

Abstract
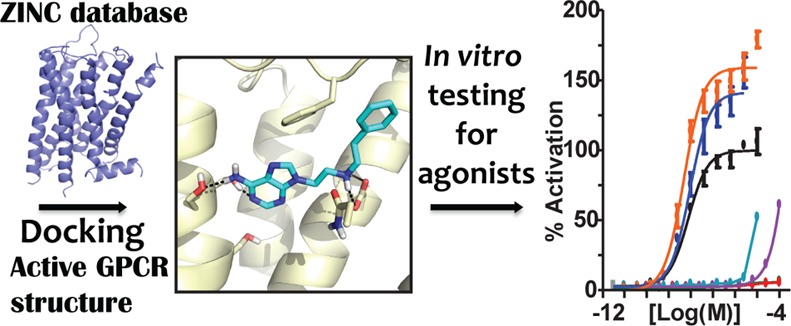
A prospective, large library virtual screen against an activated β2-adrenergic receptor (β2AR) structure returned potent agonists to the exclusion of inverse-agonists, providing the first complement to the previous virtual screening campaigns against inverse-agonist-bound G protein coupled receptor (GPCR) structures, which predicted only inverse-agonists. In addition, two hits recapitulated the signaling profile of the co-crystal ligand with respect to the G protein and arrestin mediated signaling. This functional fidelity has important implications in drug design, as the ability to predict ligands with predefined signaling properties is highly desirable. However, the agonist-bound state provides an uncertain template for modeling the activated conformation of other GPCRs, as a dopamine D2 receptor (DRD2) activated model templated on the activated β2AR structure returned few hits of only marginal potency.
The recent abundance of crystal structures of G protein coupled receptors (GPCRs) has inspired a surge of structure-based discovery campaigns against these targets. In the past three years, prospective docking screens of large chemical libraries have been prosecuted against the β2-adrenergic receptor (β2AR), the adenosine A2A receptor, the histamine H1 receptor, the dopamine D3 receptor, and the chemokine CXC-4 receptor.1−6 Despite the use of multiple docking programs by several independent groups, three unifying features have emerged: (1) hit rates are unusually high, ranging from 17% to 70% (compounds active/tested); (2) hits are unusually potent; and (3) the activity of the hits has recapitulated the activity of the co-crystallized inverse-agonist; all GPCR crystal structures used for virtual screening were solved in the inactive state, and all hits predicted by virtual screening were subsequently confirmed to be inverse-agonists.
The recently determined structure of the β2AR in an activated state revealed surprisingly subtle changes in the orthosteric binding site,7,8 supporting the idea that agonist binding and activation requires only modest conformational change in that region.9−13 The slight conformational change is subsequently translated to much larger changes at the intracellular G protein interface, nearly 40 Å away. Given the small differences between the active and inactive binding site conformation, the functional fidelity of docking hits to the state of the receptor is surprising.
Two explanations for the high hit rates and affinities of GPCR ligands predicted by docking are possible: GPCR binding sites may be unusually well suited to small molecule binding, or docking libraries may be biased toward analogues of signaling molecules.14 By extension, it may be that (1) the inverse-agonist-bound GPCR states are genuinely selective for inverse-agonists; (2) the libraries are biased toward inverse-agonists; or (3) a combination of the two. If the docking results reflect structural information encoded in the binding site conformation, one might expect agonist hits to dominate docking campaigns against the active structure. Conversely, if library bias dominates, one might expect the screen to return molecules that resemble the docking library used. In the second case, a ligand-based screen would return molecules that resemble the structure-based docking hits.
Here, we investigate the effect of binding site conformation on virtual screening by targeting the agonist/nanobody-bound activated state of the β2AR. We prospectively screen the ZINC library of 3.4 million “lead-like” and “fragment-like” molecules against this target, experimentally testing 22 high-ranking molecules for activity against the β2AR. For each docking hit, we evaluated G protein and β-arrestin mediated signaling in cells.15−19 To control for the role of library bias, in parallel we undertook a ligand-based screen of the same ZINC library, testing 30 molecules predicted by two-dimensional chemical similarity to resemble the co-crystallized ligand BI-167107 and six additional β2AR agonists.
Finally, we investigated whether the active β2AR structure can act as a modeling template to predict other active GPCR structures, that is, whether the structural information encoded in the active structure is transferrable. Previous work has suggested that GPCR structures of suitably high sequence identity in the inactive state can reliably template the modeling of other GPCRs for predictive virtual screening.20 Determining activated GPCR states will often be more challenging then inactivate states,21 and the ability to use one active structure as a model for others, as well as recapitulating the activated function in the ligands, would have wide impact.
Results and Discussion
We first carried out a retrospective docking of known β2AR ligands to the active structure. At the time we undertook this study, the agonist-bound structure of β2AR available was stabilized in the active conformation by a potent agonist, BI-167107, and by a G protein mimetic nanobody (PDB ID 3P0G, referred to as the “active structure”). This structure is almost identical in the binding site to a later active structure co-crystallized with BI-167107 and the G protein itself (PDB ID 3SN6) that we did not use due to lower resolution in the binding site. Using a set of 30 β2AR agonists and 30 β2AR inverse-agonists,22 we tested the active structure’s ability to recognize known β2AR ligands against a background of property matched decoys23 and to preferentially score agonists over inverse-agonists. We used the metric of adjusted LogAUC, which measures the ranking of true positives (known ligands) over false positives (decoy molecules) compared to what would be expected at random (an adjusted LogAUC of 0 represents the random ranking). This measure emphasizes early enrichment of ligands, as the first 0.1% of the database is weighted equally to the next 0.1–1% of the database, and to the next 1–10% and 10–100% of the database.24,25 The active structure enriched the 60 known β2AR ligands over computational decoys, with an enrichment of 23.6% adjusted LogAUC. As well as recognizing known ligands, the active structure also distinguishes agonists from inverse-agonists, with adjusted LogAUC of 35.4% for agonists and 10.6% for inverse-agonists (Supplementary Figure S1). In the top 1% of the database, 20% of agonists were found (6/30 docked agonists), while at 10% of the database, 75% (22/30 agonists) were found. Using the same set of agonists and inverse-agonists with the same decoys, the inactive carazolol-bound β2AR crystal structure (PDB ID 2RH1) found no agonists in the top 1% of the database and 13% (4/30 agonists) in the top 10% of the database. For comparison, the inactive β2AR structure enriched inverse-agonists, with 6% (2/30 inverse-agonists) in the top 1% of the database and 46% (14/30 inverse-agonists) in the top 10% of the database (Supplementary Figure S1).
