

Abstract
Transmissible spongiform encephalopathies (TSEs), or prion diseases, represent a unique form of infectious disease based on misfolding of a self-protein (PrPC) into a pathological, infectious conformation (PrPSc). Prion diseases of food animals gained notoriety during the bovine spongiform encephalopathy (BSE) outbreak of the 1980s. In particular, disease transmission to humans, to the generation of a fatal, untreatable disease, elevated the perspective on livestock prion diseases from food production to food safety. While the immediate threat posed by BSE has been successfully addressed through surveillance and improved management practices, another prion disease is rapidly spreading. Chronic wasting disease (CWD), a prion disease of cervids, has been confirmed in wild and captive populations with devastating impact on the farmed cervid industries. Furthermore, the unabated spread of this disease through wild populations threatens a natural resource that is a source of considerable economic benefit and national pride. In a worst-case scenario, CWD may represent a zoonotic threat either through direct transmission via consumption of infected cervids or through a secondary food animal, such as cattle. This has energized efforts to understand prion diseases as well as to develop tools for disease detection, prevention, and management. Progress in each of these areas is discussed.
1. Introduction
Traditionally infectious diseases were limited to the categories of bacterial, viral, fungal, and parasitic. Despite their differences, these agents are all unified by the requirement of genetic material for protein expression and replication of the agent. More recently, however, a novel infectious disease, which appeared to lack its own genome and is capable of causing devastating pathology in humans and animals, was identified. The causative agent of these diseases has been termed a proteinaceous infectious agent or “prion.” Eventually, after much resistance from the scientific community, it was established that a new class of infectious disease had been discovered.
2. History
While descriptions of prion diseases in sheep can be traced back hundreds of years, many of the pivotal experiments which defined the unique nature of the prion diseases did not occur until the 1950s. Specifically, many of the pivotal discoveries of prion diseases reflected efforts of anthropologists, pathologists, and physicians in Papua New Guinea where a novel form of neurological disease called kuru was described. Initially attributed to a “slow virus,” further research in the 1970s revealed that this new agent lacked the qualifying features (nucleic acid) necessary for its classification as a virus. The discovery of kuru stimulated research which led to the identification of a broader group of neurodegenerative disorders which were collectively referred to as transmissible spongiform encephalopathies (TSEs). Interestingly, the concept of slow viruses was first described by Sigurdsson while researching neurodegenerative disease in sheep [1], and later, in 1959, a veterinary pathologist hypothesized that kuru and this neurodegenerative disease of sheep, scrapie, were related disorders. At the same time, similar assumptions about kuru and another human neurodegenerative disease called Creutzfeldt-Jacob disease were made [2]. Ultimately work by Gajdusek's group, in which they successfully transmitted kuru to chimpanzees, changed the concepts regarding the cause of neurodegenerative disorders by indicating that a transmissible agent was responsible for the disease [3, 4].
3. Prion Diseases
Prion diseases have been shown to occur in a number of different species. Different names were attributed to each of the diseases based on the affected species: scrapie in sheep, Creutzfeldt-Jacob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) in cattle, and chronic wasting disease (CWD) in cervids. While these diseases all seem to share a common mechanism, they display a number of species-specific differences in symptoms, pathology, and transmissibility (Table 1).
Table 1.
Comparative analysis of prion diseases of food animals.
| TSE | Age of onset (yr) | Disease variants | Mode of transmission | Genetic predisposition | Clinical signs | Cross-species infectivity |
|---|---|---|---|---|---|---|
| Scrapie | 2–4 [33] | Classical atypical/Nor98 | Amniotic fluid and placenta [34] | Susceptibility 136V, 154R, 171Q [15] Reduced susceptibility 136A, 154R, 171R [16] |
Weight loss Ataxia Abnormal behavior Sensory changes Incoordination Head tremors Recumbency [35] |
Cattle, goat, mouse, hamster, rat, bank vole, deer, and elk [36–38] |
|
| ||||||
| BSE | 4-5 [39] | Classical H-type L-type |
Contaminated feed [39] | Susceptibility [40, 41] 23 bp PRNP promoter insertion/deletion 12 bp PRNP intron insertion/deletion E211K |
Weight loss Ataxia Abnormal behavior Sensory changes Incoordination Bradycardia Reduced rumination Decreased milk yield [42] |
Bison, sheep, goat, deer, pig, mink, marmoset, cat, mouse, human, nonhuman primates [43] |
|
| ||||||
| CWD | 2–4 [24] | Possible varied conformer population [44, 45] | Animal contact and environmental contamination [20] | Susceptibility [20] Elk-132MM Mule Deer-132SS–225SS Cervid rigid Loop-S170N [46] -N174T [47] Reduced susceptibility [48] White tail Deer-Q95H-G96S |
Weight loss Abnormal behavior Listlessness Incoordination Polydipsia Polyuria Hypersalivation Lowering of the head and ears Anorexia [21] |
Moose, cattle, sheep, goat, mouse, hamster, ferret, mink, and squirrel monkey [20] |
In sheep and cattle, the symptoms of prion disease are quite similar. The symptomatic phase of the disease initially presents with rubbing of the head, flanks, and buttocks, a symptom which may last well into the later stages of the disease. Also evident at the early stages is the nibbling of hair on the lower legs. General weakness follows which progresses to the inability to rise. Wasting symptoms appear, such as a loss in body weight despite normal food and water intake. Neurological motor deficits affecting gait include wide-based stance of hind limbs and high-stepping motions in the front limbs, which in severe cases makes the animals appear intoxicated. Further neurological changes include body tremors and personality/behavioural alterations which may manifest as uncooperativeness, excitability, and extreme distress. Once an animal is symptomatic, scrapie and BSE are slowly progressive and lethal within 2 to 6 months [5, 6].
Symptoms of CWD are more subtle in the early stages of disease. They include weight loss, excessive drinking, and urination as well as regurgitation. The motor deficits common to scrapie and BSE are often absent in CWD cases [7]. Despite the lack of easily observable symptoms, histopathology reveals neurological lesions similar to scrapie, BSE, and human prion diseases [8]. It takes anywhere from several weeks to many months for the infected animal to succumb to CWD once it has spread to the nervous system. Some of the more notable similarities and differences between scrapie, BSE, and CWD are presented in (Table 1).
4. Scrapie
Scrapie is almost certainly an ancient disease. Historical descriptions of scrapie can be conclusively traced back to 1732, while other sources claim there are descriptions of scrapie as early as the Roman era [9]. The term “scrapie” was first seen in a paper from 1750, but a more common term for it at the time was “distemper” [10]. The majority of written accounts were not produced by scientists or veterinarians, but by farmers, shepherds, and government employees. These individuals were immediately involved with the infected animals and sometimes went to great efforts to cover up the outbreaks because of the associated monetary/economic implications [9]. Similar to current management strategies, acknowledgment of a scrapie-infected animal meant the entire herd would be culled and pasture land quarantined indefinitely [11]. This behaviour indicated a basic understanding of scrapie as an infectious entity.
While historical descriptions of scrapie symptoms were accurate, the understanding of what caused the disease varied greatly. The range of hypotheses of causative agents include thunder, extreme atmospheric conditions, food/nutritional insufficiency, parasites, humidity in the sheep pen, tail docking, mating age, cross-breeding, and inbreeding [9]. The method of transmission was equally debated with ideas ranging from sexual transmission [10], sporadic occurrence, and genetic inheritance. In 1936, after 100 years of failed attempts to experimentally demonstrate scrapie transmissibility, two French scientists successfully infected sheep and goats intracerebrally, intraocularly, epidurally, and subcutaneously using brain and spinal cord suspensions from scrapie-infected sheep [12]. Their experiment confirmed a number of facts that scrapie was infectious, that infection required a long incubation period (18 months), and that the incubation period was affected by species. The infected sheep demonstrated a quicker symptom onset (11–22 months) than goats (25-26 months) [13].
Even within species, sheep possess a unique attribute with regards to TSE transmission. Their susceptibility is influenced by polymorphisms at three different codons (136, 154, and 171) within the PrP sequence [14]. It has been observed that susceptibility to scrapie strongly correlates with valine (V) at position 136, arginine (R) at 154, and glutamine (Q) at 171 (VRQ) [15], whereas resistance is attributed to alanine (A) at 136, with R at 154 and 171 (ARR) [16]. Prion disease is rare in heterozygous ARR animals and virtually undetected in animals homozygous for ARR [17]. These varying susceptibilities to scrapie infection led to a breeding strategy in Europe where sheep flocks were bred to attain the ARR genotype in an attempt to eradicate the transmission of scrapie among herds [18]. Concerns have been raised, however, that sheep selected from these breeding programs may have greater susceptibility to nontraditional forms of scrapie. Specifically atypical forms of scrapie have been reported in resistant sheep, and a novel form of scrapie, Nor98, primarily affects sheep resistant to conventional scrapie [19].
5. Chronic Wasting Disease
CWD was first identified at a research station in Fort Collins, Colorado in 1967 [20]. It was not until 1978, however, that the relationship between CWD and other TSEs was recognized following a comparative analysis of neuronal lesions [21] and the accumulation of PrP aggregates was observed [22]. During the 1970s and 80s, CWD existed primarily in the Colorado Rocky Mountains and an area extending along the river valleys draining into Wyoming [23]. However, in 1996, the disease was also detected on an elk farm in the Canadian prairies [7]. To date, CWD has been detected in wild cervids in 17 US states and 2 Canadian provinces. CWD is now known to be one of the most contagious TSEs and can reach a prevalence of 30% in wild populations and as high as 100% in captive cervids [20].
Strategies for containing CWD are extremely problematic because the afflicted animals primarily consist of wild, free-ranging populations. Other complicating factors include the scavenging of contaminated deer carcases by predators ranging from mountain lions to vultures, persistence of CWD prion protein in soil, and a poor understanding of the natural routes of disease transmission [7]. Although oral cross-species transmission to humans has not been directly observed, it was suspected in a few cases of human CJD that the disease may have contracted through eating CWD-infected deer meat [24].
There is also considerable concern that environmental contamination through the increasing prevalence of CWD in wild cervid populations may serve as a reservoir for CWD transmission to farmed deer and cattle. The close ecological and phylogenetic relationship of elk and deer to cattle supports the potential for eventual disease transmission [7]. While the BSE outbreak of the 1980s was successfully addressed through improved feeding practices, it would be more difficult to manage sources of infection from wild animals and/or environmental contamination. It is reassuring that cattle orally inoculated with CWD-infected brains, as well as cattle housed with CWD-positive deer, do not contract a TSE [8]. That cattle inoculated intracerebrally with CWD material do develop a TSE [9, 10] indicates the potential, given the right circumstances, for CWD to overcome this species barrier. Additionally, in the wild, there may be a greater opportunity to overcome this species barrier through intermediate species linking cervids to cattle. While it is difficult to predict the precise nature of the disease that would occur in cattle should CWD transmit from cervids to cattle, the very perception of the disease would be certain to have devastating consequences to the livestock industry.
6. Bovine Spongiform Encephalopathy
Since the first BSE diagnosis in 1986 more than 179,000 positive cases have been identified in Great Britain [25] triggering the culling of 750,000 animals [26, 27]. Experts suggested that as many as 3 million BSE-infected animals entered the human and animal food chains undetected [26]. While the recycling of BSE-infected material through cattle feed was likely the primary factor in disease amplification, the source of the original BSE agent remains unknown [28]. It has been suggested that BSE developed spontaneously in cattle via a somatic or germ-line mutation of PrP or that cattle may have been infected by another species such as sheep [29]. Twenty years of rigorous surveillance following the first BSE outbreak have indicated that disease transmission is under control in most of Europe [30]. Mass screening of bovine tissues as part of the surveillance initiatives has identified two new BSE strains, L-Type and H-Type, which are distinct from the classic BSE [31]. These strains are more rare than classic BSE and are usually detected in older animals, thus representing a possible sporadic form of TSE in cattle [32]. Both strains are infectious, maintain their original molecular phenotype upon transmission, and cause distinct neuropathology. In mouse infection studies, the L-type has been observed to be more virulent, replicating faster than both classical BSE and the H-type strain [31].
Discovering the link between BSE and the vCJD outbreak of 1996 brought TSEs to the attention of the public and health professionals. While confirmed cases of vCJD are only at 188 worldwide [49], the potential spread via contaminated blood or tissue products taken from asymptomatic carriers is of great concern.
7. Creutzfeldt-Jacob Disease
Prion diseases of livestock animals are the priority of this report. However, given the demonstrated zoonotic potential of BSE, and the concern that CWD may also represent a zoonotic disease, a brief overview of human prion diseases is appropriate to provide context of the different attributes of the various prion diseases. In the 1950s, an epidemic spreading amongst the Fore people of Papua New Guinea gained the attention of health officials. At the peak of the epidemic, as many as 10% of the local Fore population was infected [50]. The disease appeared to affect the central nervous system and displayed symptoms such as progressive ataxia or loss of motor coordination [51]. Derived from the appearance of these symptoms, the disease was given the name kuru, which in the Fore language means “to shiver” [52]. William Hadlow, a veterinary pathologist specialising in TSEs, made the connection between scrapie and kuru in 1959. He noted that epidemiologically, clinically, and pathologically, the two diseases were remarkably similar [53]. This led to further investigations by Gajdusek who was able to demonstrate that kuru was transmissible by successfully infecting chimpanzees [3].
Epidemiological studies suggested that kuru was transmitted by the cannibalistic Fore funeral rituals [54]. These ceremonies involved the children and women consuming nervous tissue from the deceased relative [55]. The number of kuru cases declined steadily following the discontinuation of the funeral ceremonies. Complete eradication of kuru has taken much longer than anticipated due incubation periods lasting as long as four decades [52].
Currently, the most common form of prion disease in humans is sporadic CJD (sCJD) with a frequency of approximately 0.5–1 case per million people [56]. Unlike the familial forms of CJD, which are caused by autosomal dominant traits linked to mutations on the Prnp gene [57], it is unknown whether sCJD is caused by endogenous or exogenous factors [58]. Countries with active surveillance programs, such as Switzerland, often report higher CJD incidence rates with 3 cases per million people [59]. Most of those cases are, however, due to iatrogenic spread rather than ingestion of infected material [60]. Symptoms in sporadic cases usually appear around the age of seventy and are lethal within weeks or months. Symptoms often include rapidly progressing dementia, ataxia, muscle twitching, and uncoordinated speech [56].
Following the BSE crisis in the United Kingdom in the 1980s, a novel form of CJD emerged. Given the name “variant CJD” (vCJD), this new form of prion disease appeared to be linked to the consumption of BSE contaminated meat products. The two main differences between sporadic and vCJD are the dramatic decrease in age at onset of symptoms (29 years) and the increased duration of symptoms (16 months) prior to death [61]. Some additional symptoms that are typically rare in sporadic CJD like involuntary movements and sensory symptoms have been observed in 100% and 50% of symptomatic vCJD patients, respectively [62].
