

Abstract
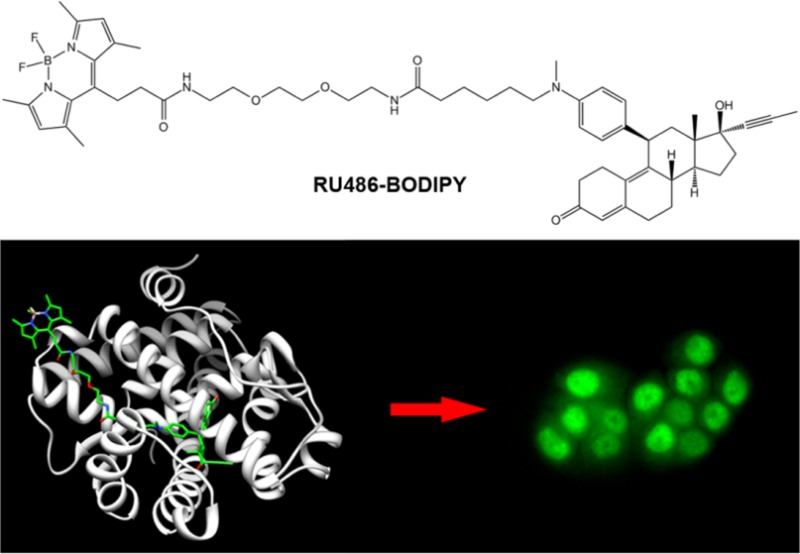
We employed molecular modeling to design and then synthesize fluorescent ligands for the human progesterone receptor. Boron dipyrromethene (BODIPY) or tetramethylrhodamine were conjugated to the progesterone receptor antagonist RU486 (Mifepristone) through an extended hydrophilic linker. The fluorescent ligands demonstrated comparable bioactivity to the parent antagonist in live cells and triggered nuclear translocation of the receptor in a specific manner. The BODIPY labeled ligand was applied to investigate the dependency of progesterone receptor nuclear translocation on partner proteins and to show that functional heat shock protein 90 but not immunophilin FKBP52 activity is essential. A tissue distribution study indicated that the fluorescent ligand preferentially accumulates in tissues that express high levels of the receptor in vivo. The design and properties of the BODIPY-labeled RU486 make it a potential candidate for in vivo imaging of PR by positron emission tomography through incorporation of 18F into the BODIPY core.
The progesterone receptor (PR) is a ligand-activated steroid receptor that belongs to the nuclear receptor superfamily of transcription factors.1,2 PR is expressed at low levels in most physiological systems but peaks in the female reproductive system and in the central nervous system.3 Thus, it plays a central role in reproductive events and sexual behavior. PR dysfunction has been indicated in multiple disorders including reproductive conditions,4 neurological syndromes,5 and cancer (breast,6 ovarian,7 endometrial8). As such, considerable effort has been focused on understanding PR functions and their underlying mechanisms in normal and pathological conditions. The human PR is encoded by a single gene that is expressed as two isoforms, PR-A and PR-B, which share most of the functional elements but have distinct functions. While PR-A remains predominantly in the nucleus, PR-B resides mostly in the cytosol as part of a multiprotein complex, which modulates its activity. According to current understanding, upon ligand binding PR-B dissociates from at least part of the complex, dimerizes, and translocates to the nucleus, where it recruits coregulating proteins and binds specific DNA sequences to exert its transcriptional effect. Recently, fusions of fluorescent protein tags to PR and its regulators have enabled their imaging with high spatial and temporal resolution, significantly improving understanding of dynamic processes such as localization, cell cycle dependence, and recycling.9−11 However, this approach requires genetic manipulation, expression of non-native PR, and often the use of cells that do not express PR endogenously. Complementary to receptor labeling, fluorescent ligands offer advantages such as receptor imaging in endogenously expressing cells, quantification of ligand–receptor interactions, and measurement of receptor–ligand complex diffusion rates.12 While biologically functional fluorescent ligands for many G protein-coupled receptors,13 retinoic acid receptor,14 and estrogen receptor15 have been reported, efforts to develop fluorescent ligands for PR were either unsuccessful16 or have not been applied to receptor imaging.17,18 The only functional fluorescent PR-ligand in mammalian cells was reported almost a decade ago, when fluorescein labeled RU486 (Mifepristone), a PR antagonist, was demonstrated to concentrate in the nuclei of PR expressing cells.19 However, it required prolonged incubation time and cells had to be fixed prior to imaging. Recently, an elegant procedure for fluorine displacement in boron-dipyrromethene (BODIPY) dyes has been described20 which was later used to introduce a 18F radioisotope into a BODIPY scaffold to generate a dual fluorescence/positron emission tomography (PET) imaging reagent.21 Other chemistries for rapid incorporation of a PET isotope into a strong fluorophore exist, e.g., a near-infrared-absorbing cyanine dye with a pendant fluoborate,22 but the size of that dye and its polar substituents would probably prevent membrane permeation. With this in mind, we sought to develop a PR fluorescent ligand based on a BODIPY dye that could be used for fluorescent imaging of PR in vitro and potentially be translated into a PET tracer for PR imaging in vivo, without modifying the original structure.
