 8a (50.0 mg, 0.216 mmol), and was obtained as a deep red solid (42.0 mg, 85%). 1H NMR (300 MHz, CDCl3): δ 7.41-7.37 (m, 2H), 7.15-7.12 (m, 2H), 4.75 (d, J = 3.0 Hz, 2H), 3.64 (t, J = 3.0 Hz, 1H). 4-(4-(2-Azidoethoxy)phenyl)-3H-1,2,4-triazole-3,5(4H)-dione (9b). The title compound 9b was prepared from 8b (49.0 mg, 0.187 mmol), and was obtained as deep red oil (39.6 mg, 81%). 1H NMR (300 MHz, CDCl3): δ 7.40-7.35 (m, 2H), 7.10-7.06 (m, 2H), 4.20 (t, J = 3.0 Hz, 2H), 3.64 (t, J = 3.0 Hz, 2H). 4-(4-(2-Oxopropoxy)phenyl)-3H-1,2,4-triazole-3,5(4H)-dione (9c). The title compound 9c was prepared from 8c (47.0 mg, 0.189 mmol), and was obtained as deep purple solid (34.9 mg, 81%).1H NMR (300 MHz, CDCl3): δ 7.42-7.38 (m, 2H), 7.05-7.02 (m, 2H), 4.61 (s, 2H), 2.31 (s, 3H). 4-(4-Azidophenyl)-3H-1,2,4-triazole-3,5(4H)-dione (9d). The title compound 9d was prepared from 8d (40 mg, 0.183 mmol), and was obtained as deep red solid (34.1 mg, 86%). 1H NMR (300 MHz, CDCl3): δ 7.51-7.46 (m, 2H), 7.22-7.17 (m, 2H). 4-(4-Ethynylphenyl)-3H-1,2,4-triazole-3,5(4H)-dione (9e). To a solution of compound 9e (4.43 mg, 0.022 mmol) in MeCN (44 μL) was added 1,3-dibromo-5,5-dimethylhydantoin (6.29 mg, 0.022 mmol) at room temperature. The resulting solution was stirred at room temperature for 10 min. Completion of the reaction was monitored by the change of reaction solution color to a characteristic deep red color. The obtained material decomposed quickly at room temperature. Therefore, the 0.5 M MeCN reaction solution was used for next step without purification.
Synthesis of Aplaviroc-urazole
Aplaviroc-alkyne (26): To a solution of Azido-alkyne (See SI) (200 mg, 0.670 mmol) in diethylether (2 mL) was added triphenylphosphine (264 mg, 1.00 mmol) at 0 °C and stirred at room temperature for 3 hours. Then, deionized water 200 mL was added to reaction mixture and stirred for 12 h. 10 % HCl aq. was added, followed by wash with diethylether. The aqueous layer was basified to pH 10 with 5N NaOH and extracted with dichloromethane/iPrOH (4:1) 5 times. The organic layer was dried over Na2SO4, and concentrated in vacuo. The crude amine 25 was used to the next reaction without further purification. To a solution of Aplaviroc 24(58) prepared by the previously reported method (300 mg, 0.513 mmol) in DMF (10 mL) was added BOP (174 mg, 0.639 mmol), triethylamine (362 mL, 2.596 mmol) and amine-alkyne 25 (174 mg, 0.639 mmol) at room temperature and stirred for 12 hours. Then, dichloromethane were added and was separated and washed sat. NaHCO3 aq. and brine. The organic solution was dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (Dichloromethan/MeOH = 9/1) to give 26 (181 mg, 42%) as thin yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.83-7.79 (m, 2H), 7.34-7.28 (m, 2H), 7.02-6.96 (m, 5H), 6.47 (br t, 1H), 6.42 (br s, 1H), 4.01 (dd, J = 1.4, 5.7 Hz, 1H), 3.68-3.60 (m, 10H), 3.54 (t, J = 4.8 Hz, 2H), 3.46-3.38 (m, 4H), 2.97-2.84 (m, 4H), 2.74-2.69 (m, 2H), 2.54-2.47 (m, 2H), 2.41-2.27 (m, 2H). 2.20-2.07 (m, 4H), 2.02 (t, J = 2.6, 1H), 1.98-1.89 (2H), 1.73-1.53 (m, 8H), 1.40-1.13 (m, 8H), 0.96-0.85 (t, J = 7.2, 4H). 13C NMR (125 MHz, MeOD-d4): δ 173.02, 171.97, 168.41, 165.92, 160.57, 156.64, 132.35, 129.57, 119.81, 117.95, 82.69, 78.94, 70.50, 70.27, 70.23, 69.69, 69.64, 69.55, 61.42, 59.37, 56.98, 53.94, 49.77, 47.27, 42.87, 40.10, 39,98, 39.42, 36.09, 34.98, 32.17, 31.65, 31.54, 30.02, 29.63, 26.53, 26.02, 20.46, 14.75, 13.30. HRMS: calcd for C46H65N5O9 (MH+) 832.4855, found 832.4854. Aplaviroc-urazole (27): To a solution of 8b (20 mg, 0.763 mmol) and 27 (70 mg, 0.0839 (458 mL, 0.0229 mmol, 50 mM solution mmol) in tert-BuOH/H2O (3 mL/1 mL) was added THPTA(59) in H2O), Copper sulfate 5 hydrate (114 mL, 0.0229 mmol, 50 mg/mL solution in H2O) and Sodium ascorbate (91 mL, 0.0229 mmol, 50 mg/mL solution in H2O) at room temperature and stirred for 30 min. Then, chloroform was added and washed with sat. NaHCO3 aq. and brine. Combined organic layer was dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (EtOAc/MeOH = 4/1) to give 27 (43 mg, 52%) as thin yellow solid. 1H NMR (400 MHz, MeOD-d4): δ 7.82-7.80 (m, 3H), 7.45-7.43 (m, 2H), 7.26-7.24 (m, 2H), 7.03-7.01 (m, 4H), 6.97-6.95 (m, 2H), 4.74 (m, 2H), 4.48 (m, 2H), 4.13 (m, 1H), 3.95 (m, 2H), 3.68-3.60 (m, 10H), 3.54 (t, J = 4.8 Hz, 2H), 3.46-3.38 (m, 4H), 2.97-2.84 (m, 2H), 2.74-2.69 (m, 2H), 2.54-2.47 (m, 2H), 2.41-2.27 (m, 2H). 2.20-2.07 (m, 4H), 2.02 (t, J = 2.6, 1H), 1.98-1.89 (2H), 1.73-1.53 (m, 8H), 1.40-1.13 (m, 8H), 0.96-0.85 (t, J = 7.2, 4H). 13C NMR (125 MHz, MeOD-d4): δ173.51, 171.84, 168.40, 165.80, 160.35, 158.03, 157.15, 155.25, 154.88, 52.16, 46.76, 132.64, 129.53, 128.05, 123.27, 119.67, 118.08, 115.07, 79.13, 70.53, 70.27, 70.13, 69.54, 69.47, 66.93, 56.87, 49.92, 49.88, 40.1221, 39.96, 39.13, 35.30, 31.49, 26.49, 25.96, 21.46, 20.41, 13.15. HRMS: calcd for C56H75N11O12 (MH+) 1094.5669, found 1094.5660.