As of 2003, the number of autopsy-confirmed cases of vCJD was 153, and currently the overall number of individuals infected is estimated at 200 (http://www.bseinfo.org/). While these numbers are low, the number of people actually infected with vCJD and not showing symptoms is likely much higher. Due to incubation times that can last decades, there is concern that there may be a population of asymptomatic carriers harbouring infectious PrP in the absence of observable symptoms. Without an accepted premortem diagnostic tests, health professionals are unable to identify these individuals. This creates a potentially dangerous situation as evidence suggests that vCJD can be transmitted via contaminated blood products [63], infectious cadaveric tissues, and contaminated surgical instruments [64].
8. Understanding Prion Diseases
Subsequent to the observation that TSE neurodegeneration reflects an infection, an exhaustive search for the traditional signs of infectious agent began. Populations of T and B cells were analyzed in mouse models of scrapie infection, but no differences between the experimental and control groups were detected [65]. Experiments looking at the potential influence B cell maturation on scrapie infections showed no difference in disease incubation or progression between wild-type mice and mice deficient in mature B cells [66]. Attempts to detect a scrapie-induced humoral response also failed [67]. It was determined, however, that scrapie infections involved the lymphoreticular system, independent of the site of inoculation, as high scrapie titers were detectable in spleen and other lymphoid organs [68, 69]. Thus, while many researchers had shown scrapie to be transmissible disease, initial efforts to characterize it from an immunological perspective indicated scrapie behaved in a fashion distinct from traditional infectious agents.
With scrapie infection studies producing perplexing results, Stanley Prusiner focused on characterizing the infectious component. Sedimentation experiments demonstrated that the scrapie agent aggregated with cellular components [70] and was stable in nonionic and anionic detergents [71]. Other experiments revealed it to be a hydrophobic molecule, a characteristic that complicates purification procedures and allows for extreme heat stability [72]. Several key pieces of evidence indicated that one of the required components of infectivity was a protein; digestion with proteinase-K or protein denaturation via sodium dodecyl sulfate (SDS), trypsin, phenol, urea, and chaotropic salts all led to a loss of infectivity [73].
Early attempts to identify the genetic component associated with the scrapie agent began with pH stability studies. Infectious material was inactivated by alkali pH, whereas acidic treatments had no effect [71]. Because alkali conditions disrupt both proteins and nucleic acid, the results were unable to rule out the presence of nucleic acids in association with the scrapie agent [71]. Further experiments, in particular ones that determined the scrapie agent was resistant to nuclease digestion and ultraviolet irradiation, suggested there was no nucleic acid content in the scrapie agent [71]. Collectively, this extensive molecular analysis determined the unique nature of the scrapie agent. Specifically, its resistance to nucleic acid degradation treatments, small size, and resistance to heat inactivation suggested the scrapie agent was a new type of infectious agent. Prusiner labelled these agents as “prions” which he defined as “small proteinaceous infectious particles which are resistant to inactivation by most procedures that modify nucleic acids” [71].
9. Cellular PrP
The human version of PrPC is a 253 amino acid (aa) protein which is encoded by the prion gene Prnp located on the short arm of chromosome 20. In addition to regulatory regions such as heat shock elements, Prnp is composed of three exons [74]. The discovery that the entire open reading frame is contained within a single exon (the third one) helped eliminate the possibility that the infectious form of the protein results from alternative RNA splicing [75]. PrPC mRNA is constitutively expressed in many different tissues with the highest levels of expression in neurons [76], but there are also significant levels of expression in the heart [77], skeletal muscle [78, 79], lymphoid tissue/white blood cells [80, 81], gut tissues [82], and reproductive tissues such as the testes and uterus [83]. Little is known regarding the regulation of PrPC expression other than the entire first intron is required for full promoter activity [84], and heat shock elements are responsible for upregulating expression during periods of cellular stress [85].
10. PrPC Structure
The cellular prion protein (PrPC) is a highly conserved glycosyl-phosphatidyl-inositol (GPI-) anchored membrane protein. Before PrPC is delivered to the outer cell membrane, and it is targeted to the endoplasmic reticulum (ER) where the N-terminal signal peptide is cleaved and a GPI anchor is added to the C-terminus at serine 231, creating a 209 amino acid protein [86]. At the outer membrane, PrP associates with lipid rafts and is cycled between the cell surface and the endocytic compartment at approximately 60-minute intervals with 95% of the internalized protein being recycled back to the cell surface [87]. Nuclear magnetic resonance (NMR) studies have determined that PrPC has a flexible N-terminal unstructured domain which contains four octapeptide-repeat regions and a globular C-terminal domain consisting of three α-helices and two antiparallel β-sheet structures [88]. There is a hydrophobic sequence in the middle of the protein from aa 113–135 which some suggest may serve as a transmembrane domain in some prion isoforms [89]. Additionally, there is a single disulphide bond between cysteine residues 179 and 214 and two N-glycosylation sites at asparagine residues 182 and 198 [31].
Prion structural comparisons among species indicate great similarity, which is anticipated with 90% sequence homology [90]. Cervid PrP, however, contains a unique and well-defined rigid loop located between α-helix 2 and the connecting β-strand (aa 166–175). This same region in other species is flexible and disordered. Further investigations revealed that the rigidity of the loop in cervids is due to a two-amino acid substitution S170N and N174T [7]. Structural differences in PrPC have been shown to influence a species susceptibility to PrPSc. Consequently, it has been hypothesized that the rigid loop structure in cervid PrP may be a contributing factor to the rapid horizontal CWD transmission [7].
11. PrPC Function
Despite the highly conserved sequence across species and ubiquitous expression within the body, little is known about the function of PrPC. It has been hypothesized that PrPC may be involved in multiple functions: copper metabolism, due to its binding affinity for copper; neuroprotective function due to its antiapoptotic activity [91, 92]; signal transduction [93, 94]; synapse formation/function [95]; neuritogenesis [96].
Copper has an important physiological role in all organisms, functioning as a cofactor in processes such oxidative stress protection, blood clotting, normal cell growth and development, respiration, and iron transport [97]. Copper deficiencies in humans have been linked to serious diseases which include Menkes syndrome [98], and various neurodegenerative disorders such as Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), and transmissible spongiform encephalopathies (TSEs) [99]. Numerous studies have shown that the histidine-containing octapeptide repeat region of the PrPC protein is capable of binding up to four Cu2+ ions [100]. These copper ions stimulate a conformational change in PrPC resulting in a structure that has some characteristics which are similar to the infectious conformation, including a higher β-sheet content, increased protease resistance, and propensity to aggregate, but the structure is distinct from the PrPSc isoform [101]. It was also found that micromolar concentrations of copper induce endocytosis of cell-surface-associated PrPC [102]. Consequently, it has been suggested that PrPC has a role in copper uptake/efflux and may also serve as a copper reservoir at the cell surface without stimulating endocytosis [103].
One model which has been used to explain the pathogenesis of several neurodegenerative disorders is chronic oxidative stress. Impairment of mitochondrial function, increased oxidative damage, defects in the ubiquitin-proteosome system, protein aggregation, altered iron metabolism, excitotoxicity, and inflammation have all been linked to oxidative stress in the brain [104]. Studies of oxidative stress in neurons suggest that PrPC protects against reactive oxygen species (ROS) [105]. Brown et al. proposed three possible mechanisms by which this may occur. One explanation is that the PrPC acts directly on the ROS via a copper-dependent superoxide dismutase (SOD) activity [106]. SODs are enzymes which remove ROS by converting them into a more stable H2O2 [104]. A second theory is that PrPC acts indirectly by upregulating activities of molecules like Cu-Zn SOD [103]. The last hypothesis posits that PrPC may prevent activation of ROS-induced apoptosis [104].
In vitro signalling analysis and prion knockout (PrP0/0) studies indicate that PrPC mediates interaction between several signalling transduction pathways including protein kinase A, Fyn, phosphatidylinositol 3-kinase (PI3K)/Akt, and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK). These pathways are known to induce neuronal survival [94, 107]. Studying induced ischemic injuries in neuronal tissue of PrP0/0 and wild type (WT) mice, it was demonstrated that the absence of PrPC exacerbates ischemic brain injury. The authors hypothesized this was due to reduced activation of the antiapoptotic pathway PI3K/Akt which in turn resulted in increased caspase-3 activity [108].
Protein localization studies determined that PrPC was present in the presynaptic region of the axon terminus [109]. This observation is supported by evidence of synapse loss, PrPSc accumulation at the synaptic terminal [110], and abnormal electrophysiological recordings during prion disease progression [111]. In the terminal stages of disease, accumulation of PrPSc in synaptosomes was found to coincide with alterations in the gamma-aminobutyric acid (GABA) system which is involved in inhibition of excitatory glutamatergic transmission [112]. The synaptic involvement of PrPC may also be linked to circadian rhythms [113] and the impaired hippocampal-dependent spatial learning phenotype in PrP-deficient mice [114].
12. Infectious PrPSc
The infectious component of prion diseases appears to be an insoluble β-sheet-rich version of the normal, nonpathogenic α-helical PrPC. While the primary structures of the two conformations are identical, differences in secondary and tertiary structure account for the unique properties of PrPSc [115]. Unlike PrPC, the infectious isoform is resistant to protease K (PK) digestion and readily forms insoluble aggregates [116]. These aggregates are composed of hydrolyzed fragments of the PrPSc protein, referred to as PrP 27–30 to reflect their molecular weights that range between 27 to 30 kDa. While X-ray and NMR structures have been determined for PrPC, the tendency for PrPSc to aggregate renders the isomer unsuitable for most types of structural analysis. Through techniques such as circular dichroism, which offer less specific structural information, it has been determined that there is an increase in β-sheet content from 3% to 42% as the conformation changes from PrPC to PrPSc [117].
Although PrPSc is generally described as the infectious conformation, subtle variations exist which make identifying the exact infectious component more challenging. One variation, referred to as PrPSensitive (PrPSen), is more susceptible to PK digestion than another variation, PrPResistant (PrPRes). While most PrPSc-infected tissues contain PrPRes, its presence is not an absolute requirement for infectivity [118, 119]. PrPSen has also been identified in scrapie-infected tissue, making it difficult to discern which version is responsible for infectivity [120, 121]. There is evidence for the existence of multiple PrPSc isoforms, also referred to as strains, which possess unique infectivity, pathology, neurotropism, and biophysical traits.
13. Prion Strains
Strains within traditional pathogens, such as bacteria and viruses, are determined by nucleic acid sequences in their genome. The term “strain,” when used in the context of prion disease, refers to phenotypic differences such as variations in the pattern of aggregate deposition, incubation times, neuronal tissue tropism, and pathological morphology [122]. The mechanism of strain formation in prions is not attributable to differences in the encoding of primary PrP structure. Rather, these differences are thought to result from the conformational flexibility of the PrPSc structure which leads to exposure of distinct cleavage sites, differing stability in the presence of denaturing compounds [123], and altered ratios of di-, mono-, and unglycosylated forms [124]. The strain of an incoming infecting PrPSc molecule appears to be imprinted in all of the subsequent PrPSc proteins produced during the conversion of the host's cellular PrP [125]. It has been demonstrated that different strains can be passaged serially through inbred mice with identical PrP gene sequences [126]. Strains have also been reisolated in mice following passage through other species with differing PrP primary structures [127].
It has been shown that prion strain characteristics contribute to species barriers. Species barrier is a concept whereby the transmission of prion diseases between different species is much less efficient than within species [126]. For example, when species B is infected with prion from species A, the infectivity rate is low and disease progression, in successfully infected animals, is slow and unpredictable. In contrast, when species A is infected with prions from species A, infection rates are high and progression is more rapid with a more consistent disease course among animals. Interestingly, following the passage of prion from species A to B, subsequent passages of the same prion to another B animal now have the same disease progression as if the prion had originated from a species B animal. The type of strain influences how easily this species barrier is broken. For example, the prion strain responsible for the BSE crisis was transmissible to a variety of species, including humans [127].
14. Transmission of Infectious PrP
Natural transmission of TSEs likely occurs though ingestion of contaminated material (skin, dirt, decomposed carcasses, urine, contaminated placentas, and faeces) or by direct contact between animals [128]. Transmission of prion diseases between different species has been shown to be much less efficient than within the same species. This suggests that TSEs are limited by a species barrier further trans-species experiments have demonstrated that transmission does occur but that incubation times may be greatly extended so that clinical disease may or may not occur during the natural lifespan of the affected animal [129]. The concept of prion strains complicates the understanding of TSE transmission in that certain strains of PrPSc derived and transmitted within the same species can result in either the development of an asymptomatic carrier or the onset of clinical disease [130].
15. Pathogenesis of Prion Diseases
The formation of PrPSc is thought to occur through an interaction whereby an infectious PrPSc particle induces refolding/misfolding of PrPC to PrPSc. PrPC is an absolute requirement for infection and disease. Challenge of PrP-deficient mice with PrPSc material produces no accumulation of infectious material or disease. When PrPC expression is restored, typical prion disease pathology is observed [131]. Transgenic mice engineered such that PrP was produced without the GPI anchor failed to develop clinical symptoms following scrapie inoculation. Unlike PrP knock-out mice, however, the GPI anchor transgenic mice still produced PrPSc and amyloid plaque aggregation [132]. This illustrates the need for membrane-anchored PrPC for pathology to develop.
One research group investigated whether PrPC expression was necessary on all types of brain cells (neurons, astrocytes, and oligodendrocytes) for formation of PrPSc, development of disease, and transmission of infectivity. Several transgenic mouse lines were created with PrPC expression restricted to specific cell lineages. Mice with only neuronal PrP expression supported prion infection and developed clinical disease [133]. In contrast, when PrPC expression was restricted to oligodendrocytes, then prion disease failed to develop [134]. Mice with PrP expression restricted to astrocytes also developed disease symptoms despite the lack of neuronal PrPC [135]. It was suggested that deposition of PrPSc in close proximity to neurons and their processes was sufficient to induce pathology [136]. This would seem to contradict the work with soluble PrPC which suggested that membrane-anchored PrP was necessary for neuronal pathology [132]. Alternatively, PrPSc may have indirect effects on neurons by altering the function of associated cells.
Because the definitive function of PrPC has yet to be determined, the pathological mechanisms of PrPSc are also uncertain. One argument offers that PrPSc causes a “gain of function” whereby the presence of the misfolded protein adds a neurotoxic function. The opposing hypothesis is that PrPSc causes a “loss of function.” Some research has also shown that PrPSc itself is not neurotoxic due to the lack of correlation between PrPSc deposition and disease severity. It has been suggested that the conversion of PrPC to PrPSc is the key component in pathogenesis rather than the accumulation of PrPSc aggregates [137].
There are two main theories about the mechanism by which PrPSc induces the misfolding of PrPC, the template-directed refolding model, and the nucleated polymerization model. The first model postulates that incoming PrPSc initiates a catalytic cascade using PrPC, or a PrP intermediate, as a substrate for conformational conversion into a new β-sheet-rich protein. This new PrPSc protein then serves to convert the next PrPC molecule as the cycle continues. The refolding mechanism is kinetically controlled by a high activation energy barrier which prevents spontaneous conversion at detectable rates. It is thought that this energy barrier is lowered by the formation of an intermediate PrPC-PrPSc heterodimer complex which facilitates the full conversion of PrPC, with the help of a chaperone molecule, into the new PrPSc conformation [124].