RU486 is a synthetic 19-nor steroid that acts as a competitive antagonist to PR (Figure 1). It has high affinity for PR (∼5 times lower Kd compared to the natural agonist progesterone23), and upon binding to PR, it preserves many of the processes initiated by progesterone binding, i.e., dissociation of PR from the multiprotein complex, dimerization, translocation to the nucleus, and DNA binding. The main functional difference is the inability of the receptor to recruit coactivators required for transcriptional activation when bound to RU486.24 These attributes make RU486 an attractive PR ligand for fluorescence labeling. Additionally, RU486 can tolerate various modifications of the dimethylamino group without significantly compromising its binding affinity and biological activity.25 This property has been recently exploited to develop an RU486-based MRI contrast agent.26 Therefore, we designed a BODIPY-labeled RU486, where the dye is separated from the ligand by a linker, intended to decrease both steric hindrance from the bulky dye as well as hydrophobicity of the conjugate (RU486-BODIPY, Figure 1a). For labeling, we chose a BODIPY structure that was demonstrated to be amenable to 18F introduction.21 Molecular docking of BODIPY-labeled RU486 with human PR showed that the labeled ligand is oriented similarly to unlabeled RU486 inside the binding pocket and that the linker extends outward through the binding pocket access channel (Figure 1b). Important contacts between the ligand and key amino acids are maintained for the labeled ligand (SI Figure S1). The model also predicted that the linker is sufficiently long (16 atoms from the aniline nitrogen to the BODIPY attachment point) to place the bulky BODIPY well outside the protein, minimizing its steric hindrance (Figure 1c). To test whether this labeling strategy might be extended to other dyes and bulky groups, we labeled RU486 with 5-carboxytetramethylrhodamine (RU486-TAMRA, Figure 1a) in a similar design. The conjugates were prepared by first oxidative N-demethylation of RU486, followed by alkylation with 6-bromohexanoic acid. Then, N-Boc-2,2′-(ethylenedioxy)diethylamine was conjugated to the carboxylic acid, followed by TFA-mediated Boc-deprotection and conjugation to the respective dye (6% and 7% overall yields for RU486-BODIPY and RU486-TAMRA, respectively; SI Scheme S1).
Figure 1.
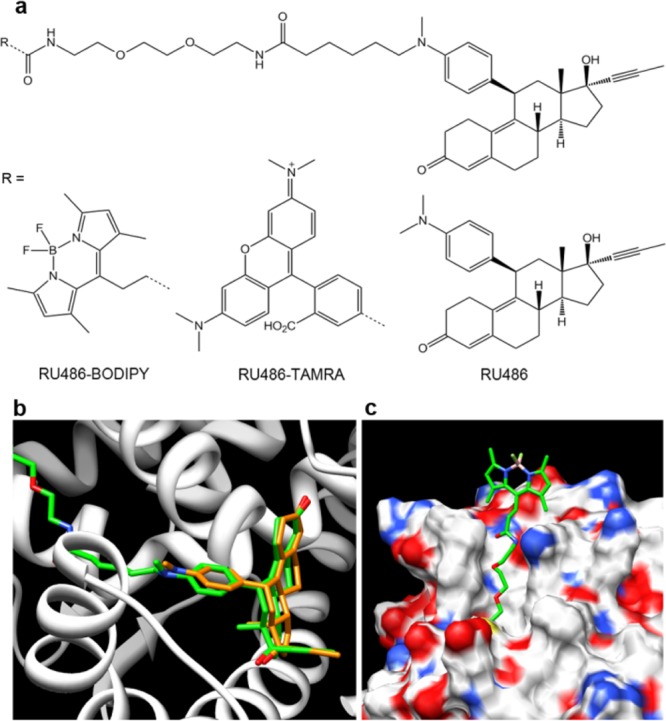
Fluorescent ligands of progesterone receptor. (a) Chemical structures of RU486 and its fluorescently labeled derivatives RU486-BODIPY and RU486-TAMRA. Molecular modeling of RU486-BODIPY (green) bound to human PR showing (b) orientation inside the ligand binding pocket compared to RU486 (orange) (amino acids 712–720 have been removed for clarity) and (c) the linker extends out of the binding pocket, placing the BODIPY dye well outside the protein shell.