To ensure that docking enriched known agonists for the right reasons, we confirmed that they not only scored well but also were docked in reasonable poses. Residues Ser2035.42, Ser2045.43, and Ser2075.46 in TM5 are proposed to be important for interaction with agonists and activation in mutagenesis studies,26,27 and the greatest structural change between active and inactive β2AR is centered around those residues. We evaluated the docked poses of known β2AR agonists by two criteria: (1) the aminergic group should salt-bridge with the key residue Asp1133.32, and (2) the polar head groups should hydrogen bond with at least one of the three TM5 serine residues (Figure 1A). The high retrospective enrichment of known ligands, high ranking of agonists, and reasonable docked poses encouraged us to move forward with a prospective virtual screen.
Figure 1.
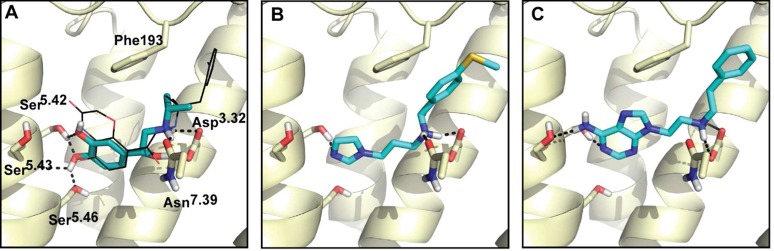
Two partial β2AR agonists with new activating chemotypes in their docked poses. (A) The docked pose of isoproterenol, with a salt-bridge between the amine group and key residue Asp1133.32 and hydrogen bonds to Ser2035.42, Ser2045.43, and Ser2075.46 in TM5. The co-crystal ligand BI-167107 is shown in black sticks. Previously unreported (B) imidazole, compound 10, and (C) amino-purine, compound 14, polar head groups make activating hydrogen bonds with TM5.
For the prospective virtual screen, we used DOCK 3.6 to virtually screen the 2.7M “lead-like” and 400K “fragment-like” molecules of the ZINC database (July 2011).28,29 Essentially this represents commercially available molecules with molecular weights below 350, logP less than 3.5, and 7 or fewer rotatable bonds. Molecules were screened to both the active and inactive crystal structure. The ZINC subsets (lead or fragment-like) were ranked separately, and only those that ranked at the top 0.2% to the active structure were considered. To select for agonists, we only considered molecules with higher ranking in the active structure than the inactive structure, as this would reflect the structural bias we found in the active structure. Molecules were filtered for a rank difference of at least 5000 between the active and inactive screen. An automatic filter was applied to select for molecules that posed well, namely, having (1) a positive charge, (2) an amine interaction to Asp1133.32, and (3) a hydrogen bond interaction to Ser2035.42, Ser2045.43, or Ser2075.46. Inverse-agonists in the inactive structure also make the amine to Asp1133.32 interaction and hydrogen bond to Ser2035.42, so we do not believe this filter unfairly biased the results. Docking ranks reported here reflect the rankings prior to filtering.
After visual inspection, 22 molecules were selected for experimental testing from the top ∼0.2% of each subset: 17 lead-like molecules ranked in the top 5000 (out of 2.7M) and 5 fragment-like molecules ranked in the top 400 (out of 400K). Molecules were selected for chemical diversity (Supplementary Table S1) and, as is typical, for criteria missing from the DOCK scoring function (detailed criteria may be found in ref (6) and are described in the Supporting Information Methods). These molecules were experimentally tested in HEK293 cells stably transfected with the human β2AR, measuring Gs protein activation through cAMP formation using the GloSensor assay30 and β-arrestin recruitment using the Tango assay.31 The sensitivity of the Tango assay is improved by using a β2AR mutant (β2/V2R) that has its carboxy-terminal tail replaced with that of the vasopressin 2 receptor. This receptor has higher affinity for binding to β-arrestins while retaining the ligand binding properties of the native β2AR.31
Six compounds of the 22 tested (27%) considerably increased cAMP formation, consistent with agonist activity, and four out of these six compounds also significantly increased β-arrestin recruitment (Table 1, Figure 2). The experiments were repeated for all 22 compounds with the addition of either 2 nM isoproterenol for cAMP formation or 200 nM for β-arrestin recruitment to test for antagonism; none of the compounds significantly inhibited activation by isoproterenol (data not shown). In summary, six hits were found, four full agonists (compounds 1, 4, 12, 22) and two partial agonists (compounds 10, 14). The two partial agonists are not predicted to interact with both Ser2035.42 and Ser2075.46 in TM5, perhaps leading to only partial agonism. Radioligand competition binding assay was carried out to confirm binding of the six hits to the β2AR using [125I]cyanopindolol with crude membrane fractions containing the overexpressed β2AR (Supplementary Figure S2). The binding affinity of agonists is relatively weaker compared to their affinities in the functional assays presumably due to the absence of G protein or β-arrestin. Their engagement is essential for stabilizing a high affinity state of the agonist binding conformation. In addition, affinity measured by the different assays cannot be directly compared, as differences in receptor reserve and amplification must be taken into account.32 Second-messenger assays such as the GloSensor assay have significant amplification, whereas the Tango and direct binding assays do not.
Table 1. Hits Found in Virtual Screening of the Active β2AR Structure.