Synthesis of model compounds used in stability studies
1-(2-Hydroxy-5-methylphenyl)-4-phenyl-1,2,4-triazolidine-3,5-dione (29). To a solution of p-cresol (80 mg, 0.740 mmol) in THF (5 mL) was added NaH (35.5 mg, 0.885 mmol) at 0 °C. After 20 min, PTAD (127 mg, 0.725 mmol) was added at 0 °C and stirred at room temperature for 3 h. Then, EtOAc and 10% HCl were added. The organic layer was separated and washed once with brine. The resulting aqueous layer was extracted once with EtOAc. The combined organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (CHCl3/MeOH) to give 29 (158 mg, 75%) as a white solid. 1H NMR (600 MHz, DMSO-d6): δ 9.86 (br, 1H), 7.53-7.49 (m, 4H), 7.43-7.40 (m, 1H), 7.19 (d, J = 2.0 Hz, 1H), 7.10 (dd, J = 8.3, 2.0 Hz, 1H), 6.87 (d, J = 8.3 Hz, 1H), 2.24 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ151.98, 151.64, 151.46, 131.86, 130.98, 129.56, 128.80, 127.91, 127.72, 126.03, 122.89, 116.57, 19.67. HRMS: calcd for C15H14N3O3 (MH+) 284.1030, found 284.1028. Benzyl-3-(N-phenylsuccinimide)-thioether (30). To a solution of N-phenylmaleimide (2 eq., 279 mg, 1.610 mmol) in EtOH (2.85 mL)/pH7.0 HEPES buffer (2.85 mL) was added benzylthiol (1 eq., 100 uL, 0.805 mmol) in MeCN (2.85 mL) at room temperature. After 12 h, EtOAc and H2O were added. The organic layer was separated and the resulting organic layer was extracted twice with EtOAc. The combined organic layer was dried over Na2SO4, and concentrated in vacuo. The residue was precipitated and washed by Et2O/EtOAc to give 30 (140 mg, 58%) as white solids. 1H NMR (400 MHz, CDCl3): δ7.51-7.26 (m, 10H), 4.29 (d, J = 13.6 Hz, 1H), 3.90 (d, J = 13.6 Hz, 1H), 3.64 (dd, J = 3.7, 9.3 Hz, 1H), 3.16 (dd, J = 9.3, 18.9 Hz, 1H), 2.58 (dd, J = 3.7, 18.9 Hz, 1H).
Peptide modification
Labeled peptide (11a) To a 2 mM solution of custom-synthesized peptide 10 (1.82 mL, 3.82 μmol) in 100 mM pH 7.0 NaH2PO4/Na2HPO4 buffer was 100 mM solution of PTAD 9a (114 μL, 11.5 μmol) in MeCN was added (9.55 μL ×12 timies, interval 1 min.) at room temperature. The resulting solution was stirred at room temperature for 30 min. The crude reaction was analyzed directly by ESI-LC/MS at 254 nm UV absorption and corresponding MS. The reaction mixture was then diluted with MeCN (1.00 mL). The obtained crude material was purified by reversed phase HPLC (mobile phase; gradient of MeCN/0.1% TFA water, 30:70 to 50:50 over 30 min, Rt; 14.5 min, detection; UV 254 nm) to give 11a (4.40 mg, 61%) as white amorphous solid. HRMS: calcd for C73H93N21O17 (MH+) 1536.7131, found 1536.7125. Reversed phase HPLC purity 95.1 % (mobile phase; gradient of MeCN/0.1% TFA water, 0:100 to 100:0 over 30 min, Rt; 15.8 min, detection; UV 254 nm). Labeled peptide (11b). The compound 11b was prepared from custom-synthesized peptide 10 (5 mg, 3.82 μmol) and 9b (2.99 mg, 11.5 μmol), and was obtained as white amorphous solid (4.40 mg, 60%). Reverse phase HPLC conditions for isolation: mobile phase - gradient of MeCN/0.1% TFA water, 30:70 to 50:50 over 30 min, Rt 15.6 min, detection at UV 254 nm). HRMS: calcd for C72H94N24O17 (MH+) 1567.7301, found 1567.7236. Reversed phase HPLC purity 91.3 % (mobile phase;: gradient of MeCN/0.1% TFA water, 0:100 to 100:0 over 30 min, Rt 15.9 min, detection at UV 254 nm). Labeled peptide (11c). The compound 11c was prepared from custom-synthesized peptide 10 (5 mg, 3.82 μmol) and 9c (2.84 mg, 11.5 μmol), and was obtained as white amorphous solid (4.60 mg, 63%). Reverse phase HPLC conditions for isolation: mobile phase - gradient of MeCN/0.1% TFA water, 30:70 to 50:50 over 30 min, Rt 14.8 min, detection at UV 254 nm). HRMS: calcd for C73H95N21O18 (MH+) 1554.7236, found 1554.7220. Reversed phase HPLC purity 93.6 % (mobile phase: gradient of MeCN/0.1% TFA water, 0:100 to 100:0 over 30 min, Rt 15.2 min, detection at UV 254 nm).
Sequential bioconjugation of albumin using three orthogonal chemistries
(Step 1) Reaction of albumin with dansyl derivative
To a 1.5 mL centrifuge tube were added 300 μL of filtered albumin solution in H2O (pH 6.0) containing 1.5 mg albumin followed by addition of 10 μL of 140 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride in H2O (pH 6.0) and 5 μL of 136 mM 11-(dansylamino) undecanoic acid in DMSO. Reaction tubes were gently shaken at 250 rpm and incubated for 14.5 hours at 37°C. The reaction mixtures were applied to Micro Bio-Spin Chromatography Columns (Bio-Gel P-6 Gel, Bio Rad) and exchanged into nanopure water. The purified albumin construct was characterized by MALDI-TOF MS. The fluorescence signal of the modified albumins 12 and 13 were recorded by a Microplate spectrophotometer (Spectra Max Gemini; Molecular Devices) using wavelength of excitation 320 nm and emission 555 nm.
(Step 2) Reaction of dansyl-albumins with PTAD derivative
The dansyl-albumin solutions 150 μL were applied to Micro Bio-Spin Chromatography Columns (Bio-Gel P-6 Gel, Bio Rad) to exchange the buffer to 100 mM Na Phosphate buffer pH 7.4. The resulting solutions were diluted to 30 μM concentration. To a 1.5 mL centrifuge tube were added 99 μL of 30 μM of dansyl-albumin solution followed by addition of 1 μL of 100 mM PTAD solution in MeCN. Reactions were gently shaken and incubated at 25 °C for 15 minutes. The reaction mixtures were applied to Micro Bio-Spin Chromatography Columns (Bio-Gel P-6 Gel, Bio Rad) to exchange the buffer to SSC buffer pH 7.0. The purified albumin constructs (HSA: 14a, 14b and 14c or BSA: 15a, 15b and 15c) were characterized by MALDI-TOF MS.