The antagonistic activity of the ligands was assessed by their ability to inhibit progesterone-induced increase in alkaline phosphatase activity in intact T47D cells.27 T47D are human hormone-dependent epithelial cells isolated from ductal carcinoma of the breast, which naturally express high level of PR. While the potency of RU486-TAMRA was ∼5-fold lower than RU486, that of RU486-BODIPY was ∼1.5-fold higher (Table 1 and SI Figure S2). The difference in potency between the two fluorescent ligands might arise from their distinct hydrophobic/hydrophilic nature (Table 1). The higher polarity of RU486-TAMRA could hinder its membrane permeability, leading to lower intracellular effective concentration. Conversely, the BODIPY dye could be engaged in additional hydrophobic interactions with the receptor, slightly increasing its affinity. The labeled ligands showed spectroscopic properties characteristic of their respective dyes (Table 1 and SI Figure S3a,b). Interestingly, the quantum yield of both compounds increased in a dose-dependent manner when bovine serum albumin (BSA) was added to the measurement buffer (0.04 up to 0.29 and 0.04 up to 0.08 for RU486-BODIPY and RU486-TAMRA, respectively; SI Figure S3c), suggesting that the ligand’s brightness might also increase when bound to the receptor. Based on this BSA-dependent increase in quantum yield, the binding constants of the ligands to BSA were measured to be K = 6349 ± 544 and 31 348 ± 2063 M–1 (RU486-BPDIPY and RU486-TAMRA, respectively; SI Figure S3d). Taken together, these results show that RU486-BODIPY and RU486-TAMRA can bind PR as high affinity antagonists with spectroscopic properties suitable for fluorescence imaging.
Table 1. Antagonistic and Spectroscopic Properties of RU486 and Its Fluorescent Derivatives.
| IC50a | RBAb | log Pc | λexd | λemd | Φfl | |
|---|---|---|---|---|---|---|
| RU486 | 1.70 ± 0.24 | 1 | (4.72) | |||
| RU486-BODIPY | 1.10 ± 0.22 | 1.5 | 3.51 ± 0.24 | 495 | 505 | 0.04–0.29 |
| RU486-TAMRA | 7.55 ± 1.34 | 0.2 | 0.95 ± 0.14 | 550 | 578 | 0.04–0.08 |
Determined from dose–response curve of progesterone-induced alkaline phosphatase activity inhibition. Units are in nM.
Relative biological activity as determined by ratio of IC50(RU486) to IC50 of tested compound.
Measured using shake flask method. In parentheses: from DrugBank [DB00834].
Units are in nm.
Next, we evaluated the fluorescent ligands for imaging endogenously expressed PR in live cells. In T47D cells incubated with 5 nM RU486-BODIPY, fluorescence was almost entirely confined to the nuclei and excluded from the nucleoli (Figure 2a). Low levels of fluorescence were also detectable in the cytoplasm. Interestingly, the nuclear distribution of the fluorescent ligand was retained even 24 h after a brief incubation (SI Figure S4). This nuclear accumulation can be reversed by applying unlabeled RU486 (SI Figure 5), thus representing specific binding, whose persistence may be a result of the inhibitory effect of antagonist binding on PR processing.28 Staining with anti-human PR antibody revealed colocalization of RU486-BODIPY and PR, along with a positive correlation between the fluorescent ligand accumulation level and the antibody labeling intensity (SI Figure S6). Incubation with increasing concentrations of RU486-BODIPY revealed that above 5 nM, nonspecific accumulation starts to appear, eventually surpassing the nuclear signal (SI Figure S7). When MDA-MB-231, an epithelial breast cancer cell line that does not express PR, was similarly treated with RU486-BODIPY, fluorescence was completely excluded from the nuclei but was observed in the cytoplasm (Figure 2a). The cytoplasmic retention of RU486-BODIPY in the absence of its target binding site (i.e., PR) represents nonspecific binding which is probably a result of the molecule’s hydrophobicity (log P = 3.5). Another possible consequence of the hydrophobicity of RU486-BODIPY is the extended time required for PR nuclear translocation process to complete (∼1 h). Antiprogestins, such as RU486, have been found to bind to both the PR and the glucocorticoid receptor (GR) with high affinity. We therefore tested the specificity of RU486-BODIPY nuclear accumulation in T47D cells by competing it with 20-fold excess of either progesterone (PR selective) or dexamethasone (GR selective). While excess progesterone completely inhibited accumulation of fluorescence in the nuclei, dexamethasone had no observable effect (Figure 2b), demonstrating the specificity of the fluorescent ligand to PR in this experimental setting. In addition, this result establishes that RU486-BODIPY binds PR through the ligand binding domain (LBD) and not through allosteric sites.