The second model is based on a thermodynamically controlled nucleated polymerization reaction. This is a noncatalytic event whereby conversion of PrPC to PrPSc is technically a reversible process. At equilibrium, however, the natural cellular PrP isoform is highly favoured. The isoform conversion only occurs when a native PrP protein monomer comes into contact with an already formed PrPSc crystal seed or aggregate. Successful refolding into the PrPSc isoform is stabilized once the new protein is added onto the preformed seed. This model implies that infectivity requires the presence of PrPSc in oligomer form and that monomers are not infectious [124].
16. Peripheral Amplification
TSE infections are usually established following oral ingestion of infectious PrPSc material. Once in the digestive tract, it is proposed that there are three potential routes for PrPSc to cross the mucosal barrier. The first utilizes a specific subset of intestinal epithelial cells called microfold cells or M cells. These cells possess a thin apical membrane made up of mircofolds and an invaginated basal membrane which houses various immune cells. The thin membrane allows the transport of many types of bacteria, viruses, and other proteins, including prions, across the epithelial layer where they then encounter lymphocytes and dendritic cells [138]. This interaction between the immune cells and pathogenic antigen usually serves to stimulate a mucosal immune response. While the M cells facilitate uptake and transfer of Ag to the immune system, they may also serve as a portal of entry for pathogens like prions which are not degraded and do not stimulate an immune response. Therefore, prions are left to continue on their route of infection and interact with other cells, alternatively, while in the intestinal lumen, PrPSc can be degraded by digestive enzymes into smaller protease-resistant fragments. These fragments may form complexes with ferritin which are transported across the intestinal epithelium by a ferritin-dependent endocytosis mechanism [139]. A third mechanism of uptake has been proposed based on observations made during bacterial research and has yet to be directly linked to TSE transport. This involves direct sampling of prion protein by migratory bone marrow-derived dendritic cells which can travel from the blood vessels to the inner surface of the intestinal wall. These DCs then sample luminal contents, including prions, and trapped protein is transported to the lymphoreticular system which includes the draining mesenteric lymph nodes [140].
Immunohistochemistry reveals that once across the mucosal barrier, PrPSc accumulation occurs in a variety of lymphoid tissues including palatine tonsils, spleen, lymph nodes, and Peyer's patches [141]. More specifically, PrPSc replication and accumulation occurs within tingible body macrophages located in B-cell follicles and later on the plasmalemma of the follicular dendritic cells (FDCs) [16]. This amplification phase, particularly within the Peyer's patches, is important for PrPSc transfer to the nervous system.
Kitamoto et al. were instrumental in establishing the relationship between B cells, FDCs, and prion disease progression. B-cell-deficient mice were successfully infected with prion material via intracranial (IC) challenge, but failed to show any signs of disease following intraperitoneal (i.p.) challenge. Further investigation of the i.p.-inoculated mice revealed that PrPSc failed to accumulate in the spleen and brain despite having normal levels of FDCs [142]. Based on previous studies which illustrated that FDC development was a B-cell-dependent process, the Kitamoto group concluded that removal of the B-cell population had prevented maturation of the FDC population. It became evident that FDCs are essential for prion replication and disease progression. This was verified by reconstituting B cells in B-cell deficient mice, which had been given prion material i.p., and restoring susceptibility to prion disease.
Following the linkage of FDCs and prion replication, studies discovered a link between the inflammatory process (involving FDCs, B cells, macrophages, and other immune cells associated with germinal centers) and accumulation of PrPSc. In the presence of inflammation, the upregulation of FDC cytokines such as lymphotoxin resulted in an ectopic induction of PrPC-expressing FDCs [60]. As a result, nonlymphoid organs undergoing inflammation, for example, kidney, liver, and pancreas, show signs of prion accumulation. In contrast, these organs are prion-free under normal conditions [143]. Experiments have shown that in symptomatic and asymptomatic scrapie-infected mice, with nephritis, prion infectivity was also detectable in the urine [144]. Similar experiments in sheep with mastitis showed that infectivity was present in colostrum and milk [60]. This concept is problematic in the context of wild/farmed animal populations where PrPSc-infected individuals suffering from inflammatory processes in excretory organs may shed PrPSc material into the environment and increase disease transmission.
17. Neuroinvasion
The mechanism by which PrPSc travels from lymphoid tissue to the nervous system is not well understood. Experimental conditions that either remove or increase lymphoid innervation show that increased lymphoid innervation correlates with faster neuroinvasion [145]. The proximity of PrPSc expressing FDCs to the sympathetic nerve endings has been shown to influence rates of neuroinvasion. Whether there is passive diffusion of infectious PrP molecules or a facilitated transport system between FDCs and nerve endings is unknown [58].
The enteric nervous system (ENS) has been identified as the first neural tissue to be invaded by PrPSc following the peripheral amplification phase [141]. The ENS, a member of the autonomic nervous system, innervates the gut and is made up of two main networks, Meissner's plexus, located in the gut submucosa, and Auerbach's plexus, which runs between the circular and longitudinal muscle layer and extends from the esophagus to the rectum [146]. As a component of the autonomic nervous system, the ENS is directed by the sympathetic and parasympathetic input from the central nervous system via efferent nerves which connect to the enteric plexi [146]. Following infection of the ENS, PrPSc travels to the brain using mainly the sympathetic pathway via the Vagus nerve to the dorsal motor nucleus in the medulla oblongata, or the parasympathetic pathway via the splanchnicus nerve to the intermediolateral column of the spinal cord [147].
18. Neurodegeneration
The mechanisms for prion-induced neurodegeneration are closely related to the proposed functions of PrPC. This is to be expected as the TSE disease process is essentially the conversion of PrPC to PrPSc which leads to a reduction in the number of functioning cellular prion proteins, thus sparking the debate about whether prion pathology is due to a gain of a neurotoxic function or the loss of a neuroprotective function. Hur et al. proposed five mechanisms of prion neurodegeneration: increased oxidative stress and mitochondrial dysfunction; disruption of iron metabolism, altered calcium metabolism; increased inflammatory activity of cytokines, chemokines, and nuclear factor-kappa; finally, apoptosis [148].
It has been observed in TSE-infected animals that there is reduced activity of manganese SOD. This mitochondrial enzyme is responsible for converting superoxide anions into less reactive species. The resulting accumulation of ROS leads to ischemic cell injury and subsequently apoptosis or necrosis. Additional mitochondrial deficits, such as decreased cytochrome-c oxidase and ATPase activity, increased lipid peroxidation, and structural abnormalities, have been detected in hippocampal and cerebral cortical neurons of TSE-infected hamsters [149].
Increased iron metabolism within the brain (favouring a redox state of Fe+3), as a result of a TSE infection, is involved in exacerbating ROS injury by converting harmful free radicals, such as a superoxide anion, into an even more highly reactive hydroxyl radical [148].
Proper calcium regulation is essential for the normal functioning of the CNS. Variations in intracellular calcium concentrations facilitate the coordination of electrochemical signals, neurite growth, synaptogenesis, synaptic transmission, cell survival, and plasticity [150]. In vitro experiments showed that in PrPSc-infected cells, there was a downregulation of N-type voltage-gated Ca2+ channels resulting in reduced cytosolic Ca2+ [151]. PrPSc aggregates that accumulate in the synaptic cleft may physically interrupt synaptic transmission of electric potentials created by Ca2+-activated potassium channels, or compromise the stability of newly formed synapses [150]. This can result in excitotoxicity [152] and ischemic brain damage [153]. The interference with the development of long-term potentiation also has negative effects on processes such as learning and memory [150].
19. Prion Diagnostics
With the lack of accepted premortem diagnostic tests, health professionals have to rely on clinical examinations for diagnosis of TSEs [154]. Definitive confirmation, however, can only be made by postmortem histological or biochemical analysis of brain tissue [155]. While histological assays are accurate, they are time consuming, labour intensive, and low throughput. Development of new biochemical tests has allowed multiple samples to be processed in a few hours. The majority of the tests have detection limits ranging from 0.5 pmol to 20 pmol and utilize immunodetection of the PrPSc isoform or proteolysis to distinguish PrPC from the infectious conformation [154].
To enable the detection of peripheral PrPSc in preclinical cases, Saborio et al. developed a method of PrPSc amplification called protein misfolding cyclic amplification (PMCA) [156]. Analogous to the polymerase chain reaction method used to amplify DNA, PrPC of the same species is added to a sample suspected to contain small quantities of PrPSc and incubated to allow for the expansion of PrPSc aggregates. These new aggregates are broken up using sonication to form more PrPSc seed molecules and are reincubated in the presence of added PrPC. After several amplification cycles, the amount of PrPSc is several millionfold higher than in the original sample [157], allowing detection using standard molecular techniques like Western blots. Concerns have been raised regarding the specificity of this technique after it was shown that PrPSc can be generated from samples lacking PrPSc [158]. A lack of specificity would limit the usefulness of PMCA as a diagnostic test.
The discovery of PrPSc infectivity in blood from vCJD patients [158, 159] emphasized the need for a blood test for prions. The first detection system was developed in 1996 and consisted of capillary electrophoresis and a competitive immunoassay which detected a PK-resistant C-terminal sequence of PrP [160–162]. Despite several revisions, the method was not suitable for routine testing [163]. More recent initiatives involve the use of a 15B3 prion-specific antibody developed by Prionics [164, 165] and a Seprion resin manufactured by Microsense. Both systems concentrate PrPSc so it is detectable by ELISA and flow cytometry. Another group designed fluorescent labelled palindromic peptides to discriminate PrPC from PrPSc. When the labelled peptide binds to PrPSc, a shift in conformation modifies the fluorescent properties of the label [166]. Despite the advances in blood diagnostics, the strict guidelines governing diagnostic requirements prevent their acceptance for use in an official capacity.
20. Historical Approach to Prion Therapy
For the last fifty years, there have been ongoing efforts to identify effective prion therapeutic agents. Typically, these agents have targeted either PrPC, PrPSc, or the conversion process. These efforts were energized with the emergence of vCJD in the 1990s and the discovery that prion infectivity can be transmitted via blood, surgical instruments, and transplant tissues from asymptomatic carriers [167]. Investigations of potential therapies have been primarily conducted in three experimental systems: cell-free in vitro conversion assays, cell-based models, or animal models [137]. The cell-free method involves the addition of a PrPSc seed molecule to PrPC substrate in the presence of a potential therapeutic agent. Therapeutic potential is evaluated based on the ability of the molecule to interfere with the conversion process. Cell based research utilizes cell lines such as N2a (mouse neuroblastoma cells) to assess the ability of a given therapeutic agent to either prevent infection of the cells by prions, or to clear PrPSc from a chronically infected cell line like ScN2a. These cell models allow for the analysis of some very basic aspects of prion pathogenesis such as isoform conversion, cytotoxicity, apoptosis, and abnormal cell signalling. Animal models, which make up the third experiment system, tend to be the most expensive, yet informative, model as many characteristics of a potential therapeutic can be analyzed. These include its ability to influence incubation phase of infection, severity of symptoms, pathology, immune response, and survival time and rate. Animal models are complicated, however, by issues such as genetic variability between and within species, inconsistent models of infection, and lengthy incubation periods.
Dating back to the 1960s, the main area of antiprion research was focused on chemotherapeutic agents [137]. As it was initially thought that prion diseases were the result of a slow virus, this biased the nature of the therapeutic agents which were investigated. Typical TSE targets/goals for chemotherapeutic agents have been sterilization of sources of infection; prion prophylaxis; interruption of PrPC conversion during peripheral amplification; prevention of neuroinvasion; reduction of PrPSc accumulation [168]. Countless agents have been tested including polysulphated polyanionic compounds, glycosaminoglycans, sulphonated dyes, quinacrines, metal chelators, tetrapyrroles, polylene antibiotics, tetracyclic compounds, and β-sheet breaker peptides [137, 168]. Very few therapies have shown significant success in vivo, particularly if delivered after symptoms had already developed [169]. This likely reflects the inability of many of these molecules to cross the blood-brain barrier which is required to interfere with prion-related pathogenesis in the CNS. However, even compounds which are able to penetrate the CNS, like curmunin, quinacrine, quinolones and polyene antibiotics, failed to improve symptoms of late-stage TSE disease [168].
The appreciation that prion diseases result from the misfolding of a self-protein, rather than a slow virus, impacted the efforts to develop therapeutic agents. Conceptually with a traditional infectious disease, there is a clear distinction between the host and pathogen. In contrast, the prion diseases, which result from the subversion of a normal and omnipresent component of the body, are conceptually closer to a cancer than a traditional infectious. Similar to challenges faced in cancer therapeutics, the issue of specificity takes on elevated importance as the distinction between host and pathogen becomes less clear.
21. Immunotherapy of Prion Diseases
While prion diseases represent a novel paradigm of infectious disease, there nevertheless appears to be an opportunity to apply traditional medicinal approaches to impact disease susceptibility and progression. In particular, there is proof-of-principle evidence from a number of models that a vaccine may be an effective tool to control prion diseases. There are numerous attempts to develop a prion vaccine which have focused on different epitopes, strategies of formulation/delivery which have been tested in a number of models of disease (Figure 1). For clarity, these efforts are grouped here according to investigations which evaluate the therapeutic potential of particular antibodies with in vitro systems and in vivo systems, which include subcategories of passive and active immunizations. The critical findings of these studies are presented in (Tables 2 and 3) for in vitro and in vivo investigations, respectively. An illustration of the various regions of the PrP protein which have been targeted in the various investigations is presented in (Figure 2).
Figure 1.
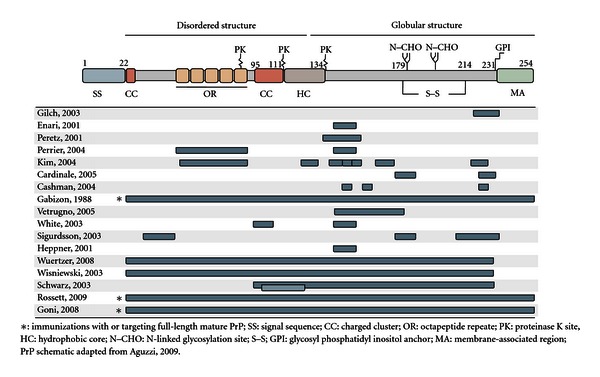
Regions of PrP targeted in the various immunotherapeutic investigations.
Table 2.