(Step 3) Reaction of dual labeled albumins with fluorescein-5-maleimide
Labeling with fluorescein-5-maleimide (Thermo SCIENTIFIC, Catalog No.: 46130) was performed according to manufacturer’s procedure. The reaction mixtures were applied to Micro Bio-Spin Chromatography Columns (Bio-Gel P-6 Gel, Bio Rad) to exchange buffer to H2O. The purified albumin constructs was characterized by MALDI-TOF MS. The fluorescence signal of the modified albumins (HSA: 16a, 16b and 16c or BSA: 17a, 17b and 17c) were recorded by a Microplate spectrophotometer (Spectra Max Gemini; Molecular Devices) using wavelength of excitation 485 nm and emission 538 nm.
Pegylation of chymotrypsinogen A
Synthesis of PEG-urazole (22)
In the 1.5 ml Eppendorf tube were mixed 5k PEG-alkyne 21 (See SI) (15 μl of 48 mM solution in DMF, 0.72 μmoles, 1 eq) and 1,2,4-triazolidine-3,5-dione azide 20(45) (30 μl of 24 mM solution in DMF, 0.72 μmoles, 1 eq) followed by addition of a small piece of copper wire and copper sulfate (0.72 μl, 100 mM solution in DI water). The reaction mixture was vortexed gently and kept at 37 °C for 2 hrs with intermittent vortexing. Copper wire was removed and copper ions were scavenged from the reaction mixture using “CupriSorb” resin (Seachem) over night at room temperature. The Cuprisorb resin was filtered and product polymer was precipitated out with cold ether, centrifuged and ether decanted. The resulting white solid as PEG-urazole (22) was washed with cold ether two times and dried. Isolated yield 4.0 mg, 95%. MALDI-TOF MWav.= 5921.
Pegylation of Chymotrypsinogen A with PTAD-PEG
To the 0.65 ml Eppendorf tube containing 5k PEG-PTAD precursor 22 (50 μl, 10 mM solution in DMF) was added 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione (0.49 μl, 100 mM solution in DMF). The reaction mixture was vortexed gently and formation of the light cranberry red color was observed, characteristic for the presence of desired PTAD reagent. The reagent was kept on ice and used for the protein modification immediately. To the 1.5 ml Eppendorf tube containing chymotrypsinogen A (MW 25kDa purchased from ImmunO) (50 μl of 1mg/ml solution; 2 nM, 1 eq) was added freshly prepared 5k PEG-PTAD solution (2 μl of 10 mM solution in DMF, 20 nM, 10 eq). The reaction was vortexed briefly and kept at room temperature for 30 min. The product pegylated chymotrypsinogen A 23 was purified by gel filtration using 7k MWC Zeba Spin Desalting column (Pierce) and characterized by MALDI-TOF and gel electrophoresis.
Bioconjugation of herceptin with aplaviroc-PTAD
To the 0.65 ml Eppendorf tube containing urazole-Aplaviroc 27 (10 ul, 6 mM solution in DMF) was added 1,3-dibromo-5, 5-dimethylimidazolidine-2,4-dione (10 ul, 6 mM solution in DMF). The reaction mixture was vortexed gently and formation of the light cranberry red color was observed, characteristic for the presence of desired PTAD reagent. The reagent was kept on ice and used for the protein modification immediately. To the 0.65 ml Eppendorf tube containing 70 uL of 1.0 mg/mL Trastuzumab (herceptin) in 100 mM pH7.4 Na phosphate buffer was added freshly prepared 3.92 uL of 3 mM PTAD-Aplaviroc in DMF in 5 aliquots at 2-sec intervals (* use fresh pipet tip for each addition). After 15 min, the resulting solution was purified by gel filtration using 7k MWC Zeba Spin Desalting column (Pierce). The concentration of recovered protein was 0.75 mg/mL (NanoDrop, IgG mode). The increase of molecular weight of 1,376 was observed by MALDI-TOF MS analysis, that corresponds to an average 1.3 aplaviroc molecules per molecule of herceptin. Herceptin-apraviroc conjugate (28) was used for HIV neutralization assay without additional purification.
HIV neutralization assay
Replication-incompetent HIV-1 enveloped pseudovirus was generated by cotransfection of 293T cells with JR-FL HIV-1 Env-expressing plasmid and pSG3ΔEnv as previously described.(60) Serial dilutions of samples (50ul) along with wt b12, 2D7, 2G12 and an isotype control antibody, DEN3, were added to TZM-bl target cells (50ul) and preincubated for 1hr at 37°C. Following incubation 250TCID50 of pseudovirus (100ul) was added to each well and incubated at 37°C. Luciferase reporter gene expression was evaluated 48 hrs post infection. The percentage of virus neutralization at a given antibody concentration was determined by calculating the reduction in luciferase expression in the presence of antibody relative to virus-only wells. The antibody dilution causing 50% reduction (50% inhibitory concentration [IC50]) was calculated by regression analysis using GraphPad Prism.
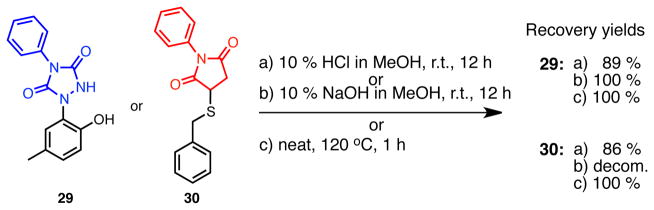
Chemical stability study
(1) Acidic or basic conditions
The solution of compound 29 or 30 (0.0353 mmol) in 10% HCl (0.5 mL) in MeOH (1.5 mL) and in 10% NaOH (0.5 mL) in MeOH (1.5 mL) was stirred at room temperature for 12 h, respectively. Then, EtOAc and water were added. In the case of basic condition, EtOAc was added after acidification with 10% HCl up to pH 3. The organic layer was separated and washed once with water. The resulting aqueous layer was extracted once with EtOAc. The combined organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (EtOAc) to recover compounds as white solids. The recovery of 29 (8.9 mg, 89 %) or 30 (9.0 mg, 86 %) under acidic conditions and 29 (10.2 mg, quant.) under basic conditions. 30 decomposed under basic hydrolysis conditions within 15 min, as monitored by TLC. The decomposed compounds were purified and analyzed by 1H-NMR and LC-MS. Hydrolysis products constituted the yield of 85 % (See the structures above).
(2) Stability study of a modified p-cresol in thermal condition
The compound 29 (4.00 mg, 0.0141 mmol) or 30 (4.00 mg, 0.0135 mmol) was heated at 120 °C for 1 h according to literature.(56) The recovery amounts were both 4.00 mg (quant.). No decomposition has been detected by 1H NMR.