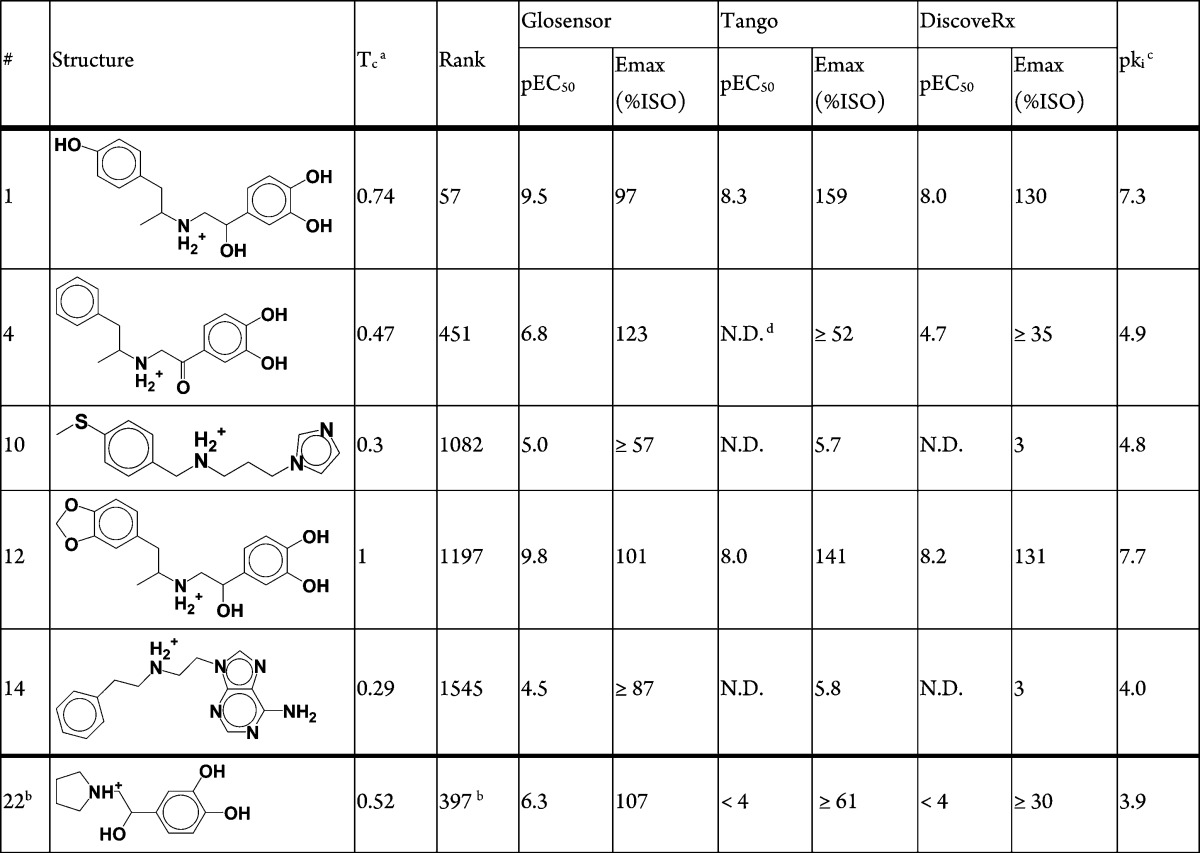
Tanimoto coefficient (Tc) calculated for all known β2 adrenergic receptor ligands in the ChEMBL 15 database.
“Fragment-like” screen.
Binding assays performed with [125I]cyanopindolol.
Not determined.
Figure 2.

Functional assays for β2AR agonists. Six compounds considerably increased cAMP formation and β-arrestin recruitment, consistent with agonism (compounds 1, 4, 10, 12, 14, and 22 as indicated colors). (A) Dose–respone curves measuring G-protein activation through cAMP formation using the GloSensor assay. (B) Known β2AR agonists used as controls in the GloSensor assay: isoproterenol (ISO, black), epinephrine (Epi, green), hydroxybenzylisoproterenol (HBI, blue), and BI-167107 (BI, red). (C) Dose–response curves measuring β-arrestin recruitment using the β2 V2R Tango assay. For compound 4, a connected line of each data point is presented instead of its dose–response curve since its fitting was not converged. (D) The control β2AR agonists in the Tango assay as described for panel B. Each data point represents mean ± SE obtained from three independent experiments done in duplicates. Dose–response curves for each compound were obtained using the nonlinear iterative curve-fitting computer program Prism.
An important advantage of structure-based virtual screening is the ability to identify wholly new chemotypes. To measure novelty, we assess chemical similarity to known ligands in ChEMBL1533 using ECFP4 topological fingerprint and Tanimoto coefficient (Tc).34 The four full agonists predicted contain the classical activating catecholamine moiety, supporting the notion that this is a privileged scaffold (Table 1). Compound 12 was later found to be the known agonist protokylol; however, this was not known to us at the time of the screening. Likewise, compound 1 is also very similar to the known agonist hydroxybenzylisoproterenol (HBI), with Tc of 0.74. Encouragingly, the two partial agonists (compound 10, 14) we predicted are novel, interacting with TM5 through previously unknown chemical moieties: an imidazole and an amino-purine (Figure 1B,C), with a Tc of 0.3 to any previously known ligand (Table 1). A total of 4745 ligands are reported for the human β2AR in ChEMBL15, reflecting the extensive medicinal chemistry surrounding this target.