Comparative analysis of in vitro and ex vivo immunotherapeutic investigations of prion diseases.
| Region | Model | Efficacy | Reference | |
|---|---|---|---|---|
| mAb 3F4 (109–113) pSera; 23–33, 90–104, 143–156, 219–232 |
Cell-free conversion | pSera 219–232 disrupted formation of new PrPSc | Gilch et al. 2003 [178] | |
| mAb 6H4 (144–152) | ScN2a cell line | Clearance of PrPSc from cell line following antibody treatment | Enari et al. 2001 [177] | |
| Fab D18 (132–156) | ScN2a cell line | Blocked PrPSc formation and cleared existing PrPSc | Peretz et al. 2001 [179] | |
| In vitro | SAF34 (octarepeat) SAF61 (144–152) | ScN2a cell line overexpressing PrPC | Both Abs decreased PrPSc and PrPC in infected and uninfected cells | Perrier et al. 2004 [180] |
| Panel of antibodies to PrPC and PrPSc generated in PrP0/0 mice | ScN2a cell line | Capacity for protection independent of epitope but dependent on ability to bind cell surface PrPC to prevent internalization | Kim et al. 2004 [181] | |
| ER targeted scFv of 8H4 (175–185) and 8F9 (225–231) | Expression in PC12 cell line | Inhibition of PrPC translocation to cell surface; prevented PrPSc accumulation | Cardinale et al. 2005 [182] | |
| mAbs to “YYR” motif | ScN2a cell line | Reduced content of PrPSc | Cashman and Caughey 2004 [168] | |
|
| ||||
| Ex vivo | PrPC poly Ab ex vivo neutralization | Golden hamster | 2-log reduction | Gabizon et al. 1988 [174] |
| Fab D18 (132–156) Ex vivo neutralization |
ScN2a cell line | Mice lived 3 months longer (3-log reduction of infectivity) | Peretz et al. 2001 [179] | |
Table 3.
Comparative analysis of in vivo immunotherapeutic investigations of prion diseases.
| In vivo | ||||
|---|---|---|---|---|
| Treatment | Region | Model | Efficacy | Reference |
| Passive | scFv 8H4 (145–180) | Lysates from scFv PC12 cells i.c. injected into mice 35 days after scrapie exposure | 2/10 mice protected from 8/10 infection mice symptom-free after 300 days | Vetrugno et al. 2005 [183] |
|
| ||||
| Passive | Anti-PrP IgG Abs ICSM 35 (94–105) ICSM 18 (144–152) |
Mice challenged i.p. with scrapie, Ab treatment twice weekly | Signficant delay in prion symptoms. Reduction in splenic PrPsc and delayed transfer to brain | White et al. 2003 [184] |
|
| ||||
| Passive | mAb 8B4 (34–52) mAb 8H4 (175–185) 8F9 (205–233) |
Mice challenged i.p. with scrapie, weekly treatment with Ab | 8H4 and 8B4 10% longer incubation period at high challenge dose. At low challenge dose, 8B4 prevented disease in 10% of animals | Sigurdsson et al. 2003 [185] |
|
| ||||
| Passive (engineered) | Transgenic mice expressing 6H4 (144–152) as single chain-Ab | Mice challenged i.p. with scrapie | Prolonged survival by 120 days | Heppner et al. 2001 [186] |
|
| ||||
| Passive (engineered) | PrPC
specific scFv |
Expression specifically in CNS with recombinant adenoassociated vector type 2 viral vector platform | Delayed onset in peripherally inoculated mice | Wuertzer et al. 2008 [187] |
|
| ||||
| Active | rPrPC
23–230 |
s.c. vaccination of mice | 10% increase in incubation time; correlated with Ab titres | Sigurdsson et al. 2003 [188] |
|
| ||||
| Active | Peptide 105–125 rPrPC 90–230 | Mice immunized and orally challenged with infected brain homogenate | Peptide improved survival by 23 days; protein had no effect | Schwarz et al. 2003 [189] |
|
| ||||
| Active (engineered) | Human PrPC | Transfer of adenotransduced dendritic cells. Mice challenged i.p. | Prolonged survival times | Rosset et al. 2009 [190] |
|
| ||||
| Active (mucosal) | Salmonella expressing PrP | Four oral vaccinations with live salmonella; 2 with dead Challenge via oral lavage |
100% of mice expressing high IgA and IgG and 33% of mice with high-IgG and low-IgA symptom-free after 400 days | Goñi et al. 2008 [191] |
Figure 2.
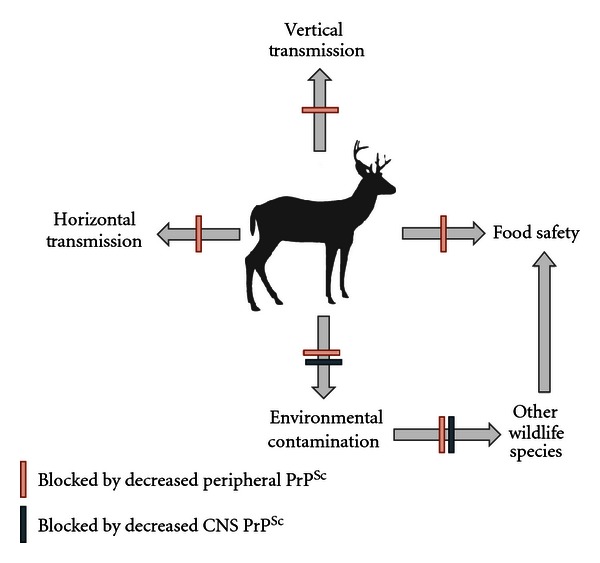
Potential points of impact of a prion vaccine. An effective CWD vaccine should decrease peripheral and CNS loads of individual animals, leading to decreased transmission both within and outside cervid populations, and decreased environmental shedding. These factors would lead to an overall reduction in available infectious prion and benefit both wild and food animals, as well as humans.
22. In Vitro Immune Therapy
Conversion of PrPC to PrPSc occurs at, or near, the cell surface [170, 171]. This provides an opportunity for antibodies to bind and prevent the interaction and conversion of PrPC by PrPSc. Such a strategy might be used as immunoprophylaxis to prevent infection of animals exposed to PrPSc or as therapy to treat infected animals, serving to delay or stop the progression of disease and minimize the shedding of infectious PrPSc. This approach is challenged by the fact that PrPC is a ubiquitously expressed, endogenous protein making it very difficult to stimulate an immune response. This is the result of an immunological phenomenon known as tolerance which occurs when the immune system recognizes a protein as being “self.” Consequently, immune cells, such as T and B cells, which have receptors specific to that particular protein, are deleted or prevented from initiating an immune response towards that particular protein [172]. Overcoming tolerance to PrPC remains one of the biggest challenges in prion vaccine research [173].
Some of the first demonstrations that antibodies could be used to neutralize prions came from experiments that demonstrated that ex vivo incubation of a prion inoculum with anti-PrPC polyclonal antibody, prior to inoculation, resulted in a 2-log reduction in infectivity [174]. Further work was done to identify the region of PrP involved in the interaction with PrPSc resulting in isoform conversion. This was done through the application of mAb 3F4 (specific for aa 109–113) [175] as well as polyclonal sera to peptides (corresponding to aa 23–33, 90–104, 143–156, and 219–232) in efforts to prevent PrPRes formation in a cell-free conversion system [174]. Antibodies to the region 219–232 were found to disrupt the formation of new PrPRes. The same effect was not observed during incubation with PrPRes seed. Furthermore, when PrPSen Abs and peptide 219–232 were coincubated, the Ab bound to the peptide enabling the uninterrupted formation of PrPRes. This illustrated that the inhibition of PrPRes formation was due to the direct binding of the Ab to the 219–232 region [176].
Preliminary immunotherapy research in 2001 showed that prolonged exposure (6 weeks) of ScN2a cells to mAb 6H4 (specific for aa 144–152 on the prion protein) resulted in the complete clearance of PrPSc offering evidence that anti-PrP Abs have the potential to interfere with the prion disease process [177].
That same year, Peretz et al. used recombinant antibody antigen-binding fragments (Fabs) to prevent PrPC conversion in ScN2a cells infected with PrPSc. They observed a direct relationship between the amount of antibody binding to surface PrPC and the inhibition of PrPSc formation. Their most effective Fab, D18, not only stopped PrPSc formation but also cleared preexisting PrPSc infection. In related challenge experiments, mice infected with scrapie-infected cells preincubated with the antibodies survived more than three months longer than those receiving untreated cells. The success of D18 was attributed to its ability to bind cell surface PrPC molecules [179].
In 2004, two antibodies, SAF34 (specific for the octarepeat region of PrPC) and SAF61 (specific for 144–152 of both PrPC and PrPSc), were studied in neuroblastoma cells overexpressing PrPC with and without PrPSc infection [180]. When used independently, each antibody decreased levels of PrPC and PrPSc in uninfected and infected cells, respectively and when used together had a synergistic effect. The authors suggested that SAF61s acted by promoting clearance of PrPC to remove the substrate required for PrPSc production. SAF34, although showing a similar end result, functioned through a different mechanism by blocking interaction between PrPC and PrPSc [177, 179]. The epitopes recognized by SAF34 and SAF61 correspond to a region of PrP thought to be involved in an interaction with the laminin receptor [192]. Consequently, the binding of SAF antibodies to PrP may also interfere with this process [180].
Using PrP0/0 mice, Kim et al. produced a panel of antibodies to either recombinant PrP or PrPSc [181] and evaluated the ability of these antibodies to protect ScN2a cells from infection. They concluded that antibody-mediated protection was not in the specificity of the epitope, but rather in the capacity of antibody to bind cell surface PrPC [193]. At the end of its natural cycle, surface-expressed PrPC becomes internalized via clathrin vesicles and then enters the degradation pathway [194, 195]. It is during this internalization process, at the level of the cell membrane, that the conversion from PrPC to PrPSc is believed to occur [171, 196]. The authors hypothesized that the binding of mAb to surface PrPC prevents internalization to inhibit the conversion process [193].
In contrast to targeting surface PrP, Cardinale et al. developed antiprion single-chain antibody fragments (scFv) with an endoplasmic reticulum retention signal to target intracellular PrP. Stable expression of these scFvs in nerve growth factor differentiating PC12 cells inhibited PrPC translocation from the ER to the cell surface and prevented PrPSc accumulation [182]. They later applied this concept to an in vivo model. Lysate from wild-type and scFv (8H4)-expressing PC12 cells was injected intracerebrally into C57BL mice, 35 days after exposure to scrapie. Mice that received wild-type PC12 cells succumbed to prion disease, whereas only 2/10 mice in the scFv (8H4) group were infected. At 300 days after infection, the eight mice of the experimental group remained symptom-free with no detectable histopathology [183].
23. In Vivo Immunization
The necessity for extracerebral PrPSc to undergo amplification in the periphery prior to neuroinvasion provides another opportunity for prophylaxis. However, due to immunological tolerance to PrPC, attempts to stimulate humoral responses against prion proteins in vivo have proven challenging. In 1993, Prusiner's group demonstrated that it is possible to raise antibodies to PrP in Prnp 0/0 mice. Aguzzi et al. speculated that the use of genes encoding high-affinity anti-PrP antibodies produced in Prnp 0/0 mice might be able to redirect B-cell responses in mice expressing PrPC. They took Prnp 0/0 and Prnp +/0 populations of mice and transgenically introduced genetic sequences which encoded for the heavy and light chains of 6H4, an mAb specific for PrPC. By four weeks of age, the Prnp 0/0 mice consistently produced high anti-PrP titers. In contrast, mice expressing PrP (Prnp +/0) developed anti-PrP antibodies more slowly. Unresponsiveness of B cells likely corresponds to the level of self-antigen presented [197] and B-cell receptor avidity/affinity [198]. Therefore, the delay in production of anti-PrP in Prnp +/0 mice may be due to negative selection of B-cells expressing 6H4 epitopes with high avidity for PrPC [186]. Consequently, the 6H4 produced by Prnp +/0 mice may have different binding properties than 6H4 produced by the Prnp 0/0 mice. All mice were inoculated i.p. with RML scrapie. Spleens from the nontransgenic Prnp +/0 group showed PrPSc accumulation, while the Prnp +/0-6H4 group did not. Brain samples, used to assess the ability of PrPSc to spread to the CNS, showed identical results to that of the spleen. To rule out unintended mechanisms of PrPSc protection, Western blots were used to determine whether the presence of 6H4 was causing reduced expression of PrPC. Equal levels of PrPC protein in both the transgenic and nontransgenic Prnp +/0 mice indicated this was not the case. Instead, it appears that protection was occurring via masking of PrPC sites critical for interaction with PrPSc. Interestingly, as PrPC is an abundant self-protein, the risk of inducing autoimmunity is always a concern; however, no obvious signs of autoimmune disease were detected in this study [186].
While the presence of anti-PrP antibodies in the peripheral compartments does not appear to have any negative consequences, Solforosi et al. investigated the effect of anti-PrPC antibodies on neuronal cells in vivo. PrPC D13 mAbs were injected into the hippocampus of C57BL/10 mice. Within 24 hrs, antibody-induced cross-linking of cell-surface PrPC resulted in extensive apoptosis of neurons in the hippocampal and cerebellar regions [199]. That administration of monovalent Fab fragments from D13, via the same methods, produced apoptosis at a much reduced rate suggests that the cross-linking event was most likely the cause of apoptosis. In contrast to the previous results, experiments with a D18 mAb specific for a region involved in PrPC-PrPSc interaction did not trigger apoptotic cell death. This may be because D18 was ineffective at cross-linking PrPC, or it obscured the region of PrPC which binds to a cofactor molecule necessary for apoptosis signalling [199].
24. Passive Immunization
Further enthusiasm for antiprion immunotherapy emerged when White et al. demonstrated that the passive transfer of anti-PrP IgG antibodies to wild-type mice, challenged i.p. with PrPSc, resulted in a significant delay in symptoms [184]. The effects were most noticeable when a high antibody dose was administered biweekly during the splenic amplification phase. The treatment decreased splenic PrPSc at 60 days after infection, and by 250 days, PrPSc was still undetectable in the brain. An unfortunate limitation, however, was the inability, or limited ability, of the anti-PrP antibodies to cross the blood-brain barrier which restricted protection by passive vaccination to the extraneural compartments. This conclusion was inferred from results showing that passive antibody transfer had no effect on prion disease progression following intracranial challenge.
Also in 2003, Sigurdsson et al. tested the ability of mAbs 8B4 (residues 34–52), 8H4 (175–185), and 8F9 (205–233) to provide passive protection against PrPSc challenge. Mice were inoculated intraperitoneally with scrapie at either a 10-fold or 1000-fold dilution. Beginning immediately after challenge, mice were injected weekly with one of the three mAbs. In the group receiving the 1 : 10 diluted challenge material, both 8H4 and 8B4 provided a 10% incubation prolongation, with the 1000-fold dilution, and disease prevention was observed in 10% of mice treated with 8B4. The ineffective response to 8F9 was likely due to its lower affinity for both PrPC and PrPSc [185].
The effectiveness of passive immunization appears to be restricted to the peripheral compartments as several experiments have shown development of prion disease following intracerebral inoculation despite peripheral treatment with anti-PrP antibodies. There is, however, some potential application of antibody for treatment of individuals accidentally exposed to prions. Although anti-PrP antibodies are rapidly cleared from the blood, their brief presence does not seem to stimulate any detectable autoimmune reaction. Whether this remains true for active vaccination strategies, where anti-PrP antibodies are present for much longer time periods, is a question to be addressed.