(3) Physiological stability in human blood plasma:(61)
To the 2 mL Eppendorf tube containing 1,900 mL of fresh whole human blood plasma (Normal Blood Donor Program at TSRI) was added 100 mL of 1 mg/mL 29 or 30, followed by the incubation at 37 °C (Final volume: 2 mL, final concentration: 50 mg/mL, DMSO content: 5%). 100 mL Aliquots were withdrawn at the following timepoints: 3 min, 15 min, 30 min, 1h, 4h, 8h, 24h, 48h, 120h and 168h. Each 100mL aliquot was precipitated by addition of 600 mL of MeCN. Samples were centrifuged and 600 mL of supernatant were transferred into a new 1.5 mL Eppendorf tube. The solution was centrifugally evaporated followed by addition of 100 mL of 2 mg/mL methyl 4-hydroxybenzoate was added as a internal standard (IS) for the subsequent HPLC assay. 80 mL of supernatants were taken and 40 mL of each sample solution were injected and analyzed by reverse phase HPLC under optimized conditions [(Analysis time: 20 min, Flow rate: 1.0 mL/min, Column: Phenomenex (4.6 × 250), Detection wavelength: 254 nm, Isostatic: 40 to 80 % MeCN in 1% TFA/H2O), TR = 14.70 min (30), TR = 6.23 min (IS), TR = 7.40 min (29)].
Results and Discussion
In order to develop a versatile family of PTAD based conjugation chemistries, we have focused on the design, preparation, and utility of PTAD analogs possessing readily derivatizable linker arms compatible with widely used bioorthogonol coupling chemistries; click chemistry, Staudinger ligation chemistry, and oxime/hydrazone chemistry. The coupling reaction between ethyl hydrazinecarboxylate 4 and anilines 5 or 6 was performed by Method A or B (Scheme 2) depending on the nucleophilicity of aniline. The anilines used were commercially available or were synthesized from commercially available compounds by the methods detailed in Scheme 2. Method A was used in the reaction of 4 with aniline 5: After activation of 4 by treatment with CDI, aniline 5 was reacted with the activated ester in THF at room temperature to afford coupling intermediate 15. The reaction of the less nucleophilic aniline 6 was carried out via Method B. Firstly, 6 was converted to the corresponding activated ester using 4-nitrophenyl chloroformate, then reacted with 4 to afford coupling intermediate 7 in THF at room temperature. Obtained intermediate 7 was cyclized in the presence of K2CO3 in MeOH under reflux without isolation. Finally triazolidine 8 was converted to desired triazole 9 by oxidization of the N-N single bond to an N-N double bond with 1,3-diboromo-5,5-dimethylhydantoin 10 according to a literature procedure.(57) Side products derived from 10 and unreacted starting material were removed by scavenging with silica sulfuric acid (SiO2-OS3H), because PTAD products 9 were unstable during silica-gel column chromatography. The oxidation reaction was readily monitored by observing the change in the reaction mixture from colorless to deep red. Reagents, 9a, 9b, 9c, and 9d were obtained as solids or oils but solutions were relatively unstable. The products 9e and 9f were unstable to isolation at room temperature. Purity of products was confirmed by 1H-NMR.
Scheme 2.

Synthesis of PTAD derivatives
The reactivity of the PTAD analogs was evaluated in reaction with N-acyl tyrosine methyl amide 1, and results are shown in Table 1. All reactions were complete in less than 5 min based on color change, however, the reaction products were characterized after a more extended reaction time of 30 min. Electronically-neutral reagents 9d and 9e gave the same results as PTAD with comparable percent conversions and without generation of side products. In contrast, significant amounts of uncharacterized side products were generated in the reaction with electronically-poor reagent 9f. The reagents substituted with electronically-rich moieties, 9a, 9b, and 9c, efficiently modified 1. These results suggest that an electron donating substitution on the phenyl ring provides stability in water without compromising reactivity toward the phenolic side chain of tyrosine.
Table 1.
Reactivity of PTAD derivatives with N-acyl tyrosine methyl amide 1
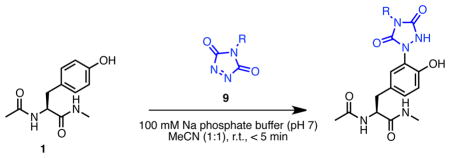
 9a
9a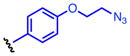 9b
9b
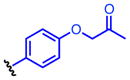 9c
9c
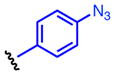 9d
9d
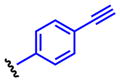 9e
9e 9f
9fReactions were performed in 100 mM NaH2PO4-Na2HPO4 (pH 7)/MeCN (1:1) at room temperature.
Percent conversion was determined by 1H-NMR.
0.5 M PTAD in MeCN mixture solution.
Reaction conditions were 100 mM NaH2PO4-Na2HPO4 (pH 7)/MeCN (1:1.5).
In order to explore the potential of the tyrosine click reaction using these reagents for labeling of more complicated targets, we synthesized dodecapeptide 10, H2N-VWSQKRHFGY-CO2H, which has a tyrosine at the C-terminus and contains the potentially reactive amino acids Trp, Ser, Glu, Lys, Arg, and His (Scheme 3). Because this peptide is designed to present amino acids with potentially reactive and competing side-chains, it serves as a stringent test of the chemoselectivity of this reaction. The reaction was performed using 3.0 equivalents of PTAD or PTAD derivative in 6% MeCN/phosphate buffer (pH 7) at room temperature. After purification using reversed-phase HPLC, labeled peptides were obtained in approximately 60% yield (Scheme 6). Significantly, a single Tyr modified compound was observed in each reaction by LC-MS (Supporting Information) and HRMS and MS/MS data indicated tyrosine-selective modification. In MS/MS analyses, products obtained using all tested PTAD analogs showed similar fragmentation patterns and all daughter ions contained the ions of modified tyrosine peptide (Supporting Information). In our initial report of this reaction,(45) we had shown that both Trp and Lys can react, albeit inefficiently, with PTAD in 50% MeCN/phosphate buffer (pH 7) at room temperature when studied in isolation. However, when N-acyl tyrosine methyl amide 1 was mixed with the corresponding Trp and Lys amino acid derivatives and reacted with PTAD under these conditions, only Tyr modification was observed. Other competitive labeling studies provided the same outcome; selective Tyr labeling (see SI, ref 45). These studies, together with the study of labeling of peptide 10 (which bears potentially competing functional groups in the same molecule) here demonstrate that the highly selective reaction of Tyr with PTAD derivatives is significantly favored under aqueous buffered conditions. As we initially reported, labeling with PTAD derivatives is effective in PBS, 2-amino-2-hydroxymethyl-propane-1,3-diol (Tris), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and a variety of other buffers. In some cases, for example where tyrosines of the target compound or protein are less accessible or reactive, we have noted that an isocyanate decomposition product of the PTAD may be formed that is promiscuous in its labeling and that the products of this type of side-reaction are observed. This is the case for chymotrypsinogen labeling with PTADs (see SI). This problem, however, is readily solved by using Tris buffer or by simply adding a small quantity of Tris to form a mixed buffered solution. The primary amine of the Tris buffer we believe then acts to scavenge the isocyanate decomposition product of PTADs to minimize production of the side-reaction product (see Supporting Information).