In addition to signaling through G proteins, GPCRs can also stimulate β-arrestin mediated signaling, and certain ligands can have different signaling efficacies for these distinct signaling pathways.19,35 A “β-arrestin biased” ligand will have better efficiency in recruitment of β-arrestin than in G protein activation, when compared to an unbiased reference agonist that signals with equal efficacy through G protein and β-arrestin dependent pathways. Among the six agonists discovered, compounds 1 and 12 both show some bias toward β-arrestin recruitment (Table 1, Figure 2). Compared to the unbiased agonist, isoproterenol,32 similar levels of G protein activation were measured, with log(EC50) values in the GloSensor assay of −9.5, −9.8, and −10.1 and Emax of 97%, 101% and 100% for compound 1, compound 12, and isoproterenol, respectively. On the other hand, these compounds showed higher levels of β-arrestin recruitment when compared to isoproterenol, with log(EC50) of −8.0, −8.3, and −8.1 and Emax of 159%, 141%, and 100% in the Tango assay for compound 1, compound 12, and isoproterenol, respectively. These β-arrestin recruitment results were confirmed using another independent assay (DiscoveRx PathHunter β-arrestin assay, Table 1, Figure 3A). There have been several ways reported to calculate such a bias of a ligand based on its potencies and efficacies in G protein activation and β-arrestin recruitment assays.32 In order to determine the degree of bias of the compounds 1 and 12, we calculated their bias factors from the data sets in Figure 2 and Figure 3A,B as well as binding affinity values (Supplementary Figure S2) using the operational model.32 The bias factors of the compounds 1 and 12 are around 1 when calculated from both Tango and DiscoveRx β-arrestin against Glosensor cAMP data sets (Figure 3C). These values indicate that they are approximately 10 times more efficacious in β-arrestin recruitment than in G protein-mediated cAMP production when compared to isoproterenol, the unbiased reference. In fact, BI-167107 itself shows stronger bias toward β-arrestin recruitment with a bias factor around 1.5, indicating that it promotes β-arrestin recruitment about 30 times more efficaciously than cAMP production (Figure 3C). These data indicate that our virtual screening with the active conformation of the β2AR/BI-167107 co-crystal structure not only detected agonists but also identified weakly biased agonists that have similar signaling bias toward β-arrestin as BI-167107. Attempts to find β-arrestin biased β2AR agonists have been almost intractable; to our knowledge these are the first partially biased agonists to emerge from virtual screening.
Figure 3.
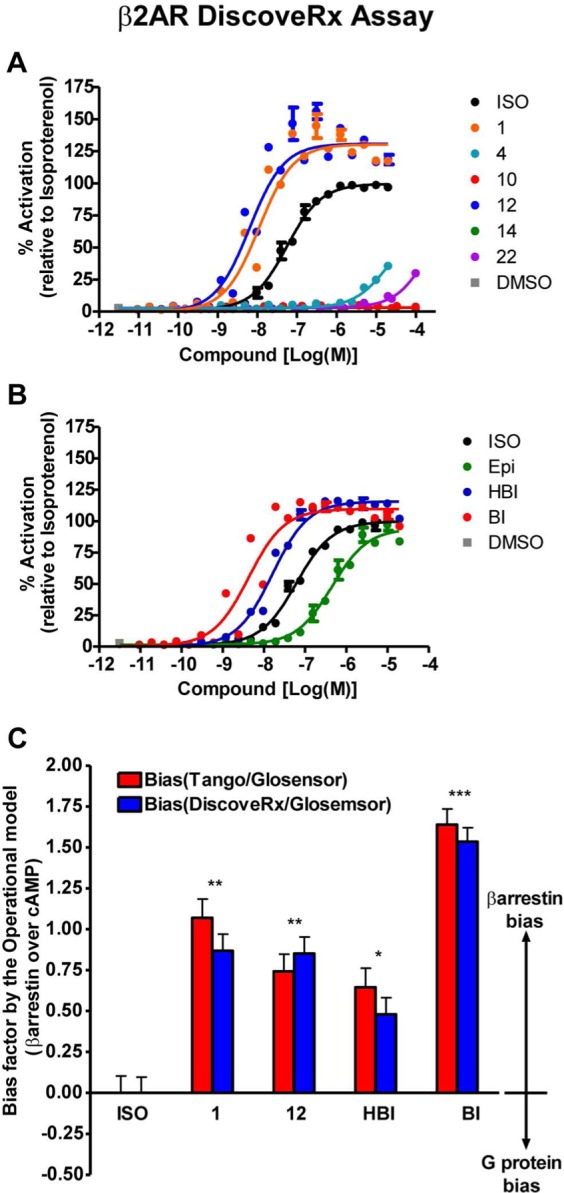
An additional DiscoveRx PathHunter β-arrestin recruitment assay and bias factors calculated using the operational model. (A) Dose–response curves for the six β2AR agonists discovered in the virtual screen. (B) Control compounds as described for Figure 2B. Each data point represents mean ± SE, and dose–response curves for each compound were obtained from three independent data sets. (C) The bias factors of indicated compounds were calculated from the Tau value analysis by the Operational Model32 using the data sets in Figure 2 and panels A and B of this figure, as well as the binding affinity values obtained in Supplementary Figure S2. Each bar represents mean ± SE. The statistical analysis was performed using one-way ANOVA with Bonferroni’s multiple comparison post-test. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared to the reference value of isoproterenol (ISO).
To deconvolute the influence of structure from library bias in the docking, we screened the ZINC library using 2D chemical similarity for additional β-arrestin biased agonists. We used the Similarity Ensemble Approach (SEA), a statistical model that ranks the significance of chemical similarity of a query molecule to a set of ligands for a target.36 We used SEA to search the ZINC database for molecules similar in 2D to a set of β2AR agonists that are partially β-arrestin biased (unpublished data and ref (32), as well as BI167107 (Supplementary Table S2)). From the most significantly similar molecules, we visually selected a diverse set of 30 compounds to test (compounds SEA1–30, Supplementary Table S3). Of these, 11 were active in the GloSensor assay (36%, Supplementary Figure S3). The 36% hit rate is consistent with the ability of ligand-based screens to recall a known chemotype.37 Seven of the new agonists resembled known adrenergic agonists (ECFP4 Tc > 0.35, Tc values for the 11 hits ranged from 0.31 to 0.70, Supplementary Table S3). Perhaps less anticipated, and in contrast to the β-arrestin biased molecules against which they were selected, most of the predicted agonists did not induce measurable β-arrestin recruitment (Supplementary Figure S3; note that sensitivity in the Tango assay is approximately 2 orders of magnitude weaker than that in the Glosensor assay, making β-arrestin recruitment undetectable by Tango for a majority of the hits that have substantially weak potencies in the Glosensor cAMP assay). It was also surprising that the relatively modest log(EC50) values were obtained in the Glosensor assay for the ligand-based agonists, which ranged from −7.3 to −4.7. These observations suggest that 2D chemical similarity alone did not lead to the β-arrestin biased compounds found by docking. We note that the partially β-arrestin biased agonists used to construct the query set do not rank well to the active structure, with none in the top 10% of the lead-like ZINC database. This is likely because they have a larger number of rotatable bonds, with 9.5 rotatable bonds on average for the 7 query-set partially biased molecules. Likewise, the compounds selected on the basis of 2D similarity did not rank highly to the active crystal structure when they were later docked to it, with only one compound scoring in the top 10% of the database.