Figure 2.
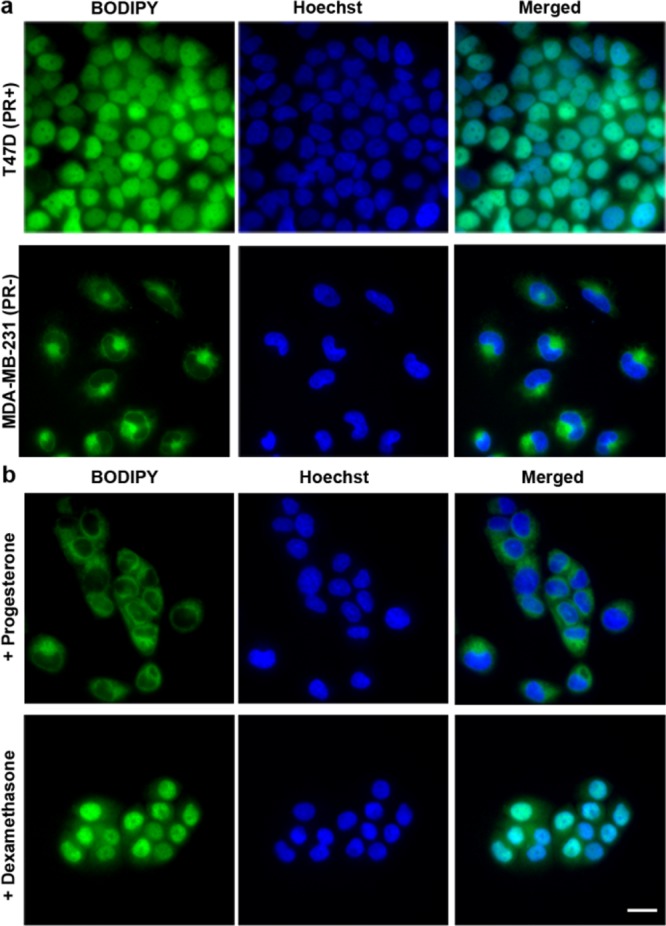
RU486-BODIPY nuclear accumulation is PR dependent. (a) RU486-BODIPY accumulates in the nuclei of PR positive cells but not PR negative cells. T47D (PR positive) or MDA-MB-231 (PR negative) cells were incubated with 5 nM RU486-BODIPY for 15 min, washed, and imaged after 45 min. (b) Nuclear accumulation of RU486-TAMRA in T47D cells can be competed off with PR agonist but not with GR agonist. T47D cells were coincubated with 5 nM RU486-BODIPY and 100 nM progesterone (PR agonist) or dexamethasone (GR agonist) for 15 min, washed, and imaged after 45 min. Scale bar 20 μm.
RU486-TAMRA showed similar accumulation patterns as RU486-BODIPY, concentrating in the nuclei of T47D cells but not in MDA-MB-231 cells. Nuclear localization was similarly specific to PR and persisted for at least 24 h (SI Figure S8a–c). In contrast to RU486-BODIPY’s tendency to accumulate in membranes in the absence of PR, RU486-TAMRA was easily washed out, maintaining a high ratio of nuclear-to-cytosol fluorescence even at high concentrations (SI Figure S9), probably due to its higher hydrophilicity. In addition, it accumulated in the nucleus at a much faster rate than RU486-BODIPY (∼18 min, SI Figure S10). However, a higher concentration was required to observe its effect (SI Figure S9). Altogether, these results demonstrate that the fluorescent ligands specifically bind human PR in T47D cells, causing PR to translocate to the nucleus and slow down the receptor’s recycling process, thus mimicking the biological effects of unlabeled RU486.