Scheme 3.

Tyrosine click reaction of designed decapeptide with PTAD derivatives to test the chemoselectivity and efficiency of the reaction.
Scheme 6.

Tyrosine click bioconjugation of trastuzamab antibody with a small molecule HIV entry inhibitor.
In our earlier study, we established the efficiency of protein labeling using a PTAD-rhodamine dye derivative and found that bovine serum albumin (BSA) was efficiently labeled in buffered solutions containing minimal organic cosolvent at pH ranging from 2 to 10 with 37–98% labeling efficiency (see ref. 14 SI). To explore the potential of the tyrosine click for protein multi-functionalizations, here we studied tri-functionalization at tyrosine, cysteine, and lysine residues of bovine serum albumin (BSA) and human serum albumin (HSA). BSA contains 60 lysines, 21 tyrosines, and 35 cysteines, whereas HSA has 55 lysines, 19 tyrosines, and 35 cysteines. Cysteine and lysine were modified with a fluorescein maleimide and 11-(dansylamino) undecanoic acid, respectively (Scheme 4). Labeling of albumins at lysine was performed using 50 equivalents of 11-(dansylamino) undecanoic acid and 100 equivalents of N-[3-(dimethylamino)propyl]-N′-ethylcarbodiimide hydrocholide (EDC HCl) in water at 37°C for 14.5 hours. After the gel filtration, MALDI TOF analysis revealed that the modified HSA (12) and modified BSA (13) had 3.8 and 4.1 dansyl residues, respectively (Table 2). Next, tyrosines were reacted in 1 % MeCN/100 mM phosphate buffer (pH 7.4) using 30 equivalents of PTAD derivatives 9a, 9b, and 9c gave products with 4 to 8 modified residues (Table 2). Final labeling at cysteine in SSC buffer (pH 7.0) was achieved using 1 mM fluorescein-5-maleimide in DMSO at room temperature for 2 hours (Table 2). As noted in Table 2, each of the desired labels could be efficiently installed onto the target proteins. Fluorescence properties and molecular masses of the modified BSA and HSA are given in the Supporting Information. Thus, PTAD derived reagents bearing bioorthogonal alkyne, azide, and ketone groups were readily prepared and efficiently modified small molecules, peptides, and proteins that can subsequently be further functionalized using click chemistries and other well established bioorthogonal reaction chemistries.
Scheme 4.

Triple-labeling of albumins at lysine, cysteine, and tyrosine
Table 2.
Triple-labeling of BSA and HSA through three different bioconjugation chemistries. Compounds numbers are shown in parenthesis. Estimated numbers of surface accessible amino acids (AAs) from ThermoScientific or estimated by PyMOL ver 1.5.
| Protein | Estimated number of available AAs/Total | Number of residues labeled by Amidation | Number of residues labeled by Tyrosine-click | Number of residues labeled by Michael reaction |
|---|---|---|---|---|
| HSA | Lys 30/55 | 4.1 (12) | 6.5 (14a) | 1.8 (16a) |
| Tyr 15/19 | 6.7 (14b) | 1.2 (16b) | ||
| Cys 1/35 | 4.6 (14c) | 1.6 (16c) | ||
| BSA | Lys 35/60 | 3.8 (13) | 8.3 (15a) | 1.3 (17a) |
| Tyr 15/21 | 7.4 (15b) | 2.3 (17b) | ||
| Cys 20/35 | 3.9 (15c) | 2.7 (17c) |
One of the most important protein conjugation reactions in the pharmaceutical industry concerns the introduction of poly(ethylene) glycol chains onto proteins (PEGylation) to modify their pharmacokinetic properties.(62, 63) Protein PEGylation is most commonly employed using maleimide- or N-hydroxysuccinimide-based PEGylation reagents. The high abundance of the lysine moieties on protein surfaces often results in generation of multiple PEG addition products and necessitates complicated separation procedures. Conjugation at cysteine residues typically requires the introduction of surface accessible cysteine residues by mutation of the parental protein sequence. To explore the potential of the tyrosine click reaction for protein PEGylation we prepared a 5-kDa PEG-PTAD precursor reagent 22 (Scheme 5, see details in Supporting Information). We used this reagent to study the modification of Chymotrypsinogen A, which contains four tyrosine and fourteen lysine residues. Reactions were performed in buffer (pH 7.4) with 10 equivalents of freshly oxidized PEG-PTAD or freshly dissolved PEG-NHS. Reactions were kept at room temperature for one hour with intermittent mixing. Excess reagents were removed using 7-kDa MWCO Zeba Spin Desalting columns, and the products were characterized by gel electrophoresis and MALDI-TOF MS (Supporting Information). We observed formation of mono-, bis-, tri-, and tetra- PEG addition products upon NHS conjugation and predominant formation of mono-PEG addition products upon PTAD conjugation. We have noted low reactivity of the chymotrypsinogen tyrosine residues accounting for the limited labeling of this protein (Supporting Information). Starting material was not completely consumed in either reaction but could be recovered and recycled.
Scheme 5.

Protein PEGylation with a linear PEG5000 PTAD reagent
Because of the significance of antibody drug conjugates in cancer and other therapeutic applications,(64–66) we studied the conjugation of the CCR5 antagonist aplaviroc (24) with a monoclonal antibody. As a model monoclonal antibody, we used the well-characterized antibody trastuzumab.(67) Aplaviroc was prepared as we previously reported.(58) The carboxylic acid moiety of aplaviroc was condensed with an alkyne linker (25) to give aplaviroc having alkyne moiety (26), then the click reaction with azide-urazole (9c) gave the PTAD-aplaviroc precursor (27). This precursor was oxidized to produce the PTAD moiety, then immediately used for labeling of trastuzumab in 0.1 M phosphate buffer (pH 7) at room temperature. After the removal of the small molecule by gel filtration, a product with a single aplaviroc was observed by MALDI-TOF MS spectrum (Supporting information).
In order to study the bioactivity of conjugated 28, HIV neutralization activity of the aplaviroc-trastuzumab conjugate (28) was tested in TZM-bl cells expressing CCR5 and clade B pseudovirus JR-FL as shown in Figure 3. The IC50 value of aplaviroc-trastuzumab conjugate 28 was 11.3 nM; trastuzamab alone had no neutralization activity. Interestingly, this value was very close to that of aplaviroc (24) alone (5.6 nM) indicating that the tyrosine click conjugation chemistry did not negatively impact the activity of the drug. No significant loss in trastuzumab binding was noted as determined by ELISA. This study and our earlier study(45) concerning the bioconjugation of a peptide to trastuzumab indicates that the tyrosine click reaction is applicable the preparation of antibody drug conjugates and supports a chemical approach to multi-specific antibodies.(68, 69)
Figure 3.

Stability in human plasma at various timepoints.