The ability to homology model active GPCR structures would be a boon to the field, as agonists alone cannot fully stabilize the active conformation and are consequently harder to crystallize.21 To test whether the active β2AR structure could template a closely related active GPCR structure, we modeled and virtually screened an active dopamine D2 receptor (DRD2) structure. Our homology modeling protocol, described previously,6,20 produces many models and selects the best model based on retrospective enrichment of docked known binders from a background of property matched decoys.23 A total of 500 models with identical backbone conformations and different side chain orientations were generated using MODELER v9.838 based on the active β2AR structure (alignment shown in Supplementary Figure S4). No ligand is present during the homology modeling. The final homology model was chosen because it enriched known DRD2 ligands and ranked agonists more highly then antagonists. The side chain angle of the critical serine residues Ser1925.42, Ser1935.43, or Ser1965.46 was enforced in all models to match that of the active β2AR structure. To select a model we used retrospective enrichment of known DRD2 ligands, and particularly agonists, from a set of computationally derived decoy molecules. We docked a chemically diverse set of 50 agonists and 50 inverse-agonists as well as 6400 decoy molecules. The average LogAUC for all generated models was 7.6% (±4.3%) for agonists and 6.7% (±2.9%) for inverse-agonists, with the selected model having LogAUC of 16.9% and 10.5% for agonists and inverse-agonists, respectively, far lower than the retrospective enrichments found for the active β2AR (Supplementary Figure S1). A previous virtual screen of a DRD3 homology modeled on the inactive β2AR structure predicted ligands with hit rates, novelty, and potency equaling that of the DRD3 crystal structure,20 confirming the transferability of structural information, at least with high sequence similarity, in the inactive state; in unpublished studies, we have observed the same for the DRD2 and serotonin 5HT2A receptors. A virtual screen of the active DRD2 model would determine if the β2AR active structure is likewise a good template. We screened the selected active DRD2 model with the lead-like and fragment-like sets of ZINC as described above, and 15 molecules were chosen from the top 0.5% of each database (Supplementary Table S4; we allowed a slightly larger slice of the database due to a prevalence of high-internal-energy molecules that ranked highly and are not penalized by the dock scoring function). These molecules were further filtered corresponding to the criteria in the original β2AR agonist screen: (1) a positive charge, (2) an amine interaction to Asp1103.32, and (3) a hydrogen bond interaction to Ser1925.42, Ser1935.43, or Ser1965.46. The reported ranks do not reflect this pose filtering. Again, only molecules with a rank difference of at least 5000 between the active and inactive screen were considered; for the inactive screen we used the DRD3 crystal structure, as DRD2 and DRD3 have 100% sequence identity in the binding site. Molecules were tested as described in GloSensor and Tango β-arrestin recruitment assays to test for agonism and in the presence of 100 nM of quinpirole to test for antagonism. Of the 15 molecules, three were active in functional assays (20% hit rate, compounds 29, 33, 34; Table 2), of which two were agonists and one was an inverse-agonist (GloSensor assay results shown in Figure 4). We do not consider the three hits novel, with Tc values of 0.36–0.48 to known dopamine receptor ligands. Moreover, the potency of the three hits was weak, suggesting that the active β2AR structure was not a good template for the active DRD2, despite high sequence homology and being a good template for the inactive conformation, where hits bound with Ki of 200 nM to 3 μM in binding affinity assays, of which several were novel.20
Table 2. Hits Found in Virtual Screening of the Active DRD2 Model.
Tanimoto coefficient (Tc) calculated for all known dopamine D2 receptor ligands in the ChEMBL 15 database.
Rank after filtering for a high-internal energy motif not captured by the DOCK scoring function.
Not determined.
Figure 4.
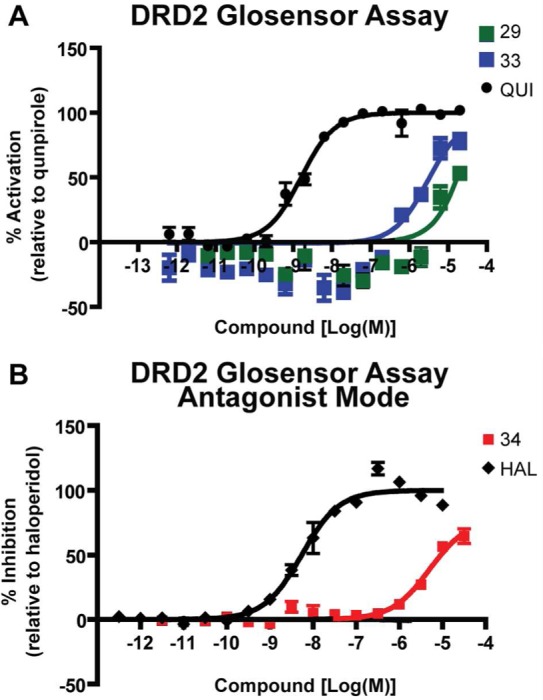
Functional assays for DRD2 agonists and inverse-agonists. (A) Two compounds (29 and 33, green and blue squares, respectively) activated Gi in GloSensor assays, consistent with partial agonism. QUI is the known agonist quinpirole (black circle). (B) The GloSensor assay was run in inverse-agonist mode with addition of 100 nM quinpirole. One compound (38, red square) inhibited Gi activation. HAL is the known inhibitor haloperidol (black diamond).