25. Active Immunization
Overcoming tolerance to PrPC is one of the greatest challenges for active PrP immunization. Numerous investigations of different carrier systems [keyhole limpet hemocyanin [200], bacterial heatshock protein DnaK [201], and multiple antigen peptide display [188]] and adjuvants [CpG ODN [202], Freund's complete or incomplete [203, 204], Montanide [189], and TitreMax [178]] are attempts to overcome the inherent lack of immunogenicity for PrPC [205]. Notably many of these potent adjuvants are not suitable for use in humans or animals due to toxicity issues [205]. Despite limitations posed by tolerance to PrPC, experiments have shown a relationship between antibody titers and protection from disease, therefore supporting the potential benefit of immunotherapeutic approaches.
The first group to show a delay in prion disease onset following active immunization with recombinant PrPC (residues 23–230) was Wisniewski et al. 2003. The results were modest with a 10% increase in the incubation time in mice vaccinated (s.c.) at two-week intervals starting 14 weeks prior to inoculation (i.p.). Despite a delay in symptom onset, all mice died as a result of disease progression. Those that were vaccinated at the time of inoculation did not benefit from the treatment [188]. The authors noted that high titers corresponded to prolonged incubation times, but following disease onset, there was no discernable histopathological difference among groups [188].
Other studies, which immunized against different regions of PrPC, demonstrated that different epitopes of the protein may have unique therapeutic value [205]. Experiments by Schwarz et al. 2003 compared the effectiveness of vaccinating with a short synthetic peptide (residue 105–125) to recombinant mouse PrP90-230 covalently linked to keyhole limpet hemocyanin. Immunization with PrP105–125 significantly prolonged survival by an average of 23 days over the adjuvant control animals. Monoclonal antibodies with the same specificity had previously been shown to bind both PrPC and PrPSc. They also prevented amyloid aggregation and dissolved preformed aggregates [200, 204]. In contrast to the previous work by Wisniewski et al. with PrP23–230, the recombinant PrP90–230 immunization was not successful in delaying or preventing disease [189]. They followed up with epitope mapping experiments to determine which epitopes, within PrP90–230, produced antibodies with the highest reactivity. Antibodies to the region between 159 and 211 were the most highly detected by ELISA. The authors suggest that only antibodies specific for residues 105–125 and 144–152 were effective in preventing prion disease [189].
26. Mucosal Immunization
As the majority of TSEs are orally transmitted, mucosal immunization may be an effective immunotherapeutic strategy. Mucosal immunity is generally humoral and noninflammatory in nature and is therefore typically a safer alternative to subcutaneous or intramuscular immunization [206]. This may be of considerable significance as immunological issues, such as those observed in a clinical trial of an Alzheimer's vaccine (AN1792), illustrate a potential complication for the use of systemic vaccine delivery for neurological protein misfolding diseases. The AN1792 vaccine stimulates an immune response to the misfolded Aβ protein found in amyloid plaques which are thought to be responsible for causing Alzheimer's disease symptoms/pathology. The case report published by Weller et al. described a patient in the trial who rapidly declined and died six weeks following her last vaccination. Postmortem analysis revealed CD4+ lymphocytic meningoencephalitis accompanied by extensive macrophage infiltration of the cerebral white matter, indicative of a significant cell-mediated (Th1) inflammatory response [207]. While only one patient died, 6% of participants experienced side effects sufficiently serious to bring the trial to a premature conclusion. Experts suggested that the vaccine may have stimulated too strong an immune response which weakened the barriers that normally protect the CNS from bacterial or viral attack, or that T-cell and microglial overactivation may have induced neuroinflammation [208].
Progress towards stimulating prion mucosal immunity was demonstrated by in vivo protection of mice following mucosal vaccination with PrP expressing Salmonella typhimurium [191]. The vaccination schedule consisted of weekly administration of four oral vaccinations using a live Salmonella construct followed by two injections of the killed Salmonella construct. Seven weeks after the first vaccination mice were exposed, via oral gavage, to 139A scrapie strain. They found that 100% of mice expressing high IgA and IgG titers, and 33% of mice with high IgG and low IgA were symptom-free 400 days after inoculation. Histological and Western blot analysis verified a lack of PrPSc in the brains of asymptomatic animals [191]. It was anticipated that the level of protection would be directly linked to the magnitude of IgA production, but the 33% survival rate observed in the low IgA/high IgG group suggested that other factors may also be important in determining vaccine efficacy [191]. They concluded that anti-PrP IgA is likely important in preventing prion uptake in the gut, while systemic anti-PrP IgG interferes with the isoform conversion process. One advantage of this system is the ability of Salmonella to target M cells, the key sites of prion uptake within the digestive tract [205]. Despite having only been tested in mice, the authors have provided insight into the type of immune response (noninflammatory Th2) which may be necessary for providing protection against oral prion infections.
27. Immunization Using Plasmid and Viral Vectors
Tolerance represents a considerable barrier for immunotherapeutic strategies targeting PrPC. Despite anergy, or deletion of most B- and T-cell expressing receptors specific for PrPC, it may be possible to stimulate the limited anti-PrPC antibody repertoire using potent immunogenic delivery systems [205, 209]. Nickles et al. hypothesized that recombinant virus-like particles (VLPs) would function as more efficient B-cell immunogens than monovalent recombinant proteins and consequently developed a retrovirus (murine leukemia virus)-based PrP vaccine construct expressing residues 121–231 (PrP111) [210]. Using three different groups of C57BL/6 mice with PrP genotypes (Prnp 0/0, 0/+, +/+) vaccination with PrP111 produced PrPC-specific responses with IgG titers inversely proportional to the number of PrP alleles expressed. The authors were surprised, however, by the similarity of IgM titers between the three genotypes in the early time points. While class switching of PrPC-specific IgM to IgG antibodies was less pronounced in wild type animals compared to Prnp 0/0, use of the murine leukemia virus delivery system appeared to provide antigenic T-helper determinants enabling class switching in the wild-type animals [210]. Whether the level of antibodies produced by this construct is sufficient to be of therapeutic value remains to be determined.
Heppner et al. engineered heterozygous prion knockout (Prnp +/o) transgenic mice to expressed 6H4 as a single chain Ab. These mice were infected with Rocky Mountain Laboratory (RML) strain of mouse-adapted scrapie via an intraperitoneal challenge and monitored for prion disease. Mice expressing 6H4 survived 120 days longer than the control mice [186].
In 2007, Kirnbaur's group incorporated a PrPC B-cell epitope (144–152) into a capsid protein component of the bovine papillomavirus type 1 which was expressed by recombinant baculovirus technology. This carrier system is advantaged in its ability to self-assemble into VLPs which display the inserted PrP epitope at a density up to 360 copies per particle. Additionally, VLP vaccines that continuously induce anti-PrP antibody result in a more sustained antibody response over time; consequently, fewer vaccinations may be required [211, 212]. The epitope, also recognized by mAb 6H4 and other commercial antibodies, was selected based on previous studies linking that region with PrPSc-induced conformational changes [211]. Rabbits and rats were vaccinated with the virus particles, along with either Freund's incomplete or CpG, four times at 2–4-week intervals. Anti-PrP IgG antibodies were detected in both species two weeks after the last boost. In vitro experiments using ScN2a and rabbit anti-PrP IgG demonstrated the ability of the antibodies to prevent de novo formation of PrPSc.
Rosset et al. compared a plasmid expressing PrP as a vaccine in wild-type and Prnp 0/0 C57BL/6 mice. On its own, PrP pDNA stimulated anti-PrP antibodies directed against the N-terminal region in the knock-out mice but failed to induce a response in the wild-type animals. The addition of CpG oligodeoxynucleotides, an adjuvant that helps stimulate dendritic cells [213] and B-cells [214], to the PrP pDNA formulation helped to successfully induce anti-PrP antibodies in the wild-type mice [202]. The epitopes recognized by these antibodies were unique to those produced in Prnp 0/0 mice and specific for the C-terminal portion of PrP. Another difference was that antibodies in Prnp 0/0 mice recognized membrane-bound PrPC, whereas none of the antibodies from wild-type animals did. The authors suggested that antibodies were specific for different locations on PrP despite identical immunizations because B cells specific for native PrPC epitopes are strongly tolerized [209]. It was also observed that Prnp 0/0 antibodies were primarily of the IgG1 isotype, whereas antibodies from wild-type mice were mainly IgG2b, an indication that the immune response in wild-type animals was skewing toward a Th1-cell-mediated response. The authors concluded that antibodies stimulated by PrP pDNA + CpG, while unable to bind PrPC, might be effective in blocking PrPSc replication by targeting regions uniquely exposed in the PrPSc conformation [209].
More recently, it was suggested that the use of xenogenic antigens may be effective in circumventing tolerance to autoantigens such as PrPC. Although 90% of the prion amino acid sequence is conserved among mammals, it was demonstrated that vaccination of mice with bovine PrP stimulated the production of anti-PrP antibodies [215]. Tang et al. implemented xenogenic prion (human) expression via an adenovirus vector system which had been shown previously, in cancer studies, to be a potent generator of antibody (CD4+) and cell-mediated (CD8+) responses to both the viral capsid proteins as well as the transgene [216]. In C57BL/6 mice, the authors compared dendritic cell-mediated delivery with direct administration of the adenovirus vector. Only the dendritic cell mediated treatment was able to break B-cell tolerance against murine PrP, producing antibodies which increased the survival of prion-infected mice by an average of 37 days (n = 5) without stimulating T-cell-directed autoimmunity [190].
28. PrPSc-Specific Immunotherapy
The understanding about the physiological function of the PrPC is still limited. For this reason, it may not be the safest strategy to target an immune response towards such a commonly expressed protein. It has previously been demonstrated that antibodies which crosslink PrPC in neural tissue cause apoptosis [199]. The systemic presence of autoreactive PrPC antibodies may also lead to an impairment of the natural function of PrPC, inappropriate cell signal activation, or stimulation of suppressor T-cell lymphocytes [205]. An alternative immunotherapeutic strategy is to stimulate an immune response specific for the PrPSc conformation. While this approach is challenged by the lack of PrPSc structural information, it may present a more viable method for circumventing the body's tolerance mechanisms to PrPC and avoid induction of autoimmunity.
In 2007, Pilon et al. created three peptide vaccine constructs ranging in length from 13 to 29 aa. One construct contained a tyrosine-tyrosine-arginine motif on α-helix 1 which was predicted by Paramithiotis et al. to be selectively exposed on the PrPSc conformation [217]. Results from vaccination trials showed all three constructs to be moderately immunogenic with the α-helix 1 group displaying higher median titers than the other two groups. In a scrapie challenge experiment, the best result observed was a 20-day increase in survival when compared to the control group. There was, however, a lack of correlation between the longest surviving group and the group with the highest median titers. The authors concluded that a successful vaccine would need to overcome self-tolerance as well as stimulate antibodies with high affinity for the PrPSc conformation [218].
One strategy for establishing an immune response to the infectious prion conformation would be to identify a cryptic epitope specifically exposed on the PrPSc conformation. In an attempt to address this issue, it was found that during the refolding process from PrPC to PrPSc there is increased solvent exposure of tyrosine (Y) side chains [217]. The majority of these residues are located in the structured C-terminal domain, a region linked to infectivity and protease resistance [88]. Of the 11 residues in the structured region, 6 are present in bityrosine pairs, a motif that is conserved in human, sheep, mouse, hamster, cattle, and deer. Two pairs are in conjunction with a C-terminal arginine (R), one of which is located on α-helix 1 and the other on β-strand 2. It was hypothesized by Cashman et al. that a YY-motif located on a β-sheet may present an epitope specific for the PrPSc conformation. Testing of this hypothesis resulted in the production of rabbit antisera that selectively immunoprecipitated (IP) PrPSc from infected mouse brain and not PrPC from uninfected brains [217]. These results indicate that the YYR epitope on β2-sheet may represent an appropriate vaccine target for the stimulation of PrPSc-specific immune response. Despite use of an aggressive vaccination protocol which utilises the commonly used vaccine carrier molecule KLH and potent adjuvants such as Freund's complete, this strategy has been limited by poor immunogenicity where IgM was the only antibody isotype detected [217].
Using the YYR epitope as a starting point for vaccine development, our lab was able to translate this weakly immunogenic epitope into a vaccine that induces robust and consistent PrPSc immune responses in a variety of species, including sheep [219]. Specifically through dual optimization of the epitope sequence and length, as well as strategies of formulation and delivery, we generated a vaccine that induces consistent and sustained serum PrPSc-specific IgG antibody responses following two vaccinations. In particular, the epitope QVYYRPVDQYSNQN, presented by a Mannheimia haemolytica leukotoxin carrier protein, emerges as a promising vaccine candidate. Antigen-specific antibodies were also present within cerebral spinal fluid and mucosal secretions. Furthermore, a thermodynamic algorithm has been developed to predict regions of the prion protein which are most likely to be exposed by misfolding [220]. These epitopes, as either univalent or multivalent vaccines of other PrPSc-epitopes, may offer the opportunity for effective and targeted prion immunotherapy.
29. Peptide-Based Vaccine Design
Traditional vaccines are often designed using attenuated live or inactivated microorganisms. Culturing these organisms can be difficult, and even the safest attenuated vaccine systems can produce harmful immune responses. For diseases such as cancer, and protein misfolding diseases like TSEs, Parkinson's, Alzheimer's, and Huntington's disease, pathology is linked to altered self-proteins. As a result, normal vaccine strategies are not applicable due to robust tolerance mechanisms. Extensive research has determined that short-peptide fragments derived from residues found on the much larger self-proteins can stimulate very specific immune responses [221]. The ability to target only the “rogue” self proteins may serve as a powerful tool in preventing the transmission of these diseases or progression of symptoms/pathology in already sick individuals, without stimulating autoimmunity.
Peptide vaccine technology offers advantages in terms of safety and ease of production. Peptide preparations can be freeze-dried making their transportation and storage much easier than those vaccines requiring refrigeration. Their safety is derived from the ability to create an immune response without the requirement for infectious material which is present in attenuated vaccines. Also, the absence of genetic material eliminates the risk of genetic integration or recombination with the patient's genome, a concern posed by DNA vaccines. Furthermore, large-scale production of peptides is economical, and purity is easily analyzed using liquid chromatography and mass spectrometry [221].
Vaccinating with an exogenously administered peptide can be challenging because the exact pathway by which the peptide is processed and then presented to immune cells is thought to be different than for traditional antigens and not well understood [221]. To successfully activate B cells specific for a certain target, the peptide fragment has to represent the appropriate conformation. For this to be achieved, structural knowledge of the native antigen is very beneficial [222]. Consequently, peptide vaccine design for TSEs is made more challenging by the lack of structural data for the misfolded prion protein.
An additional challenge for peptide vaccine design is overcoming the weaker immunogenicity of the peptide fragments, a drawback attributed to their small size. This is particularly true of the cryptic YYR epitope identified by Cashman and Caughey as a potential peptide target for prion immunotherapy. There may be opportunity to address these limitations through the use of recombinant carrier proteins which help promote immune responses. Such systems have been shown to dramatically increase the magnitude of response against short, self-peptides [219]. Such systems may also hold cost savings advantages as the production of carrier-epitope recombinants in bacterial systems is less expensive than chemical peptide synthesis.