After establishing that the fluorescent ligands retain many of the biological properties of RU486, we applied RU486-BODIPY to study the dependency of PR nuclear translocation process on proteins involved in its complex. In vitro assembly studies established the identity of the proteins required for a functional PR complex as well as the order and stoichiometry of their assembly.29 Heat shock protein 90 (HSP90) is a molecular chaperone involved in many cellular processes and is a key component in PR complexes. In addition to stabilizing PR in the cytoplasm, HSP90 potentiates PR’s hormone-dependent response by binding to its LBD, causing it to adopt an open conformation that allows the hormone to enter and bind.30 This process is ATP-dependent31 and was demonstrated in binding assays to be inhibited by geldanamycin,32 a specific inhibitor of HSP90 ATPase domain. Therefore, we first tested whether 17-AAG (17-N-allylamino-17-demethoxygeldanamycin), a less toxic synthetic derivative of geldanamycin, interferes with RU486-BODIPY nuclear accumulation. Indeed, in T47D cells treated with 17-AAG for one hour prior to RU486-BODIPY application, nuclear fluorescence accumulation was inhibited in a dose-dependent manner (Figure 3a,b). The half-maximal effective concentration (EC50) of 17-AAG for PR nuclear translocation inhibition in this experimental setting was 76 ± 11 nM, much lower than its reported IC50 in T47D cells (3.82 ± 0.97 μM upon 24 h treatment33). Inhibition of histone deacetylases (HDAC) by broad spectrum inhibitors such as vorinostat (SAHA) and panobinostat (LBH-589), which leads to HSP90 hyper-acetylation and dysfunction,34 also resulted in a marked decrease of nuclear fluorescence accumulation (Figure 3c and SI Figure S11). Together, these results reinforce the importance of a functional HSP90 to PR translocation and demonstrate that RU486-BODIPY is effective in sensing perturbations to this process. HSP70 is another chaperone involved in the early steps of PR complex assembly.35 VER155008, a recently reported specific HSP70 inhibitor,36 failed to disrupt PR translocation even after 12 h of treatment. On the other hand, methylene blue, also reported to inhibit HSP70,37 significantly decreased the rate of PR translocation. However, methylene blue is not HSP70-specific; hence its effect on PR cannot be solely attributed to HSP70 inhibition. FKBP52 (FK506-binding protein 4) is an immunophilin thought to act in steroid-receptor complexes, including PR, as an adapter to the motor protein dynein to facilitate receptor shuttling along cytoskeletal tracks.38 Inhibition of FKBP52 PPIase activity (peptidylprolyl isomerase) by FK506 has been demonstrated to block PR hormone-dependent transcription activation.39 We therefore tested the effect of FKBP52-dynein inhibition on PR trafficking. Pretreatment of T47D cells with FK506 (10 μM, 12 h) had no effect on RU486-BODIPY nuclear accumulation. In addition, inhibition of dynein by erythro-9-amino-β-hexyl-α-methyl-9H-purine-9-ethanol (EHNA, 500 μM, 1 h) also failed to affect PR translocation. In combination, while these observations do not rule out PR trafficking along cytoskeletal tracks, they imply that nuclear translocation does not exclusively rely on active cytoskeletal transport and highlight the need to clarify the role of active movement machinery in PR complex shuttling. In addition, they also suggest that the requirement for FKBP52 activity in PR transcriptional activity is downstream of the nuclear translocation process.
Figure 3.

Effect of PR-multiprotein-complex modulators on RU486-BODIPY mediated PR nuclear translocation. (a) Dose–response curve of PR nuclear translocation in T47D cells treated with HSP90 inhibitor 17-AAG. Cells were incubated with 17-AAG at indicated concentrations for 1 h before treated with 5 nM RU486-BODIPY. Each data point represents mean nuclear (●) or cytoplasmic (▲) fluorescence intensity of 30 cells. Error bars represent ± SD. (b) Effect of 17-AAG on PR nuclear translocation at 0.1 and 10 μM. Scale bar 20 μm. (c) Cellular distribution of 5 nM RU486-BODIPY in T47D cells pretreated with: 0.1% v/v DMSO for 12 h (control), 10 μM 17-AAG or 500 μM EHNA for 1 h, 10 μM FK506 for 2 h, 10 μM vorinostat, panobinostat, VER-155008, or methylene blue for 12 h. Each bar represents the ratio of mean nucleus-to-cytoplasm fluorescence of 30 cells. Error bars represent ± SD. * p < 0.01 (two-tail t-test). (d) Tissue distribution of RU486-BODIPY in FVB/N female mice 4 h post I.V. injection (n = 4). Error bars represent ± SD. * p < 0.05 (one-tail t-test).