Discussion
Two principal observations emerge from this study: First, a prospective, large library screen against an activated β2AR structure returned potent agonists, essentially to the exclusion of inverse-agonists, with a high hit rate. This study therefore provides the first complement to the previous campaigns against inverse-agonist-bound structures, supporting the functional fidelity of the docking hits to the conformation of the GPCR target. Not only did we exclusively find agonists, a handful also recapitulated the partial bias toward β-arrestin signaling of the co-crystallized agonist BI-167107. The structure-to-function link is strengthened by the results of the 2D ligand-based control screen, which was designed to predict similarly arrestin biased ligands but produced none. In addition, two previously unreported agonist chemotypes were found. Second, a corollary of this functional fidelity is that, unlike the inverse-agonist-bound structure, the agonist-bound state provides an uncertain template for modeling the activated state of other GPCRs. Although we did find agonists against the D2 receptor, they were mixed with one antagonist, the hit rate was lower, the compounds had lower affinities, and the agonism was weaker.
In our screen of the activated β2AR structure, all hits mirrored the agonist activity of the co-crystal ligand. This in itself was unexpected in view of the subtle conformational changes in the binding site upon activation. An inward bulge of TM5, centered at Ser5.46, as well as rotation of the Ser5.42 and Ser5.46 side chains, allows the active structure to discriminate between docked agonists and antagonists. These activating interactions are captured in the docked poses of the novel partial agonists discovered, as well as the catecholamine hits. The inactive structure does not allow for these favorable electrostatic interactions, as the serines are pointed away from the binding site, and for this reason agonists do not rank as highly when docked to it.
Also remarkable was the observation that two of these hits recapitulated the properties of the co-crystal ligand BI-167107 in terms of G protein and β-arrestin mediated signaling. These two compounds have similar (but weaker) bias factors compared to those of BI-167107 determined by the operational model32 (Figure 3C). It is currently unknown whether the crystallized, BI-167107-bound β2AR structure represents a somewhat β-arrestin biased conformation. Compounds 1 and 12 discovered in our screen represent the first partially biased compounds to be found by virtual screening. However, we could not arrive at similarly β-arrestin biased compounds using only 2D chemical similarity to the co-crystal ligand and other agonists with comparable signaling profiles (Supplementary Figure S3). The structure-based docking may be capturing structural information encoded in the co-crystal structure; however, as the conformation was crystallized in the presence of a G protein mimetic nanobody, what if any biased structural information was exploited by virtual screening remains opaque.
The active structure led to the prediction of two unprecedented β2AR activating chemotypes, an imidazole and an amino-purine (compounds 10 and 14, Table 1, Figure 1B,C). All of the catecholamines tested (4 of the 22 compounds tested) were found to be agonists (Table 1, Figure 2). Although these were predicted by docking, we do not consider their identification remarkable in itself, as they could have been predicted by any pharmaceutical chemist familiar with β2AR agonists. In contrast, the discovery of two new agonist chemotypes for a well studied GPCR such as β2AR is in itself an interesting result and emphasizes the ability of virtual screening to predict novel chemical scaffolds even in a crowded field. Admittedly, these two scaffolds were the only ones that were active of the 16 novel molecules, those with Tc ≤ 0.35 to known β2AR ligands, that were tested. Unlike inverse agonists, agonists must not only bind to the receptor but must also make activating interactions with it, and there may be few chemotypes that can do so in our current libraries.
More generally, we asked if the active β2AR structure could act as a modeling template to predict other active GPCR structures. The ability to model active structures for agonist prediction would be particularly useful as active structures are difficult to crystallize. While inactive structures of the bioaminergic receptors have been solved in remarkably similar conformations, it is unknown whether active conformations of bioaminergic receptors are also alike. Virtually screening the active DRD2 model predicted only two weak agonists, as well as an inverse-agonist (Table 2, Figure 4); hit rates and potencies were far lower when compared to the screen of the active β2AR structure and as compared to a similar screen of an inactive DRD3 model templated on the inactive β2AR structure.20 These results indicate that despite 42% sequence identity, structural information from the active β2AR was not transferrable. The agonist state may be more particular to any given GPCR–ligand pair, reducing the transference of structures. Whether this observation is applicable to other GPCRs remains to be determined and will become clear as more active structures are determined.
Several cautions should be aired. First, we used domain knowledge of the β2AR residues important for agonist recognition to prioritize molecules, as is common practice in the field. Since these interactions are also found in inverse-agonists, they alone would not ensure us of agonists. Top ranked molecules from the large-scale docking screen were filtered on the basis of data implicating residues Ser5.42, Ser5.43, and Ser5.46 in activation. Additionally, the measured β-arrestin bias of compounds 1 and 12 remains modest, as is, in fact, that of the co-crystal ligand BI-167107. The bias was, however, confirmed independently in two separate assays, Tango and DiscoverX. Finally, the low hit rate produced by the active DRD2 model does not preclude other explanations: database bias may still play a role, although dopamine agonists are well represented in the pharmacopoeia. Dopamine agonists may simply be harder to predict or to assay. DRD2 couples to the inhibitory G protein Gi (rather than stimulatory Gs as β2AR does), and accordingly, measurement of G protein signaling is less straightforward. It may be that the lack of transferability from the active β2AR structure to active DRD2 is particular to this case or to our modeling and docking protocols.
Conclusions
A large library virtual screen of the activated BI-167107/β2AR co-crystal structure predicted exclusively agonists, just as previous virtual screens of inactive GPCR structures predicted exclusively inverse-agonists. The remarkable functional fidelity of the docking hits to the form of the receptor has important implications for drug design: small molecule GPCR ligands induce a variety of signaling behaviors, most likely through subtly different receptor conformations. As co-crystal GPCR structures with these ligands emerge, virtual screening might be used to predict new ligands with similar signaling properties. However, structural information from the activated β2AR structure was not transferrable to the closely related active dopamine D2 receptor structure, suggesting that the agonist state is more particular to a given GPCR-ligand pair.