Peak Area is from HPLC. IS: internal standard; Ethyl 4-hydroxy benzoate
Key to many bioconjugation chemistries is the stability of the designed linkage. To further stability of the tyrosine click reaction in bioconjugation chemistry, we studied the stability of C-N linkage installed using PTAD reagents in comparison with the more commonly utilized C-S linkage provided by maleimide coupling chemistry (Scheme 7). As noted in our original communication on the PTAD adduct to p-cresol (29), it is stabile to acidic or basic conditions at room temperature for 24 hours or at 120 °C for 1 h. On the other hand, 30, containing an S-C bond, was stabile in acidic conditions, but was hydrolyzed within 5 min in basic condition, although the S-C bond was not cleaved. 30 demonstrated a thermal stability similar to 29 after heat treatment for 1 hr. This study suggests that the 1,2,4-triazolidine-3,5-dione linkage is hydrolytically and thermally stabile, whereas the maleimide linkage is unstable in basic conditions. Next, stability was evaluated in human blood plasma in anticipation of the use of the tyrosine click reaction in protein conjugates, specifically antibody-drug conjugates (ADCs).(70) For this purpose, we studied the stability of 29 and 30 by incubation in fresh human blood plasma at 37 °C for one week (Figure 3). At various time points the reaction was quenched by precipitation with MeCN and analyzed by reversed-phase HPLC (Supporting Information). Compound 29 was found to be completely stabile over the course of the 7-day experiment, while 30 decomposed after an hours. This data demonstrates that the Tyr click linkage is significantly more stabile in human blood plasma than the maleimide linkage and is consistent with reports that protein conjugates prepared with maleimide undergo maleimide exchange with reactive thiols in albumin, free cysteine or glutathione.(71, 72) Furthermore, thiol-maleimide is prone to oxidation, and this facilitates the retro-Michael reaction and subsequent decomposition.(73) In view of our studies here and reports concerning maleimide based linkages, the tyrosine click reaction provides a more robust linkage for bioconjugation than maleimide based connections.
Scheme 7.
Linkage stability study
Conclusions and Implications
The studies described herein indicate that the tyrosine click reaction of PTAD derivatives is a highly efficient and chemoselective strategy for small molecule, peptide, and protein conjugation. The reactions of PTAD and designed derivatives developed here were selective for the phenolic side chain of tyrosine residues and proceeded in buffered aqueous media over a broad pH range without the requirement of added heavy metals or other reagents. The C-N linkage that is the product of the tyrosine click reaction is stabile to extremes of pH, temperature and in human serum for extended periods of time. The stability profile of this C-N linkage is significantly better than the stability profile we determined for a model maleimide linkage. While tyrosine residues are commonly found in proteins, surface accessible tyrosines are less common and provide attractive opportunities for minimal labeling using this approach. Because this reaction is highly chemoselective for phenols, it can be applied to couple small molecules, peptides, and poly(ethylene) glycol chains (PEGylation) onto proteins without issues of self reaction provided that one coupling partner lacks free phenolic groups. This provides an attractive new strategy for the preparation of protein conjugates, including drug conjugates and PEGylated products. We expect that this new methodology and the reagents developed here will find broad utility in production of novel biomolecules, labeled peptides and proteins, and a new chemistry for protein immobilization. Through an agreement with Sigma-Aldrich, alkyne 9a is now commercially available (product number: T-511544).
Supplementary Material
Figure 2.
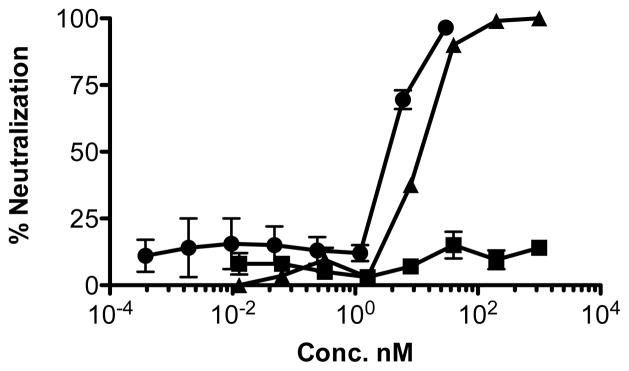
JR-FL HIV-1 pseudovirus neutralization assay: aplaviroc 24 (●), trastuzumab (■) and aplaviroc-trastuzamab 28 (▲).
Acknowledgments
Funding Sources
This study was supported by National Institutes of Health grants Pioneer Award DP1 OD006990, RO1 AI095038, and The Skaggs Institute for Chemical Biology.
We thank Prof. Dennis Burton and Angelica Cuevas for HIV-1 assays (The Scripps Research Institute). MN thanks The Uehara Memorial Foundation for a postdoctoral fellowship for research abroad. We thank Dr. Qi-Yang Hu for discussion.
ABBREVIATIONS
- CCR5
CC chemokine receptor 5
- PEG
polyethyleneglycol
Footnotes
Full experimental procedures and characterization data are available for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Best MD. Click Chemistry and Bioorthogonal Reactions: Unprecedented Selectivity in the Labeling of Biological Molecules. Biochemistry (Mosc) 2009;48:6571–6584. doi: 10.1021/bi9007726. [DOI] [PubMed] [Google Scholar]
- 2.Sletten EM, Bertozzi CR. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew Chem Int Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Devaraj NK, Weissleder R. Biomedical Applications of Tetrazine Cycloadditions. Acc Chem Res. 2011;44:816–827. doi: 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aslam M, Dent A. Bioconjugation. Protein Coupling Techniques for Biomedical Sciences Grove’s Dictionaries Inc; New York, NY: 1998. [Google Scholar]
- 5.Carrico IS, Carlson BL, Bertozzi CR. Introducing genetically encoded aldehydes into proteins. Nat Chem Biol. 2007;3:321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
- 6.Cornish VW, Hahn KM, Schultz PG. Site-Specific Protein Modification Using a Ketone Handle. J Am Chem Soc. 1996;118:8150–8151. [Google Scholar]
- 7.Rose K, Vizzavona J. Stepwise Solid-Phase Synthesis of Polyamides as Linkers. J Am Chem Soc. 1999;121:7034–7038. [Google Scholar]
- 8.O’Shannessy DJ, Dobersen MJ, Quarles RH. A novel procedure for labeling immunoglobulins by conjugation to oligosaccharide moieties. Immunol Lett. 1984;8:273–277. doi: 10.1016/0165-2478(84)90008-7. [DOI] [PubMed] [Google Scholar]
- 9.Mahal LK, Yarema KJ, Bertozzi CR. Engineering Chemical Reactivity on Cell Surfaces Through Oligosaccharide Biosynthesis. Science. 1997;276:1125–1128. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- 10.Hang HC, Bertozzi CR. Ketone Isosteres of 2-N-Acetamidosugars as Substrates for Metabolic Cell Surface Engineering. J Am Chem Soc. 2001;123:1242–1243. doi: 10.1021/ja002962b. [DOI] [PubMed] [Google Scholar]