Finally, we tested whether RU486-BODIPY will accumulate preferentially in tissues that naturally express high levels of progesterone receptor in vivo.40 We first evaluated the detectable dose by intravenous injection of either 1 or 10 nmol RU-486-BODIPY into FBV/N female mice (one mouse each) and analyzing tissue uptake by HPLC/MS/MS 4 h postinjection. While the lower dose was hardly detectable in any tissue (data not shown), at the higher dose, RU486-BODIPY was detected in most analyzed tissues. Therefore, we used the high-dose conditions (10 nmol) to similarly treat and analyze three more mice (Figure 3d). At 4 h post injection, no probe was detected in the blood or the brain. Although RU486 has a very long half-life in human, in rodents it is considerably reduced (30 vs 1 h, respectively).41 The highest uptake of RU486-BODIPY was observed in the liver, suggestive of its role in metabolism and excretion of the probe, and also in accordance with RU486 biodistribution in rodents.41 Importantly, RU486 BODIPY accumulation in uterus was consistently and significantly higher than in muscle (∼3.5-fold on average). The ovaries also showed a consistently higher uptake than muscle (∼1.2-fold), but this difference was not statistically significant.
In conclusion, we have designed and synthesized two fluorescent ligands for the human progesterone receptor. The ligands show antagonistic potency comparable to their parent RU486 in live cells and have spectroscopic properties suitable for fluorescence imaging. Both ligands triggered PR nuclear translocation in a receptor-dependent and specific manner in endogenously expressing cells. RU486-BODIPY was used to study the effect of PR complex components inhibition on its nuclear translocation process. Our results reinforce the importance of functional HSP90 in this process as both inhibition of its ATPase activity and its hyperacetylation, led to impaired PR shuttling. In addition, we found that FKBP52 activity is not essential for PR nuclear translocation, suggesting that FKBP52 plays a role in PR activation after the nuclear accumulation process. Finally, RU486-BODIPY preferentially accumulated in tissues that express high levels of PR in vivo. Thus, RU486-BODIPY’s design and properties make it a potential candidate for in vivo imaging of PR by PET through incorporation of 18F into the BODIPY fragment. Noninvasive whole-body imaging of steroid receptors could be of considerable value in classifying and staging many cancers of the endocrine and reproductive systems.
Acknowledgments
R.W. is supported by R25T CRIN training grant 5R25CA153915-03. R.Y.T. is supported by HHMI, NIH CA158448, and DOD W81XWH-09-1-0699.
Supporting Information Available
Synthetic procedures, chemical and spectral characterization, cell culture and imaging methods, alkaline phosphatase assay details, additional figures as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mangelsdorf D. J.; Thummel C.; Beato M.; Herrlich P.; Schutz G.; Umesono K.; Blumberg B.; Kastner P.; Mark M.; Chambon P.; Evans R. M. (1995) The nuclear receptor superfamily: the second decade. Cell 83, 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek F. M. (2011) What are nuclear receptor ligands?. Mol. Cell. Endocrinol. 334, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookout A. L.; Jeong Y.; Downes M.; Yu R. T.; Evans R. M.; Mangelsdorf D. J. (2006) Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 126, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conneely O. M.; Mulac-Jericevic B.; Lydon J. P. (2003) Progesterone-dependent regulation of female reproductive activity by two distinct progesterone receptor isoforms. Steroids 68, 771–778. [DOI] [PubMed] [Google Scholar]