30. Challenges to a Prion Vaccine
The current priority of livestock prion diseases is CWD. CWD is the most contagious of the TSEs and presents a significant threat to both wild and farmed deer populations with rates of infectivity as high as 30% and 100%, respectively [20]. The possibility for cross-species infectivity with humans and farmed animals, as well as its ability to persist in the environment for years, and a poor understanding of CWD transmission make containment, using current resources, unattainable [7]. Efforts in regions such as Colorado suggest that culling is not an effective approach for CWD management [223]. We believe that vaccination may be an effective strategy to reduce/eliminate PrPSc shedding and to break the cycle of disease transmission. That the threat of CWD relates to both farmed and wild populations of cervids presents an additional challenge for vaccine development, while most of the strategies which have been investigated thus far have focussed on various epitopes and methods of delivery virtually all are administered through parental injection, the traditional route of vaccine delivery. While this may present an effective tool for farmed animals this is not a viable option for control of CWD in the wild.
Efficient vaccination of wildlife for control of infectious disease has been demonstrated, most notably for rabies [224]. Through this well-documented example, it is apparent that it is possible to achieve protective immunity through oral, self-administered vaccines. The implementation of rabies vaccination programs in Europe and North America has been highly successful; similar investigations have been initiated for vaccination of wild populations for Lyme disease, plague, bovine tuberculosis, and Brucellosis [225]. The release of a vaccine into the wild is not without risks and obstacles. Vaccination of wild life is only recommended as a management tool under three circumstances.
(1) The Disease Represents a Threat to Human Health —
There are no confirmed cases of human infection from CWD, but the zoonotic potential, through direct infection of humans or indirect infection of livestock, is of sufficient concern to be considered a threat to human health.
(2) The Disease Represents an Economic Threat to the Livestock Industry —
While infection of cattle with CWD has only occurred through unnatural routes of laboratory infection, it highlights the potential for these protective species barriers to be broken. Furthermore, CWD has had a devastating impact on the cervid livestock and gaming industries.
(3) The Disease Represents a Threat to an Endangered Species —
It is difficult to predict the long-term consequences of CWD, but modelling investigations anticipate a dramatic reduction, and potential elimination, of cervid populations [226]. From these perspectives, a wildlife vaccine for CWD is justified and appropriate.
Implementation of an oral CWD vaccine for wild cervids must also carefully consider “what success looks like.” Progression of an infectious disease in wild populations depends on natural variables (herd dynamics, rates and modes of disease transmission, and population densities). Influencing disease progression through vaccination depends on experimental variables (vaccine efficacy, vaccine mechanisms, onset and duration of immunity, timing, localization, and intensity of vaccine delivery). Similarly, the unique biology of the prion diseases, in particular the phases of peripheral amplification and CNS-associated disease pathology, may offer various opportunities to impact disease progression (Figure 2).
Following oral exposure, prion diseases progress through an initial phase of asymptomatic peripheral amplification followed by eventual transmission to the CNS. The pathology and ultimately fatal outcome of the disease is associated with accumulation of PrPSc in the brain. Peripheral amplification of prions is of importance for disease propagation within individual animals as well as transmission within populations of animals. While the precise relationship between peripheral loads of PrPSc and shedding has not been defined, it would seem intuitive that animals with lower peripheral levels of PrPSc would be less likely to shed infectious material into the environment. The carcasses of animals with lower peripheral loads of PrPSc would also be expected to contribute less to environmental contamination. Thus far the efficacy of prion vaccines has been evaluated through their ability to delay the onset of pathological symptoms, which reflects PrPSc accumulation in the CNS. However, as the vast majority of the immune response is localized to the periphery—only about 1% of circulating antibodies cross the blood-brain barrier [219]—these vaccines may have greater impact on peripheral rather than central PrPSc loads. At this time, the extent to which the transition of the disease from periphery to CNS reflects peripheral loads of PrPSc or a temporal relationship which is independent of peripheral PrPSc quantities has not been defined. Theoretically, a vaccine could have significant impact on peripheral PrPSc while having only minimal impact on the timing and severity of disease pathology. While offering minimal benefit to an individual animal, such a vaccine may be valuable from a population perspective by reducing shedding and minimize disease propagation.
31. Importance of a TSE Vaccine
As a group, TSEs have had undeniable economical impact, leaving many countries with billions of dollars in lost revenue due to trade embargos and weakened consumer confidence. While key historical TSE events have provided the momentum for research initiatives aimed at understanding PrPC function, PrPSc infectivity, and designing effective premortem diagnostics and therapeutic strategies, definitive answers in these areas remain elusive. Despite the research initiatives that have been underway for decades, there are still large deficits in key areas of understanding such as PrPC function and pre-mortem diagnostics. Identifying a disease specific epitope may have significant applications in more than one of these TSE research areas. These events have provided the momentum for research initiatives aimed at understanding PrPC function, PrPSc infectivity, and designing effective diagnostic tests and therapeutic strategies.
References
- 1.Sigurdsson B. Rida, a chronic encephalitis of sheep: with general remarks on infections which develop slowly and some of their special characteristics. British Veterinary Journal. 1954;110:341–354. [Google Scholar]
- 2.Klatzo I, Gajdusek DC, Zigas V. Pathology of kuru. Laboratory Investigation. 1959;8(4):799–847. [PubMed] [Google Scholar]
- 3.Gajdusek DC, Gibbs CJ, Alpers M. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature. 1966;209(5025):794–796. doi: 10.1038/209794a0. [DOI] [PubMed] [Google Scholar]
- 4.Gibbs CJ, Jr., Gajdusek DC, Asher DM, et al. Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science. 1968;161(3839):388–389. doi: 10.1126/science.161.3839.388. [DOI] [PubMed] [Google Scholar]
- 5.Parry HB. Scrapie: a transmissible hereditary disease of sheep. Heredity. 1962;17:75–105. doi: 10.1038/hdy.1962.4. [DOI] [PubMed] [Google Scholar]
- 6.Beghi E, Gandolfo C, Ferrarese C, et al. Bovine spongiform encephalopathy and Creutzfeldt-Jakob disease: facts and uncertainties underlying the causal link between animal and human diseases. Neurological Sciences. 2004;25(3):122–129. doi: 10.1007/s10072-004-0249-9. [DOI] [PubMed] [Google Scholar]
- 7.Sigurdson CJ, Aguzzi A. Chronic wasting disease. Biochimica et Biophysica Acta. 2007;1772(6):610–618. doi: 10.1016/j.bbadis.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams ES, Young S. Neuropathology of chronic wasting disease of mule deer (Odocoileus hemionus) and elk (Cervus elaphus nelsoni) Veterinary Pathology. 1993;30(1):36–45. doi: 10.1177/030098589303000105. [DOI] [PubMed] [Google Scholar]
- 9.Schneider K, Fangerau H, Michaelsen B, Raab WH. The early history of the transmissible spongiform encephalopathies exemplified by scrapie. Brain Research Bulletin. 2008;77(6):343–355. doi: 10.1016/j.brainresbull.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 10.Comber T. Real Improvements in Agriculture. 1st edition. London, UK: Nicoll, W.; 1772. [Google Scholar]
- 11.Turner G. Extracts from a "general" view of the agriculture of the country of Gloucester; with observations on the means of its improvements; drawn up for the consideration of the board of agriculture. The Royal Bath and West of England Society. 1795;7 [Google Scholar]
- 12.Cuille P, Chelle L. Pathologie animale—la malade dite tremblante du mouton est-elle inocuable? Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences. 1936;203:1552–1554. [Google Scholar]
- 13.Cuille P, Chelle L. Le tremblante du mouton est-elle determinee par un virus filtrable? Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences. 1938;203:1687–1688. [Google Scholar]
- 14.Houston F, Goldmann W, Chong A, et al. Prion diseases: BSE in sheep bred for resistance to infection. Nature. 2003;423(6939):p. 498. doi: 10.1038/423498a. [DOI] [PubMed] [Google Scholar]
- 15.Goldmann W, Hunter N, Smith G, Foster J, Hope J. PrP genotype and agent effects in scrapie: change in allelic interaction with different isolates of agent in sheep, a natural host of scrapie. Journal of General Virology. 1994;75(5):989–995. doi: 10.1099/0022-1317-75-5-989. [DOI] [PubMed] [Google Scholar]
- 16.van Keulen LJ, Schreuder BE, Meloen RH, Mooij-Harkes G, Vromans ME, Langeveld JPM. Immunohistochemical detection of prion protein in lymphoid tissues of sheep with natural scrapie. Journal of Clinical Microbiology. 1996;34(5):1228–1231. doi: 10.1128/jcm.34.5.1228-1231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikeda T, Horiuchi M, Ishiguro N, Muramatsu Y, Kai-Uwe GD, Shinagawa M. Amino acid polymorphisms of PrP with reference to onset of scrapie in Suffolk and Corriedale sheep in Japan. Journal of General Virology. 1995;76(10):2577–2581. doi: 10.1099/0022-1317-76-10-2577. [DOI] [PubMed] [Google Scholar]
- 18.Kao RR, Gravenor MB, Baylis M, et al. The potential size and duration of an epidemic of bovine spongiform encephalopathy in British sheep. Science. 2002;295(5553):332–335. doi: 10.1126/science.1067475. [DOI] [PubMed] [Google Scholar]
- 19.Baylis M, McIntyre KM. Transmissible spongiform encephalopathies: scrapie control under new strain. Nature. 2004;432(7019):810–811. doi: 10.1038/432810a. [DOI] [PubMed] [Google Scholar]
- 20.Williams ES. Chronic wasting disease. Veterinary Pathology. 2005;42(5):530–549. doi: 10.1354/vp.42-5-530. [DOI] [PubMed] [Google Scholar]
- 21.Williams ES, Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. Journal of Wildlife Diseases. 1980;16(1):89–98. doi: 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- 22.Browning SR, Mason GL, Seward T, et al. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. Journal of Virology. 2004;78(23):13345–13350. doi: 10.1128/JVI.78.23.13345-13350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller MW, Williams ES, McCarty CW, et al. Epizootiology of chronic wasting disease in free-ranging cervids in Colorado and Wyoming. Journal of Wildlife Diseases. 2000;36(4):676–690. doi: 10.7589/0090-3558-36.4.676. [DOI] [PubMed] [Google Scholar]
- 24.Belay ED, Maddox RA, Williams ES, Miller MW, Gambetti P, Schonberger LB. Chronic wasting disease and potential transmission to humans. Emerging Infectious Diseases. 2004;10(6):977–984. doi: 10.3201/eid1006.031082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wells GA, Scott AC, Johnson CT, et al. A novel progressive spongiform encephalopathy in cattle. Veterinary Record. 1987;121(18):419–420. doi: 10.1136/vr.121.18.419. [DOI] [PubMed] [Google Scholar]
- 26.Donnelly CA, Ferguson NM, Ghani AC, Anderson RM. Implications of BSE infection screening data for the scale of the British BSE epidemic and current European infection levels. Proceedings of the Royal Society B: Biological Sciences. 2002;269(1506):2179–2190. doi: 10.1098/rspb.2002.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilesmith JW, Wells GA, Cranwell MP, Ryan JP. Bovine spongiform encephalopathy: epidemiological studies. Veterinary Record. 1988;123(25):638–644. [PubMed] [Google Scholar]
- 28.Baron T, Biacabe AG. Origin of bovine spongiform encephalopathy. Lancet. 2006;367(9507):297–298. doi: 10.1016/S0140-6736(06)68060-4. [DOI] [PubMed] [Google Scholar]
- 29.Konold T, Lee YH, Stack MJ, et al. Different prion disease phenotypes result from inoculation of cattle with two temporally separated sources of sheep scrapie from Great Britain. BMC Veterinary Research. 2006;2, article 31 doi: 10.1186/1746-6148-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Editorial-team. Fourth case of transfusion-associated vCJD infection in the United Kingdom. Eurosurveillance. 2007;12(1):E070118–E070124. doi: 10.2807/esw.12.03.03117-en. [DOI] [PubMed] [Google Scholar]
- 31.Cappai R, Collins SJ. Structural biology of prions. Contributions to Microbiology. 2004;11:14–32. doi: 10.1159/000077050. [DOI] [PubMed] [Google Scholar]
- 32.Biacabe AG, Morignat E, Vulin J, Calavas D, Baron TG. Atypical bovine spongiform encephalopathies, France, 2001-2007. Emerging Infectious Diseases. 2008;14(2):298–300. doi: 10.3201/eid1402.071141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Detwiler LA. Scrapie. Revue Scientifique et Technique: Office International des Epizooties. 1992;11(2):491–537. doi: 10.20506/rst.11.2.607. [DOI] [PubMed] [Google Scholar]
- 34.Hunter N. Scrapie and experiomental BSE in sheep. British Medical Bulletin. 2006;66:171–183. doi: 10.1093/bmb/66.1.171. [DOI] [PubMed] [Google Scholar]
- 35.Houston EF, Gravenor MB. Clinical signs in sheep experimentally infected with scrapie and BSE. Veterinary Record. 2003;152(11):334–336. doi: 10.1136/vr.152.11.333. [DOI] [PubMed] [Google Scholar]
- 36.Petrie L, Heath B, Harold D. Scrapie: report of an outbreak and brief review. Canadian Veterinary Journal. 1989;30:321–327. [PMC free article] [PubMed] [Google Scholar]
- 37.Greenlee JJ, Smith JD, Kunkle RA. White-tailed deer are susceptible to the agent of sheep scrapie by intracerebral inoculation. Veterinary Research. 2011;42(1):p. 107. doi: 10.1186/1297-9716-42-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamir AN, Miller JM, Cutlip RC, et al. Transmission of sheep scrapie to elk (Cervus elaphus nelsoni) by intracerebral inoculation: final outcome of the experiment. Journal of Veterinary Diagnostic Investigation. 2004;16(4):316–321. doi: 10.1177/104063870401600410. [DOI] [PubMed] [Google Scholar]
- 39.Bradley R. Bovine spongiform encephalopathy epidemiology—a brief review. Livestock Production Science. 1994;38(1):5–16. [Google Scholar]
- 40.Gurgul A, Słota E. Effect of bovine PRNP gene polymorphisms on BSE susceptibility in cattle. Folia Biologica. 2007;55(3-4):81–86. doi: 10.3409/173491607781492533. [DOI] [PubMed] [Google Scholar]