- 11.Cohen JD, Zou P, Ting AY. Site-Specific Protein Modification Using Lipoic Acid Ligase and Bis-Aryl Hydrazone Formation. ChemBioChem. 2012;13:888–894. doi: 10.1002/cbic.201100764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rashidian M, Song JM, Pricer RE, Distefano MD. Chemoenzymatic Reversible Immobilization and Labeling of Proteins without Prior Purification. J Am Chem Soc. 2012;134:8455–8467. doi: 10.1021/ja211308s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saxon E, Bertozzi CR. Cell Surface Engineering by a Modified Staudinger Reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 14.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. Bioconjugation by Copper(I)-Catalyzed Azide-Alkyne [3 + 2] Cycloaddition. J Am Chem Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 16.Agard NJ, Prescher JA, Bertozzi CR. A Strain-Promoted [3 + 2] Azide-Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 17.Ning X, Guo J, Wolfert MA, Boons GJ. Visualizing Metabolically Labeled Glycoconjugates of Living Cells by Copper-Free and Fast Huisgen Cycloadditions. Angew Chem Int Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jewett JC, Bertozzi CR. Cu-free click cycloaddition reactions in chemical biology. Chem Soc Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Suarez M, Baruah H, Martinez-Hernandez L, Xie KT, Baskin JM, Bertozzi CR, Ting AY. Redirecting lipoic acid ligase for cell surface protein labeling with small-molecule probes. Nat Biotech. 2007;25:1483–1487. doi: 10.1038/nbt1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dierks T, Lecca MR, Schlotterhose P, Schmidt B, von Figura K. Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J. 1999;18:2084–2091. doi: 10.1093/emboj/18.8.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rush JS, Bertozzi CR. New Aldehyde Tag Sequences Identified by Screening Formylglycine Generating Enzymes in Vitro and in Vivo. J Am Chem Soc. 2008;130:12240–12241. doi: 10.1021/ja804530w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blackman ML, Royzen M, Fox JM. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels-Alder Reactivity. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor MT, Blackman ML, Dmitrenko O, Fox JM. Design and Synthesis of Highly Reactive Dienophiles for the Tetrazine-trans-Cyclooctene Ligation. J Am Chem Soc. 2011;133:9646–9649. doi: 10.1021/ja201844c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sletten EM, Bertozzi CR. A Bioorthogonal Quadricyclane Ligation. J Am Chem Soc. 2011;133:17570–17573. doi: 10.1021/ja2072934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noren C, Anthony-Cahill S, Griffith M, Schultz P. A general method for site-specific incorporation of unnatural amino acids into proteins. Science. 1989;244:182–188. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- 26.Bain JD, Diala ES, Glabe CG, Dix TA, Chamberlin AR. Biosynthetic site-specific incorporation of a non-natural amino acid into a polypeptide. J Am Chem Soc. 1989;111:8013–8014. [Google Scholar]
- 27.Wang L, Schultz PG. Expanding the Genetic Code. Angew Chem Int Ed. 2005;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- 28.Ellman J, Mendel D, Anthony-Cahill S, Noren CJ, Schultz PG, John JL. Methods Enzymol. Academic Press; 1991. [15] Biosynthetic method for introducing unnatural amino acids site-specifically into proteins; pp. 301–336. [DOI] [PubMed] [Google Scholar]
- 29.Wong SS. Chemistry of Protein Conjugation and Cross-Linking. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- 30.Lundblad RL. Chemical Reagents for Protein Modification. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- 31.Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego, CA: 1996. [Google Scholar]
- 32.Seitchik JL, Peeler JC, Taylor MT, Blackman ML, Rhoads TW, Cooley RB, Refakis C, Fox JM, Mehl RA. Genetically Encoded Tetrazine Amino Acid Directs Rapid Site-Specific in Vivo Bioorthogonal Ligation with trans-Cyclooctenes. J Am Chem Soc. 2012;134:2898–2901. doi: 10.1021/ja2109745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, Chin JW. Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction. Nat Chem. 2012;4:298–304. doi: 10.1038/nchem.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim Y, Ho SO, Gassman NR, Korlann Y, Landorf EV, Collart FR, Weiss S. Efficient Site-Specific Labeling of Proteins via Cysteines. Bioconjugate Chem. 2008;19:786–791. doi: 10.1021/bc7002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones MW, Strickland RA, Schumacher FF, Caddick S, Baker JR, Gibson MI, Haddleton DM. Polymeric Dibromomaleimides As Extremely Efficient Disulfide Bridging Bioconjugation and Pegylation Agents. J Am Chem Soc. 2012;134:1847–1852. doi: 10.1021/ja210335f. [DOI] [PubMed] [Google Scholar]
- 36.Smith MEB, Schumacher FF, Ryan CP, Tedaldi LM, Papaioannou D, Waksman G, Caddick S, Baker JR. Protein Modification, Bioconjugation, and Disulfide Bridging Using Bromomaleimides. J Am Chem Soc. 2010;132:1960–1965. doi: 10.1021/ja908610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antos JM, Francis MB. Selective Tryptophan Modification with Rhodium Carbenoids in Aqueous Solution. J Am Chem Soc. 2004;126:10256–10257. doi: 10.1021/ja047272c. [DOI] [PubMed] [Google Scholar]
- 38.Antos JM, McFarland JM, Iavarone AT, Francis MB. Chemoselective Tryptophan Labeling with Rhodium Carbenoids at Mild pH. J Am Chem Soc. 2009;131:6301–6308. doi: 10.1021/ja900094h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joshi NS, Whitaker LR, Francis MB. A Three-Component Mannich-Type Reaction for Selective Tyrosine Bioconjugation. J Am Chem Soc. 2004;126:15942–15943. doi: 10.1021/ja0439017. [DOI] [PubMed] [Google Scholar]
- 40.McFarland JM, Joshi NS, Francis MB. Characterization of a Three-Component Coupling Reaction on Proteins by Isotopic Labeling and Nuclear Magnetic Resonance Spectroscopy. J Am Chem Soc. 2008;130:7639–7644. doi: 10.1021/ja710927q. [DOI] [PubMed] [Google Scholar]
- 41.Minakawa M, Guo HM, Tanaka F. Imines that React with Phenols in Water over a Wide pH Range. J Org Chem. 2008;73:8669–8672. doi: 10.1021/jo8017389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kodadek T, Duroux-Richard I, Bonnafous JC. Techniques: Oxidative cross-linking as an emergent tool for the analysis of receptor-mediated signalling events. Trends Pharmacol Sci. 2005;26:210–217. doi: 10.1016/j.tips.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 43.Seim KL, Obermeyer AC, Francis MB. Oxidative Modification of Native Protein Residues Using Cerium(IV) Ammonium Nitrate. J Am Chem Soc. 2011;133:16970–16976. doi: 10.1021/ja206324q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hermans J, Leach SJ, Scheraga HA. Thermodynamic Data from Difference Spectra. 1,2 II Hydrogen Bonding in Salicylic Acid and its Implications for Proteins. J Am Chem Soc. 1963;85:1390–1395. [Google Scholar]