Methods
Homology Modeling and Docking
We used DOCK 3.625 to screen the ZINC database as described (see Results). Complimentarity of each ligand pose is scored as the sum of the receptor–ligand electrostatic and van der Waals interaction energy and corrected for ligand desolvation. Partial charges from the united-atom AMBER force field were used for all receptor atoms except for Ser5.42, Ser5.43, and Ser5.46, for which the dipole moment was increased as previously described3 to boost electrostatic scores for poses in polar contact with these important residues. The hit list was automatically filtered to remove a previously known high-internal-energy motif that results in unreasonably favorable docking scores and for favorable activating interactions with the receptor, as described (the rankings reported do not reflect this further filtering). MODELER v9.838 was used for DRD2 homology model generation, based on PDB ID 3P0G as the active template.
Materials
Compounds were obtained from commercial vendors, as well as from the Developmental Therapeutics Program at the National Cancer Institute. All compounds were sourced at 95% or greater purity. All active compounds were further tested for purity by LC–MS, at UCSF, and were found to be pure as judged by peak height and identity. Bright-Glo and Glosensor reagents were obtained from Promega (Madison, WI). The Tango construct for the β2 V2R and the parental Tango cell line expressing β-arrestin2-TEV and luciferase reporter protein were provided by Gilad Barnea and Richard Axel. The stable cell line and the reagents for the β2AR DiscoveRx PathHunter β-arrestin assay were obtained from DiscoveRx (Fremont, CA).
β2AR Functional and Binding Affinity Assays
G protein activation and β-arrestin recruitment to receptor was measured in the GloSensor cAMP accumulation assay and the Tango assay, respectively, as previously described.32 To ensure that the results obtained using the Tango assay were not an artifact of overnight incubation, we also used the PathHunter β-arrestin assay from DiscoverRx, which has shorter incubation time.32 All functional assays were done using stably transfected cell lines. Radioligand binding assays were performed with crude membrane fractions from β2AR stably overexpressing HEK-293 cells, using 60 pM [I125]-cyanopindolol as a tracer.39
DRD2 Functional and Binding Affinity Assays
GloSensor, Tango, and binding affinity assays were carried out at the National Institute of Mental Health Psychoactive Drug Screening Program as previously described.40,41
Data Analysis
Calculation of EC50, binding affinitiy (Ki), and Tau values, as well as dose–response curves, were obtained using the nonlinear iterative curve-fitting computer program Prism (GraphPad Software Inc., San Diego, CA).
Acknowledgments
D.R.W. thanks B. Kobilka and D. Rosenbaum for useful discussions on understanding the activated GPCR structure and R. Coleman for discussing modeling and docking of the inactive dopamine receptor. Supported by National Institute of Health grants GM59957 (to B.K.S.), GM072970 (PI R. Altman), HL16037 (to R.J.L.), F32GM093580 (to D.R.W.), and the National Institutes of Mental Health Psychoactive Drug Screening Program (to B.L.R.). R.J.L. is an investigator with the Howard Hughes Medical Institute.
Glossary
Abbreviations
- GPCR
G protein coupled receptor
- TM
transmembrane
- β2AR
β2-adrenergic receptor
- DRD2
dopamine D2 receptor
- DRD3
dopamine D3 receptor
- Tc
Tanimoto coefficient
- cAMP
cyclic adenosine monophosphate
Supporting Information Available
Supplementary Figures S1–S4 and Tables S1–S4. Detailed methods outlining criteria by which molecules are manually rejected for testing even though they are high-ranking in the virtual screen. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sabio M.; Jones K.; Topiol S. (2008) Use of the X-ray structure of the beta2-adrenergic receptor for drug discovery. Part 2: Identification of active compounds. Bioorg. Med. Chem. Lett. 18, 5391–5395. [DOI] [PubMed] [Google Scholar]
- Kolb P.; Rosenbaum D. M.; Irwin J. J.; Fung J. J.; Kobilka B. K.; Shoichet B. K. (2009) Structure-based discovery of beta2-adrenergic receptor ligands. Proc. Natl. Acad. Sci. U.S.A. 106, 6843–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J.; Yoo L.; Gao Z. G.; Irwin J. J.; Shoichet B. K.; Jacobson K. A. (2010) Structure-based discovery of A2A adenosine receptor ligands. J. Med. Chem. 53, 3748–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V.; Jaakola V. P.; Lane J. R.; Lin J.; Ijzerman A. P.; Yeager M.; Kufareva I.; Stevens R. C.; Abagyan R. (2010) Structure-based discovery of novel chemotypes for adenosine A(2A) receptor antagonists. J. Med. Chem. 53, 1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf C.; Kooistra A. J.; Vischer H. F.; Katritch V.; Kuijer M.; Shiroishi M.; Iwata S.; Shimamura T.; Stevens R. C.; de Esch I. J.; Leurs R. (2011) Crystal structure-based virtual screening for fragment-like ligands of the human histamine H(1) receptor. J. Med. Chem. 54, 8195–8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Weiss D. R.; Ziarek J. J.; Gravel S.; Doak A. K.; Karpiak J.; Heveker N.; Shoichet B. K.; Volkman B. F. (2012) Structure-based ligand discovery for the protein-protein interface of chemokine receptor CXCR4. Proc. Natl. Acad. Sci. U.S.A. 109, 5517–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G.; Choi H. J.; Fung J. J.; Pardon E.; Casarosa P.; Chae P. S.; Devree B. T.; Rosenbaum D. M.; Thian F. S.; Kobilka T. S.; Schnapp A.; Konetzki I.; Sunahara R. K.; Gellman S. H.; Pautsch A.; Steyaert J.; Weis W. I.; Kobilka B. K. (2011) Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G.; DeVree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D.; Mathiesen J. M.; Shah S. T.; Lyons J. A.; Caffrey M.; Gellman S. H.; Steyaert J.; Skiniotis G.; Weis W. I.; Sunahara R. K.; Kobilka B. K. (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deflorian F.; Kumar T. S.; Phan K.; Gao Z. G.; Xu F.; Wu H.; Katritch V.; Stevens R. C.; Jacobson K. A. (2012) Evaluation of molecular modeling of agonist binding in light of the crystallographic structure of an agonist-bound A(2)A adenosine receptor. J. Med. Chem. 55, 538–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S.; Hall S. E.; Li H.; Vaidehi N. (2008) Ligand-stabilized conformational states of human beta(2) adrenergic receptor: insight into G-protein-coupled receptor activation. Biophys. J. 94, 2027–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf C.; Rognan D. (2008) Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic receptor. J. Med. Chem. 51, 4978–4985. [DOI] [PubMed] [Google Scholar]