- Brinton R. D.; Thompson R. F.; Foy M. R.; Baudry M.; Wang J.; Finch C. E.; Morgan T. E.; Pike C. J.; Mack W. J.; Stanczyk F. Z.; Nilsen J. (2008) Progesterone receptors: form and function in brain. Front. Neuroendocrinol. 29, 313–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obr A. E.; Edwards D. P. (2012) The biology of progesterone receptor in the normal mammary gland and in breast cancer. Mol. Cell. Endocrinol. 357, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akahira J.; Inoue T.; Suzuki T.; Ito K.; Konno R.; Sato S.; Moriya T.; Okamura K.; Yajima A.; Sasano H. (2000) Progesterone receptor isoforms A and B in human epithelial ovarian carcinoma: immunohistochemical and RT-PCR studies. Br. J. Cancer 83, 1488–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Thiel K. W.; Leslie K. K. (2011) Progesterone: the ultimate endometrial tumor suppressor. Trends Endocrinol. Metab. 22, 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayasam G. V.; Elbi C.; Walker D. A.; Wolford R.; Fletcher T. M.; Edwards D. P.; Hager G. L. (2005) Ligand-specific dynamics of the progesterone receptor in living cells and during chromatin remodeling in vitro. Mol. Cell. Biol. 25, 2406–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C. S.; Baumann C. T.; Htun H.; Xian W.; Irie M.; Smith C. L.; Hager G. L. (1999) Differential localization and activity of the A- and B-forms of the human progesterone receptor using green fluorescent protein chimeras. Mol. Endocrinol. 13, 366–375. [DOI] [PubMed] [Google Scholar]
- Narayanan R.; Edwards D. P.; Weigel N. L. (2005) Human progesterone receptor displays cell cycle-dependent changes in transcriptional activity. Mol. Cell. Biol. 25, 2885–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopoldo M.; Lacivita E.; Berardi F.; Perrone R. (2009) Developments in fluorescent probes for receptor research. Drug Discovery Today 14, 706–712. [DOI] [PubMed] [Google Scholar]
- Briddon S. J.; Kellam B.; Hill S. J. (2011) Design and use of fluorescent ligands to study ligand-receptor interactions in single living cells. Methods Mol. Biol. 746, 211–236. [DOI] [PubMed] [Google Scholar]
- Wang W.; Qiu X.; Zhang F.; Sun J.; Cameron A. G.; Wendt J. A.; Mawad M. E.; Ke S. (2011) An imageable retinoid acid derivative to detect human cancer xenografts and study therapeutic dosing to reduce its toxicity. Contr. Media Mol. Imaging 6, 200–208. [DOI] [PubMed] [Google Scholar]
- Adamczyk M.; Reddy R. E.; Yu Z. (2002) Synthesis of a novel fluorescent probe for estrogen receptor. Bioorg. Med. Chem. Lett. 12, 1283–1285. [DOI] [PubMed] [Google Scholar]
- Carlson K. E.; Coppey M.; Magdelenat H.; Katzenellenbogen J. A. (1989) Receptor binding of NBD-labeled fluorescent estrogens and progestins in whole cells and cell-free preparations. J. Steroid Biochem. 32, 345–355. [DOI] [PubMed] [Google Scholar]
- Teutsch G.; Klich M.; Bouchoux F.; Cerede E.; Philibert D. (1994) Synthesis of a fluorescent steroid derivative with high affinities for the glucocorticoid and progesterone receptors. Steroids 59, 22–26. [DOI] [PubMed] [Google Scholar]
- Sakai H.; Hirano T.; Mori S.; Fujii S.; Masuno H.; Kinoshita M.; Kagechika H.; Tanatani A. (2011) 6-arylcoumarins as novel nonsteroidal type progesterone antagonists: an example with receptor-binding-dependent fluorescence. J. Med. Chem. 54, 7055–7065. [DOI] [PubMed] [Google Scholar]
- Hodl C.; Strauss W. S.; Sailer R.; Seger C.; Steiner R.; Haslinger E.; Schramm H. W. (2004) A novel, high-affinity, fluorescent progesterone receptor antagonist. Synthesis and in vitro studies. Bioconjugate Chem. 15, 359–365. [DOI] [PubMed] [Google Scholar]
- Hudnall T. W.; Gabbai F. P. (2008) A BODIPY boronium cation for the sensing of fluoride ions. Chem. Commun. 4596–4597. [DOI] [PubMed] [Google Scholar]
- Hendricks J. A.; Keliher E. J.; Wan D.; Hilderbrand S. A.; Weissleder R.; Mazitschek R. (2012) Synthesis of [18F]BODIPY: bifunctional reporter for hybrid optical/positron emission tomography imaging. Angew. Chem., Int. Ed. 51, 4603–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting R.; Aguilera T. A.; Crisp J. L.; Hall D. J.; Eckelman W. C.; Vera D. R.; Tsien R. Y. (2010) Fast 18F labeling of a near-infrared fluorophore enables positron emission tomography and optical imaging of sentinel lymph nodes. Bioconjugate Chem. 21, 1811–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers H. C.; Versteegh J. E.; Uitdehaag J. C. (2009) The X-ray structure of RU486 bound to the progesterone receptor in a destabilized agonistic conformation. J. Biol. Chem. 284, 19572–19579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt S. A.; Edwards D. P. (2002) Mechanism of action of progesterone antagonists. Exp. Biol. Med. 227, 969–980. [DOI] [PubMed] [Google Scholar]