- 45.Ban H, Gavrilyuk J, Barbas CF. Tyrosine Bioconjugation through Aqueous Ene-Type Reactions: A Click-Like Reaction for Tyrosine. J Am Chem Soc. 2010;132:1523–1525. doi: 10.1021/ja909062q. [DOI] [PubMed] [Google Scholar]
- 46.Schroeter SH. Reaction of phenols with ethyl azodicarboxylate. The Journal of Organic Chemistry. 1969;34:4012–4015. [Google Scholar]
- 47.Mitchell H, Leblanc Y. Amination of Arenes with Electron-Deficient Azodicarboxylates. The Journal of Organic Chemistry. 1994;59:682–687. [Google Scholar]
- 48.Leblanc Y, Boudreault N. Para-Directed Amination of Electron-Rich Arenes with Bis(2,2,2-Trichloroethyl) Azodicarboxylate. The Journal of Organic Chemistry. 1995;60:4268–4271. [Google Scholar]
- 49.Yadav JSR, Kumar BVS, Madan GMC. InCl3-SiO2 Catalyzed Electrophilic Amination of Arenes: A Facile and Rapid Synthesis of Aryl Hydrazides. Synlett. 2001;11:1781–1783. [Google Scholar]
- 50.Bombek S, Lenarsic R, Kocevar M, Saint-Jalmes L, Desmurs J-R, Polanc S. ZrCl4-promoted halogen migration during an electrophilic amination of halogenated phenols. Chem Commun. 2002:1494–1495. doi: 10.1039/b203622c. [DOI] [PubMed] [Google Scholar]
- 51.Yadav JS, Reddy BVS, Veerendhar G, Rao RS, Nagaiah K. Sc(OTf)3 Catalyzed Electrophilic Amination of Arenes: An Expeditious Synthesis of Aryl Hydrazides. Chem Lett. 2002;31:318–319. [Google Scholar]
- 52.Kinart WJ, Kinart CM. Studies on the catalysis of the reaction of organotin phenoxides with diethyl azodicarboxylate by lithium perchlorate. J Organomet Chem. 2003;665:233–236. [Google Scholar]
- 53.Chee GL. Efficient Synthesis of Bifenazate. Synth Commun. 2006;36:2151–2156. [Google Scholar]
- 54.Brandes S, Bella M, Kjærsgaard A, Jørgensen KA. Chirally Aminated 2-Naphthols—Organocatalytic Synthesis of Non-Biaryl Atropisomers by Asymmetric Friedel–Crafts Amination. Angew Chem Int Ed. 2006;45:1147–1151. doi: 10.1002/anie.200503042. [DOI] [PubMed] [Google Scholar]
- 55.Desimoni G, Faita G, Righetti PP, Sfulcini A, Tsyganov D. Solvent effect in pericyclic reactions. IX The ene reaction. Tetrahedron. 1994;50:1821–1832. [Google Scholar]
- 56.Baran PS, Guerrero CA, Corey EJ. The First Method for Protection-Deprotection of the Indole 2,3-π Bond. Org Lett. 2003;5:1999–2001. doi: 10.1021/ol034634x. [DOI] [PubMed] [Google Scholar]
- 57.Zolfigol MA, Nasr-Isfahanib H, Mallakpourc S, Safaiee M. Oxidation of Urazoles with 1,3-Dihalo-5,5-dimethylhydantoin, both in Solution and under Solvent-Free Conditions. Synlett. 2005;5:761–764. [Google Scholar]
- 58.Gavrilyuk J, Uehara H, Otsubo N, Hessell A, Burton DR, Barbas CF. Potent Inhibition of HIV-1 Entry with a Chemically Programmed Antibody Aided by an Efficient Organocatalytic Synthesis. ChemBioChem. 2010;11:2113–2118. doi: 10.1002/cbic.201000432. [DOI] [PubMed] [Google Scholar]
- 59.Hong V, Presolski SI, Ma C, Finn MG. Analysis and Optimization of Copper-Catalyzed Azide–Alkyne Cycloaddition for Bioconjugation. Angew Chem Int Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. Human Immunodeficiency Virus Type 1 env Clones from Acute and Early Subtype B Infections for Standardized Assessments of Vaccine-Elicited Neutralizing Antibodies. J Virol. 2005;79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Imanishi M. Design of Artificial DNA Binding Proteins toward Control and Elucidation of Cellular Functions. Yakaugaku Zasshi. 2012;132:1431–1436. doi: 10.1248/yakushi.12-00228. [DOI] [PubMed] [Google Scholar]
- 62.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 63.Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2002;54:459–476. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 64.Lyon RP, Meyer DL, Setter JR, Senter PD, Wittrup KD, Gregory LV. Methods Enzymol. Academic Press; 2012. Chapter six - Conjugation of Anticancer Drugs Through Endogenous Monoclonal Antibody Cysteine Residues; pp. 123–138. [DOI] [PubMed] [Google Scholar]
- 65.Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol. 2005;14:529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- 66.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotech. 2005;23:1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 67.Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J. Recombinant humanized anti-HER2 antibody (Herceptin (TM)) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 1998;58:2825–2831. [PubMed] [Google Scholar]
- 68.Wuellner U, Gavrilyuk JI, Barbas CF., 3rd Expanding the Concept of Chemically Programmable Antibodies to RNA Aptamers: Chemically Programmed Biotherapeutics. Angew Chem Int Ed Engl. 2010;49:5934–5937. doi: 10.1002/anie.201001736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gavrilyuk JI, Wuellner U, Salahuddin S, Goswami RK, Sinha SC, Barbas CF., III An efficient chemical approach to bispecific antibodies and antibodies of high valency. Bioorg Med Chem Lett. 2009;19:3716–3720. doi: 10.1016/j.bmcl.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blättler WA, Lambert JM, Chari RVJ, Lutz RJ, Wong WLT, Jacobson FS, Koeppen H, Schwall RH, Kenkare-Mitra SR, Spencer SD, Sliwkowski MX. Targeting HER2-Positive Breast Cancer with Trastuzumab-DM1, an Antibody–Cytotoxic Drug Conjugate. Cancer Res. 2008;68:9280–9290. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 71.Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, Li D, Tibbitts J, Baudys J, Saad OM, Scales SJ, McDonald PJ, Hass PE, Eigenbrot C, Nguyen T, Solis WA, Fuji RN, Flagella KM, Patel D, Spencer SD, Khawli LA, Ebens A, Wong WL, Vandlen R, Kaur S, Sliwkowski MX, Scheller RH, Polakis P, Junutula JR. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotech. 2012;30:184–189. doi: 10.1038/nbt.2108. [DOI] [PubMed] [Google Scholar]
- 72.Baldwin AD, Kiick KL. Tunable Degradation of Maleimide-Thiol Adducts in Reducing Environments. Bioconjugate Chem. 2011;22:1946–1953. doi: 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lewis MR, Shively JE. Maleimidocysteineamido-DOTA Derivatives: New Reagents for Radiometal Chelate Conjugation to Antibody Sulfhydryl Groups Undergo pH-Dependent Cleavage Reactions. Bioconjugate Chem. 1998;9:72–86. doi: 10.1021/bc970136v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.