- 41.Richt JA, Hall SM. BSE case associated with prion protein gene mutation. PLoS Pathogens. 2008;4(9) doi: 10.1371/journal.ppat.1000156. Article ID e1000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harman JL, Silva CJ. Bovine spongiform encephalopathy. Journal of the American Veterinary Medical Association. 2009;234(1):59–72. doi: 10.2460/javma.234.1.59. [DOI] [PubMed] [Google Scholar]
- 43.Novakofski J, Brewer MS, Mateus-Pinilla N, Killefer J, McCusker RH. Prion biology relevant to bovine spongiform encephalopathy. Journal of Animal Science. 2005;83(6):1455–1476. doi: 10.2527/2005.8361455x. [DOI] [PubMed] [Google Scholar]
- 44.Perrott MR, Sigurdson CJ, Mason GL, Hoover EA. Evidence for distinct CWD strains in experimental CWD in ferrets. General Virology. 2012;93:212–221. doi: 10.1099/vir.0.035006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raymond GJ, Raymond LD, Meade-White KD, et al. Transmission and adaptation of chronic wasting disease to hamsters and transgenic mice: evidence for strains. Journal of Virology. 2007;81(8):4305–4314. doi: 10.1128/JVI.02474-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soto C. Constraining the loop, releasing prion infectivity. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(1):10–11. doi: 10.1073/pnas.0811625106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurt TD, Telling GC, Zabel MD, Hoover EA. Trans-species amplification of PrPCWD and correlation with rigid loop 170N. Virology. 2009;387(1):235–243. doi: 10.1016/j.virol.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 48.Johnson C, Johnson J, Vanderloo JP, Keane D, Aiken JM, McKenzie D. Prion protein polymorphisms in white-tailed deer influence susceptibility to chronic wasting disease. Journal of General Virology. 2006;87(7):2109–2114. doi: 10.1099/vir.0.81615-0. [DOI] [PubMed] [Google Scholar]
- 49.Collee JG, Bradley R, Liberski PP. Variant CJD (vCJD) and bovine spongiform encephalopathy (BSE): 10 and 20 years on: part 2. Folia Neuropathologica. 2006;44(2):102–110. [PubMed] [Google Scholar]
- 50.Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea; the endemic occurrence of kuru in the native population. The New England Journal of Medicine. 1957;257(20):974–978. doi: 10.1056/NEJM195711142572005. [DOI] [PubMed] [Google Scholar]
- 51.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annual Review of Neuroscience. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 52.Collins SJ, Lawson VA, Masters CL. Transmissible spongiform encephalopathies. Lancet. 2004;363(9402):51–61. doi: 10.1016/S0140-6736(03)15171-9. [DOI] [PubMed] [Google Scholar]
- 53.Hadlow WJ. Scrapie and kuru. The Lancet. 1959;274(7097):289–290. [Google Scholar]
- 54.Gajdusek DC. Unconventional viruses and the origin and disappearance of Kuru. Science. 1977;197(4307):943–960. doi: 10.1126/science.142303. [DOI] [PubMed] [Google Scholar]
- 55.Klitzman RL, Alpers MP, Gajdusek DC. The natural incubation period of kuru and the episodes of transmission in three clusters of patients. Neuroepidemiology. 1984;3(1):3–20. [Google Scholar]
- 56.Coulthart MB, Cashman NR. Variant Creutzfeldt-Jakob disease: a summary of current scientific knowledge in relation to public health. Canadian Medical Association Journal. 2001;165(1):51–58. [PMC free article] [PubMed] [Google Scholar]
- 57.Hsiao K, Baker HF, Crow TJ, et al. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature. 1989;338(6213):342–345. doi: 10.1038/338342a0. [DOI] [PubMed] [Google Scholar]
- 58.Aguzzi A. Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. Journal of Neurochemistry. 2006;97(6):1726–1739. doi: 10.1111/j.1471-4159.2006.03909.x. [DOI] [PubMed] [Google Scholar]
- 59.Glatzel M, Ott PM, Linder T, et al. Human prion diseases: epidemiology and integrated risk assessment. Lancet Neurology. 2003;2(12):757–763. doi: 10.1016/s1474-4422(03)00588-x. [DOI] [PubMed] [Google Scholar]
- 60.Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiological Reviews. 2009;89(4):1105–1152. doi: 10.1152/physrev.00006.2009. [DOI] [PubMed] [Google Scholar]
- 61.Will R. New variant Creutzfeldt-Jakob disease. Biomedicine and Pharmacotherapy. 1999;53(1):9–13. doi: 10.1016/s0753-3322(99)80054-7. [DOI] [PubMed] [Google Scholar]
- 62.Zeidler M, Stewart GE, Barraclough CR, et al. New variant Creutzfeldt-Jakob disease: neurological features and diagnostic tests. Lancet. 1997;350(9082):903–907. doi: 10.1016/s0140-6736(97)07472-2. [DOI] [PubMed] [Google Scholar]
- 63.Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet. 2004;364(9433):527–529. doi: 10.1016/S0140-6736(04)16811-6. [DOI] [PubMed] [Google Scholar]
- 64.Brown P, Preece M, Brandel JP, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55(8):1075–1081. doi: 10.1212/wnl.55.8.1075. [DOI] [PubMed] [Google Scholar]
- 65.Garfin DE, Stites DP, Zitnik LA, Prusiner SB. Suppression of polyclonal B cell activation in scrapie-infected C3H/HeJ mice. Journal of Immunology. 1978;120(6):1986–1990. [PubMed] [Google Scholar]
- 66.Kasper KC, Stites DP, Bowman KA, et al. Immunological studies of scrapie infection. Journal of Neuroimmunology. 1982;3(3):187–201. doi: 10.1016/0165-5728(82)90022-4. [DOI] [PubMed] [Google Scholar]
- 67.Kasper KC, Bowman K, Stites DP, Prusiner SB. Toward development of assays for scrapie-specific antibodies. In: Streilein JW, Hart DA, Stein-Streilein J, Duncan WR, Billingham RE, editors. Hamster Immune Responses in Infectious and Oncological Diseases. New York, NY, USA: Plenum Press; 1981. pp. 401–413. [Google Scholar]
- 68.Dickinson AG, Fraser H. Genetical control of the concentration of ME7 scrapie agent in mouse spleen. Journal of Comparative Pathology. 1969;79(3):363–366. doi: 10.1016/0021-9975(69)90051-6. [DOI] [PubMed] [Google Scholar]
- 69.Stites DP, Garfin DE, Prusiner SB. The immunology of scrapie. In: Prusiner SB, Hadlow WJ, editors. Slow Transmissible Diseases of the Nervous System. New York, NY, USA: Academic Press; 1979. pp. 211–221. [Google Scholar]
- 70.Prusiner SB, Garfin DE, Cochran SP, et al. Experimental scrapie in the mouse: electrophoretic and sedimentation properties of the partially purified agent. Journal of Neurochemistry. 1980;35(3):574–582. doi: 10.1111/j.1471-4159.1980.tb03693.x. [DOI] [PubMed] [Google Scholar]
- 71.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 72.Prusiner SB, Garfin DE, Cochran SP, et al. Evidence for hydrophobic domains on the surface of the scrapie agent. Transactions of the American Neurological Association. 1978;103:62–64. [PubMed] [Google Scholar]
- 73.Prusiner SB, McKinley MP, Groth DF, et al. Scrapie agent contains a hydrophobic protein. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(11):6675–6679. doi: 10.1073/pnas.78.11.6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haigh CL, Wright JA, Brown DR. Regulation of prion protein expression by noncoding regions of the PRNP gene. Journal of Molecular Biology. 2007;368(4):915–927. doi: 10.1016/j.jmb.2007.02.086. [DOI] [PubMed] [Google Scholar]
- 75.Basler K, Oesch B, Scott M, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986;46(3):417–428. doi: 10.1016/0092-8674(86)90662-8. [DOI] [PubMed] [Google Scholar]
- 76.Kretzschmar HA, Prusiner SB, Stowring LE, de Armond SJ. Scrapie prion proteins are synthesized in neurons. American Journal of Pathology. 1986;122(1):1–5. [PMC free article] [PubMed] [Google Scholar]
- 77.Brown HR, Goller NL, Rudelli RD, et al. The mRNA encoding the scrapie agent protein is present in a variety of non-neuronal cells. Acta Neuropathologica. 1990;80(1):1–6. doi: 10.1007/BF00294214. [DOI] [PubMed] [Google Scholar]
- 78.Brown DR, Schmidt B, Groschup MH, Kretzschmar HA. Prion protein expression in muscle cells and toxicity of a prion protein fragment. European Journal of Cell Biology. 1998;75(1):29–37. doi: 10.1016/S0171-9335(98)80043-5. [DOI] [PubMed] [Google Scholar]
- 79.Bosque PJ, Ryou C, Telling G, et al. Prions in skeletal muscle. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(6):3812–3817. doi: 10.1073/pnas.052707499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu T, Li R, Wong BS, et al. Normal cellular prior protein is preferentially expressed on subpopulations of murine hemopoietic cells. Journal of Immunology. 2001;166(6):3733–3742. doi: 10.4049/jimmunol.166.6.3733. [DOI] [PubMed] [Google Scholar]
- 81.Paltrinieri S, Comazzi S, Spagnolo V, Rondena M, Ponti W, Ceciliani F. Bovine doppel (Dpl) and prion protein (PrP) expression on lymphoid tissue and circulating leukocytes. Journal of Histochemistry and Cytochemistry. 2004;52(12):1639–1645. doi: 10.1369/jhc.4A6441.2004. [DOI] [PubMed] [Google Scholar]
- 82.Morel E, Fouquet S, Chateau D, et al. The cellular prion protein PrPc is expressed in human enterocytes in cell-cell junctional domains. Journal of Biological Chemistry. 2004;279(2):1499–1505. doi: 10.1074/jbc.M308578200. [DOI] [PubMed] [Google Scholar]
- 83.Tanji K, Saeki K, Matsumoto Y, et al. Analysis of PrPc mRNA by in situ hybridization in brain, placenta, uterus and testis of rats. Intervirology. 1995;38(6):309–315. doi: 10.1159/000150457. [DOI] [PubMed] [Google Scholar]
- 84.Inoue S, Tanaka M, Horiuchi M, Ishiguro N, Shinagawa M. Characterization of the bovine prion protein gene: the expression requires interaction between the promoter and intron. Journal of Veterinary Medical Science. 1997;59(3):175–183. doi: 10.1292/jvms.59.175. [DOI] [PubMed] [Google Scholar]
- 85.Shyu WC, Kao MC, Chou WY, Hsu YD, Soong BW. Heat shock modulates prion protein expression in human NT-2 cells. NeuroReport. 2000;11(4):771–774. doi: 10.1097/00001756-200003200-00023. [DOI] [PubMed] [Google Scholar]
- 86.Hope J, Morton LJ, Farquhar CF, Multhaup G, Beyreuther K, Kimberlin RH. The major polypeptide of scrapie-associated fibrils (SAF) has the same size, charge distribution and N-terminal protein sequence as predicted for the normal brain protein (PrP) EMBO Journal. 1986;5(10):2591–2597. doi: 10.1002/j.1460-2075.1986.tb04539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shyng SL, Huber MT, Harris DA. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. Journal of Biological Chemistry. 1993;268(21):15922–15928. [PubMed] [Google Scholar]
- 88.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121-231) Nature. 1996;382(6587):180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 89.Bartz JC, Bessen RA, Mckenzie D, Marsh RF, Aiken JM. Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. Journal of Virology. 2000;74(12):5542–5547. doi: 10.1128/jvi.74.12.5542-5547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alonso DO, An C, Daggett V. Simulations of biomolecules: characterization of the early steps in the pH-induced conformational conversion of the hamster, bovine and human forms of the prion protein. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences. 2002;360(1795):1165–1178. doi: 10.1098/rsta.2002.0986. [DOI] [PubMed] [Google Scholar]
- 91.Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. Journal of Biological Chemistry. 2001;276(42):39145–39149. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- 92.Diarra-Mehrpour M, Arrabal S, Jalil A, et al. Prion protein prevents human breast carcinoma cell line from tumor necrosis factor α-induced cell death. Cancer Research. 2004;64(2):719–727. doi: 10.1158/0008-5472.can-03-1735. [DOI] [PubMed] [Google Scholar]
- 93.Koch CA, Anderson D, Moran MF, Ellis C, Pawson T. SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science. 1991;252(5006):668–674. doi: 10.1126/science.1708916. [DOI] [PubMed] [Google Scholar]
- 94.Mouillet-Richard S, Ermonval M, Chebassier C, et al. Signal transduction through prion protein. Science. 2000;289(5486):1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- 95.Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annual Review of Neuroscience. 2008;31:439–477. doi: 10.1146/annurev.neuro.31.060407.125620. [DOI] [PubMed] [Google Scholar]
- 96.Graner E, Mercadante AF, Zanata SM, et al. Cellular prion protein binds laminin and mediates neuritogenesis. Molecular Brain Research. 2000;76(1):85–92. doi: 10.1016/s0169-328x(99)00334-4. [DOI] [PubMed] [Google Scholar]
- 97.Puig S, Thiele DJ. Molecular mechanisms of copper uptake and distribution. Current Opinion in Chemical Biology. 2002;6(2):171–180. doi: 10.1016/s1367-5931(02)00298-3. [DOI] [PubMed] [Google Scholar]
- 98.Harrison MD, Dameron CT. Molecular mechanisms of copper metabolism and the role of the menkes disease protein. Journal of Biochemical and Molecular Toxicology. 1999;13(2):93–106. doi: 10.1002/(sici)1099-0461(1999)13:2<93::aid-jbt5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 99.Waggoner DJ, Bartnikas TB, Gitlin JD. The role of copper in neurodegenerative disease. Neurobiology of Disease. 1999;6(4):221–230. doi: 10.1006/nbdi.1999.0250. [DOI] [PubMed] [Google Scholar]
- 100.Walter ED, Chattopadhyay M, Millhauser GL. The affinity of copper binding to the prion protein octarepeat domain: evidence for negative cooperativity. Biochemistry. 2006;45(43):13083–13092. doi: 10.1021/bi060948r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Quaglio E, Chiesa R, Harris DA. Copper converts the cellular prion protein into a protease-resistant species that is distinct from the scrapie isoform. Journal of Biological Chemistry. 2001;276(14):11432–11438. doi: 10.1074/jbc.M009666200. [DOI] [PubMed] [Google Scholar]
- 102.Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. Journal of Biological Chemistry. 1998;273(50):33107–33110. doi: 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- 103.Westergard L, Christensen HM, Harris DA. The cellular prion protein PrPc: its physiological function and role in disease. Biochimica et Biophysica Acta. 2007;1772(6):629–644. doi: 10.1016/j.bbadis.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Halliwell B. Oxidative stress and neurodegeneration: where are we now? Journal of Neurochemistry. 2006;97(6):1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 105.Milhavet O, Lehmann S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Research Reviews. 2002;38(3):328–339. doi: 10.1016/s0165-0173(01)00150-3. [DOI] [PubMed] [Google Scholar]
- 106.Brown DR, Wong BS, Hafiz F, Clive C, Haswell SJ, Jones IM. Normal prion protein has an activity like that of superoxide dismutase. Biochemical Journal. 1999;344(1):1–5. [PMC free article] [PubMed] [Google Scholar]
- 107.Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. EMBO Journal. 2002;21(13):3317–3326. doi: 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weise J, Sandau R, Schwarting S, et al. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke. 2006;37(5):1296–1300. doi: 10.1161/01.STR.0000217262.03192.d4. [DOI] [PubMed] [Google Scholar]
- 109.Fournier JG, Escaig-Haye F, Billette de Villemeur T, Robain O. Ultrastructural localization of cellular prion protein PrPc in synaptic boutons of normal hamster hippocampus. Comptes Rendus de l'Academie des Sciences: Serie III. 1995;318(3):339–344. [PubMed] [Google Scholar]
- 110.Jeffrey M, Halliday WG, Bell J, et al. Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathology and Applied Neurobiology. 2000;26(1):41–54. doi: 10.1046/j.1365-2990.2000.00216.x. [DOI] [PubMed] [Google Scholar]