- Hodl C.; Raunegger K.; Strommer R.; Ecker G. F.; Kunert O.; Sturm S.; Seger C.; Haslinger E.; Steiner R.; Strauss W. S.; Schramm H. W. (2009) Syntheses and antigestagenic activity of mifepristone derivatives. J. Med. Chem. 52, 1268–1274. [DOI] [PubMed] [Google Scholar]
- Saha P.; Hodl C.; Strauss W. S.; Steiner R.; Goessler W.; Kunert O.; Leitner A.; Haslinger E.; Schramm H. W. (2010) Synthesis, in vitro progesterone receptors affinity of gadolinium containing mifepristone conjugates and estimation of binding sites in human breast cancer cells. Bioorg. Med. Chem. 18, 1891–1898. [DOI] [PubMed] [Google Scholar]
- Di Lorenzo D.; Albertini A.; Zava D. (1991) Progestin regulation of alkaline phosphatase in the human breast cancer cell line T47D. Cancer Res. 51, 4470–4475. [PubMed] [Google Scholar]
- Horwitz K. B. (1992) The molecular biology of RU486. Is there a role for antiprogestins in the treatment of breast cancer?. Endocr. Rev. 13, 146–163. [DOI] [PubMed] [Google Scholar]
- Smith D. F.; Toft D. O. (2008) Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Mol. Endocrinol. 22, 2229–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt W. B.; Morishima Y.; Osawa Y. (2008) The Hsp90 chaperone machinery regulates signaling by modulating ligand binding clefts. J. Biol. Chem. 283, 22885–22889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. F.; Stensgard B. A.; Welch W. J.; Toft D. O. (1992) Assembly of progesterone receptor with heat shock proteins and receptor activation are ATP mediated events. J. Biol. Chem. 267, 1350–1356. [PubMed] [Google Scholar]
- Segnitz B.; Gehring U. (1997) The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J. Biol. Chem. 272, 18694–18701. [DOI] [PubMed] [Google Scholar]
- Zajac M.; Gomez G.; Benitez J.; Martinez-Delgado B. (2010) Molecular signature of response and potential pathways related to resistance to the HSP90 inhibitor, 17AAG, in breast cancer. . BMC Med. Genomics 3, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali P.; Pranpat M.; Bradner J.; Balasis M.; Fiskus W.; Guo F.; Rocha K.; Kumaraswamy S.; Boyapalle S.; Atadja P.; Seto E.; Bhalla K. (2005) Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 280, 26729–26734. [DOI] [PubMed] [Google Scholar]
- Hernandez M. P.; Chadli A.; Toft D. O. (2002) HSP40 binding is the first step in the HSP90 chaperoning pathway for the progesterone receptor. J. Biol. Chem. 277, 11873–11881. [DOI] [PubMed] [Google Scholar]
- Williamson D. S.; Borgognoni J.; Clay A.; Daniels Z.; Dokurno P.; Drysdale M. J.; Foloppe N.; Francis G. L.; Graham C. J.; Howes R.; Macias A. T.; Murray J. B.; Parsons R.; Shaw T.; Surgenor A. E.; Terry L.; Wang Y.; Wood M.; Massey A. J. (2009) Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. J. Med. Chem. 52, 1510–1513. [DOI] [PubMed] [Google Scholar]
- Wang A. M.; Morishima Y.; Clapp K. M.; Peng H. M.; Pratt W. B.; Gestwicki J. E.; Osawa Y.; Lieberman A. P. (2010) Inhibition of hsp70 by methylene blue affects signaling protein function and ubiquitination and modulates polyglutamine protein degradation. J. Biol. Chem. 285, 15714–15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies T. H.; Sanchez E. R. (2005) Fkbp52. Int. J. Biochem. Cell Biol. 37, 42–47. [DOI] [PubMed] [Google Scholar]
- Le Bihan S.; Marsaud V.; Mercier-Bodard C.; Baulieu E. E.; Mader S.; White J. H.; Renoir J. M. (1998) Calcium/calmodulin kinase inhibitors and immunosuppressant macrolides rapamycin and FK506 inhibit progestin- and glucocorticosteroid receptor-mediated transcription in human breast cancer T47D cells. Mol. Endocrinol. 12, 986–1001. [DOI] [PubMed] [Google Scholar]
- Uotinen N.; Puustinen R.; Pasanen S.; Manninen T.; Kivineva M.; Syvala H.; Tuohimaa P.; Ylikomi T. (1999) Distribution of progesterone receptor in female mouse tissues. Gen. Comp. Endrocrinol. 115, 429–441. [DOI] [PubMed] [Google Scholar]
- Heikinheimo O.; Pesonen U.; Huupponen R.; Koulu M.; Lahteenmaki P. (1994) Hepatic metabolism and distribution of mifepristone and its metabolites in rats. Hum. Reprod. 9(Suppl 1), 40–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.