- Katritch V.; Reynolds K. A.; Cherezov V.; Hanson M. A.; Roth C. B.; Yeager M.; Abagyan R. (2009) Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J. Mol. Recognit. 22, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar S.; Karpiak J.; Berk B.; Costanzi S. (2011) In silico analysis of the binding of agonists and blockers to the beta2-adrenergic receptor. J. Mol. Graphics Modell. 29, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hert J.; Irwin J. J.; Laggner C.; Keiser M. J.; Shoichet B. K. (2009) Quantifying biogenic bias in screening libraries. Nat. Chem. Biol. 5, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galandrin S.; Oligny-Longpre G.; Bouvier M. (2007) The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol. Sci. 28, 423–430. [DOI] [PubMed] [Google Scholar]
- Leach K.; Sexton P. M.; Christopoulos A. (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci. 28, 382–389. [DOI] [PubMed] [Google Scholar]
- Kenakin T.; Watson C.; Muniz-Medina V.; Christopoulos A.; Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S.; Rajagopal K.; Lefkowitz R. J. (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discovery 9, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E.; Ahn S.; Shukla A. K.; Lefkowitz R. J. (2012) Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J.; Coleman R. G.; Setola V.; Irwin J. J.; Fan H.; Schlessinger A.; Sali A.; Roth B. L.; Shoichet B. K. (2011) Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat. Chem. Biol. 7, 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M.; Zhang C.; Lyons J. A.; Holl R.; Aragao D.; Arlow D. H.; Rasmussen S. G.; Choi H. J.; Devree B. T.; Sunahara R. K.; Chae P. S.; Gellman S. H.; Dror R. O.; Shaw D. E.; Weis W. I.; Caffrey M.; Gmeiner P.; Kobilka B. K. (2011) Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 469, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S.; Vilar S. (2011) In Silico screening for agonists and blockers of the beta(2) adrenergic receptor: Implications of inactive and activated state structures. J. Comput. Chem. 33, 561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. (2012) Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N.; Shoichet B. K.; Irwin J. J. (2006) Benchmarking sets for molecular docking. J. Med. Chem. 49, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Shoichet B. K. (2010) Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 50, 1561–1573. [DOI] [PubMed] [Google Scholar]
- Strader C. D.; Candelore M. R.; Hill W. S.; Sigal I. S.; Dixon R. A. (1989) Identification of two serine residues involved in agonist activation of the beta-adrenergic receptor. J. Biol. Chem. 264, 13572–13578. [PubMed] [Google Scholar]
- Liapakis G.; Ballesteros J. A.; Papachristou S.; Chan W. C.; Chen X.; Javitch J. A. (2000) The forgotten serine. A critical role for Ser-2035.42 in ligand binding to and activation of the beta 2-adrenergic receptor. J. Biol. Chem. 275, 37779–37788. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. (2005) ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Sterling T.; Mysinger M. M.; Bolstad E. S.; Coleman R. G. (2012) ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F.; Binkowski B. F.; Butler B. L.; Stecha P. F.; Lewis M. K.; Wood K. V. (2008) Novel genetically encoded biosensors using firefly luciferase. ACS Chem. Biol. 3, 346–351. [DOI] [PubMed] [Google Scholar]
- Barnea G.; Strapps W.; Herrada G.; Berman Y.; Ong J.; Kloss B.; Axel R.; Lee K. J. (2008) The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. U.S.A. 105, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S.; Ahn S.; Rominger D. H.; Gowen-MacDonald W.; Lam C. M.; Dewire S. M.; Violin J. D.; Lefkowitz R. J. (2011) Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol. 80, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. (2011) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, 1100–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hert J.; Willett P.; Wilton D. J.; Acklin P.; Azzaoui K.; Jacoby E.; Schuffenhauer A. (2004) Comparison of topological descriptors for similarity-based virtual screening using multiple bioactive reference structures. Org. Biomol. Chem. 2, 3256–3266. [DOI] [PubMed] [Google Scholar]
- Wisler J. W.; DeWire S. M.; Whalen E. J.; Violin J. D.; Drake M. T.; Ahn S.; Shenoy S. K.; Lefkowitz R. J. (2007) A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 16657–16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser M. J.; Roth B. L.; Armbruster B. N.; Ernsberger P.; Irwin J. J.; Shoichet B. K. (2007) Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 25, 197–206. [DOI] [PubMed] [Google Scholar]
- Evers A.; Hessler G.; Matter H.; Klabunde T. (2005) Virtual screening of biogenic amine-binding G-protein coupled receptors: comparative evaluation of protein- and ligand-based virtual screening protocols. J. Med. Chem. 48, 5448–5465. [DOI] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Samama P.; Cotecchia S.; Costa T.; Lefkowitz R. J. (1993) A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 268, 4625–4636. [PubMed] [Google Scholar]
- Allen J. A.; Yost J. M.; Setola V.; Chen X.; Sassano M. F.; Chen M.; Peterson S.; Yadav P. N.; Huang X. P.; Feng B.; Jensen N. H.; Che X.; Bai X.; Frye S. V.; Wetsel W. C.; Caron M. G.; Javitch J. A.; Roth B. L.; Jin J. (2011) Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U.S.A. 108, 18488–18493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Sassano M. F.; Zheng L.; Setola V.; Chen M.; Bai X.; Frye S. V.; Wetsel W. C.; Roth B. L.; Jin J. (2012) Structure--functional selectivity relationship studies of beta-arrestin-biased dopamine D(2) receptor agonists. J. Med. Chem. 55, 7141–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.