- 111.Barrow PA, Holmgren CD, Tapper AJ, Jefferys JG. Intrinsic physiological and morphological properties of principal cells of the hippocampus and neocortex in hamsters infected with scrapie. Neurobiology of Disease. 1999;6(5):406–423. doi: 10.1006/nbdi.1999.0255. [DOI] [PubMed] [Google Scholar]
- 112.Bouzamondo-Bernstein E, Hopkins SD, Spilman P, et al. The neurodegeneration sequence in prion diseases: evidence from functional, morphological and ultrastructural studies of the GABAergic system. Journal of Neuropathology and Experimental Neurology. 2004;63(8):882–899. doi: 10.1093/jnen/63.8.882. [DOI] [PubMed] [Google Scholar]
- 113.Tobler I, Gaus SE, Deboer T, et al. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature. 1996;380(6575):639–642. doi: 10.1038/380639a0. [DOI] [PubMed] [Google Scholar]
- 114.Criado JR, Sánchez-Alavez M, Conti B, et al. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiology of Disease. 2005;19(1-2):255–265. doi: 10.1016/j.nbd.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 115.Zou WQ, Cashman NR. Acidic pH and detergents enhance in vitro conversion of human brain PrPc to a PrPSc-like form. Journal of Biological Chemistry. 2002;277(46):43942–43947. doi: 10.1074/jbc.M203611200. [DOI] [PubMed] [Google Scholar]
- 116.Prusiner SB. Prions. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(23):13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pan K-M, Baldwin M, Nguyen J, et al. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(23):10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. Journal of Virology. 1994;68(12):7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lasmézas CI, Deslys JP, Robain O, et al. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science. 1997;275(5298):402–405. doi: 10.1126/science.275.5298.402. [DOI] [PubMed] [Google Scholar]
- 120.Caughey B, Kocisko DA, Raymond GJ, Lansbury PT., Jr. Aggregates of scrapie-associated prion protein induce the cell-free conversion of protease-sensitive prion protein to the protease-resistant state. Chemistry and Biology. 1995;2(12):807–817. doi: 10.1016/1074-5521(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 121.Safar J, Wille H, Itri V, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nature Medicine. 1998;4(10):1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 122.Falsig J, Nilsson KP, Knowles TP, Aguzzi A. Chemical and biophysical insights into the propagation of prion strains. HFSP Journal. 2008;2(6):332–341. doi: 10.2976/1.2990786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Peretz D, Scott MR, Groth D, et al. Strain-specified relative conformational stability of the scrapie prion protein. Protein Science. 2001;10(4):854–863. doi: 10.1110/ps.39201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annual Review of Pathology. 2008;3:11–40. doi: 10.1146/annurev.pathmechdis.3.121806.154326. [DOI] [PubMed] [Google Scholar]
- 125.Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116(2):313–327. doi: 10.1016/s0092-8674(03)01031-6. [DOI] [PubMed] [Google Scholar]
- 126.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318(5852):930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 127.Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H. Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 1994;343(1306):405–411. doi: 10.1098/rstb.1994.0036. [DOI] [PubMed] [Google Scholar]
- 128.Krüger D, Thomzig A, Lenz G, Kampf K, McBride P, Beekes M. Faecal shedding, alimentary clearance and intestinal spread of prions in hamsters fed with scrapie. Veterinary Research. 2009;40(1):p. 4. doi: 10.1051/vetres:2008042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Race R, Raines A, Raymond GJ, Caughey B, Chesebro B. Long-term subclinical carrier state precedes scrapie replication and adaptation in a resistant species: analogies to bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease in humans. Journal of Virology. 2001;75(21):10106–10112. doi: 10.1128/JVI.75.21.10106-10112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Weissmann C. The state of the prion. Nature Reviews Microbiology. 2004;2(11):861–871. doi: 10.1038/nrmicro1025. [DOI] [PubMed] [Google Scholar]
- 131.Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302(5646):871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 132.Chesebro B, Trifilo M, Race R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308(5727):1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- 133.Race RE, Priola SA, Bessen RA, et al. Neuron-specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron. 1995;15(5):1183–1191. doi: 10.1016/0896-6273(95)90105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Prinz M, Montrasio F, Furukawa H, et al. Intrinsic resistance of oligodendrocytes to prion infection. Journal of Neuroscience. 2004;24(26):5974–5981. doi: 10.1523/JNEUROSCI.0122-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Raeber AJ, Race RE, Brandner S, et al. Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO Journal. 1997;16(20):6057–6065. doi: 10.1093/emboj/16.20.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jeffrey M, Goodsir CM, Race RE, Chesebro B. Scrapie-specific neuronal lesions are independent of neuronal PrP expression. Annals of Neurology. 2004;55(6):781–792. doi: 10.1002/ana.20093. [DOI] [PubMed] [Google Scholar]
- 137.Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain. 2006;129(9):2241–2265. doi: 10.1093/brain/awl150. [DOI] [PubMed] [Google Scholar]
- 138.Neutra MR. M cells in antigen sampling in mucosal tissues. Current Topics in Microbiology and Immunology. 1999;236:17–32. doi: 10.1007/978-3-642-59951-4_2. [DOI] [PubMed] [Google Scholar]
- 139.Mishra RS, Basu S, Gu Y, et al. Protease-resistant human prion protein and ferritin are cotransported across Caco-2 epithelial cells: implications for species barrier in prion uptake from the intestine. Journal of Neuroscience. 2004;24(50):11280–11290. doi: 10.1523/JNEUROSCI.2864-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang F-P, Farquhar CF, Mabbott NA, Bruce ME, MacPherson GG. Migrating intestinal dendritic cells transport PrPSc from the gut. Journal of General Virology. 2002;83(1):267–271. doi: 10.1099/0022-1317-83-1-267. [DOI] [PubMed] [Google Scholar]
- 141.Andreoletti O, Berthon P, Marc D, et al. Early accumulation of PrPSc in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. Journal of General Virology. 2000;81(12):3115–3126. doi: 10.1099/0022-1317-81-12-3115. [DOI] [PubMed] [Google Scholar]
- 142.Kitamoto T, Muramoto T, Mohri S, Doh-Ura K, Tateishi J. Abnormal isoform of prion protein accumulates in follicular dendritic cells in mice with Creutzfeldt-Jakob disease. Journal of Virology. 1991;65(11):6292–6295. doi: 10.1128/jvi.65.11.6292-6295.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Heikenwalder M, Zeller N, Seeger H, et al. Chronic lymphocytic inflammation specifies the organ tropism of prions. Science. 2005;307(5712):1107–1110. doi: 10.1126/science.1106460. [DOI] [PubMed] [Google Scholar]
- 144.Seeger H, Heikenwalder M, Zeller N, et al. Coincident scrapie infection and nephritis lead to urinary prion excretion. Science. 2005;310(5746):324–326. doi: 10.1126/science.1118829. [DOI] [PubMed] [Google Scholar]
- 145.Glatzel M, Heppner FL, Albers KM, Aguzzi A. Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron. 2001;31(1):25–34. doi: 10.1016/s0896-6273(01)00331-2. [DOI] [PubMed] [Google Scholar]
- 146.van Keulen LJ, Bossers A, van Zijderveld F. TSE pathogenesis in cattle and sheep. Veterinary Research. 2008;39(4, article 24) doi: 10.1051/vetres:2007061. [DOI] [PubMed] [Google Scholar]
- 147.van Keulen LJ, Schreuder BE, Vromans ME, Langeveld JP, Smits MA. Pathogenesis of natural scrapie in sheep. Archives of Virology, Supplementum. 2000;(16):57–71. doi: 10.1007/978-3-7091-6308-5_5. [DOI] [PubMed] [Google Scholar]
- 148.Hur K, Kim JI, Choi SI, Choi EK, Carp RI, Kim YS. The pathogenic mechanisms of prion diseases. Mechanisms of Ageing and Development. 2002;123(12):1637–1647. doi: 10.1016/s0047-6374(02)00099-4. [DOI] [PubMed] [Google Scholar]
- 149.Choi SI, Ju WK, Choi EK, et al. Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathologica. 1998;96(3):279–286. doi: 10.1007/s004010050895. [DOI] [PubMed] [Google Scholar]
- 150.Sorgato MC, Bertoli A. From cell protection to death: may Ca2+ signals explain the chameleonic attributes of the mammalian prion protein? Biochemical and Biophysical Research Communications. 2009;379(2):171–174. doi: 10.1016/j.bbrc.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 151.Sandberg MK, Wallén P, Wikström MA, Kristensson K. Scrapie-infected GT1-1 cells show impaired function of voltage-gated N-type calcium channels (Cav 2.2) which is ameliorated by quinacrine treatment. Neurobiology of Disease. 2004;15(1):143–151. doi: 10.1016/j.nbd.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 152.Khosravani H, Zhang Y, Tsutsui S, et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. Journal of Cell Biology. 2008;181(3):551–555. doi: 10.1083/jcb.200711002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Spudich A, Frigg R, Kilic E, et al. Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiology of Disease. 2005;20(2):442–449. doi: 10.1016/j.nbd.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 154.Soto C. Diagnosing prion diseases: needs, challenges and hopes. Nature Reviews Microbiology. 2004;2(10):809–819. doi: 10.1038/nrmicro1003. [DOI] [PubMed] [Google Scholar]
- 155.Ingrosso L, Vetrugno V, Cardone F, Pocchiari M. Molecular diagnostics of transmissible spongiform encephalopathies. Trends in Molecular Medicine. 2002;8(6):273–280. doi: 10.1016/s1471-4914(02)02358-4. [DOI] [PubMed] [Google Scholar]
- 156.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411(6839):810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 157.Saa P, Castilla J, Soto C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. Journal of Biological Chemistry. 2006;281(46):35245–35252. doi: 10.1074/jbc.M603964200. [DOI] [PubMed] [Google Scholar]
- 158.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(23):9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Llewelyn CA, Hewitt PE, Knight RS, et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363(9407):417–421. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- 160.Schmerr MJ, Goodwin KR, Cutlip RC, Jenny AL. Improvements in a competition assay to detect scrapie prion protein by capillary electrophoresis. Journal of Chromatography B: Biomedical Applications. 1996;681(1):29–35. doi: 10.1016/0378-4347(95)00541-2. [DOI] [PubMed] [Google Scholar]
- 161.Schmerr MJ, Cutlip RC, Jenny A. Capillary isoelectric focusing of the scrapie prion protein. Journal of Chromatography A. 1998;802(1):135–141. doi: 10.1016/s0021-9673(97)01120-5. [DOI] [PubMed] [Google Scholar]
- 162.Schmerr MJ, Jenny A. A diagnostic test for scrapie-infected sheep using a capillary electrophoresis immunoassay with fluorescent-labeled peptides. Electrophoresis. 1998;19(3):409–414. doi: 10.1002/elps.1150190308. [DOI] [PubMed] [Google Scholar]
- 163.Everest DJ, Waterhouse S, Kelly T, Velo-Rego E, Sauer MJ. Effectiveness of capillary electrophoresis fluoroimmunoassay of blood PrPSc for evaluation of scrapie pathogenesis in sheep. Journal of Veterinary Diagnostic Investigation. 2007;19(5):552–557. doi: 10.1177/104063870701900516. [DOI] [PubMed] [Google Scholar]
- 164.Korth C, Streit P, Oesch B. Monoclonal antibodies specific for the native, disease-associated isoform of the prion protein. Methods in Enzymology. 1999;309:106–122. doi: 10.1016/s0076-6879(99)09010-2. [DOI] [PubMed] [Google Scholar]
- 165.Nazor KE, Kuhn F, Seward T, et al. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO Journal. 2005;24(13):2472–2480. doi: 10.1038/sj.emboj.7600717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Grosset A, Moskowitz K, Nelsen C, Pan T, Davidson E, Orser CS. Rapid presymptomatic detection of PrPSc via conformationally responsive palindromic PrP peptides. Peptides. 2005;26(11):2193–2200. doi: 10.1016/j.peptides.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 167.Collinge J. Variant Creutzfeldt-Jakob disease. Lancet. 1999;354(9175):317–323. doi: 10.1016/S0140-6736(99)05128-4. [DOI] [PubMed] [Google Scholar]
- 168.Cashman NR, Caughey B. Prion diseases—close to effective therapy? Nature Reviews Drug Discovery. 2004;3(10):874–884. doi: 10.1038/nrd1525. [DOI] [PubMed] [Google Scholar]
- 169.Dormont D. Approaches to prophylaxis and therapy. British Medical Bulletin. 2003;66:281–292. doi: 10.1093/bmb/66.1.281. [DOI] [PubMed] [Google Scholar]
- 170.Borchelt DR, Scott M, Taraboulos A, Stahl N, Prusiner SB. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. Journal of Cell Biology. 1990;110(3):743–752. doi: 10.1083/jcb.110.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. Journal of Biological Chemistry. 1991;266(27):18217–18223. [PubMed] [Google Scholar]
- 172.Zinkernagel RM, Hengartner H. Regulation of the immune response by antigen. Science. 2001;293(5528):251–253. doi: 10.1126/science.1063005. [DOI] [PubMed] [Google Scholar]
- 173.Heppner FL, Aguzzi A. Recent developments in prion immunotherapy. Current Opinion in Immunology. 2004;16(5):594–598. doi: 10.1016/j.coi.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 174.Gabizon R, McKinley MP, Groth D, Prusiner SB. Immunoaffinity purification and neutralization of scrapie prion infectivity. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(18):6617–6621. doi: 10.1073/pnas.85.18.6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kascsak RJ, Rubenstein R, Merz PA, et al. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. Journal of Virology. 1987;61(12):3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Horiuchi M, Caughey B. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO Journal. 1999;18(12):3193–3203. doi: 10.1093/emboj/18.12.3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(16):9295–9299. doi: 10.1073/pnas.151242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gilch S, Wopfner F, Renner-Müller I, et al. Polyclonal anti-PrP auto-antibodies induced with dimeric PrP interfere efficiently with PrPSc propagation in prion-infected cells. Journal of Biological Chemistry. 2003;278(20):18524–18531. doi: 10.1074/jbc.M210723200. [DOI] [PubMed] [Google Scholar]
- 179.Peretz D, Williamson RA, Kaneko K, et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412(6848):739–743. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
- 180.Perrier V, Solassol J, Crozet C, et al. Anti-PrP antibodies block PrPSc replication in prion-infected cell cultures by accelerating PrPC degradation. Journal of Neurochemistry. 2004;89(2):454–463. doi: 10.1111/j.1471-4159.2004.02356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kim CL, Umetani A, Matsui T, Ishiguro N, Shinagawa M, Horiuchi M. Antigenic characterization of an abnormal isoform of prion protein using a new diverse panel of monoclonal antibodies. Virology. 2004;320(1):40–51. doi: 10.1016/j.virol.2003.10.026. [DOI] [PubMed] [Google Scholar]
- 182.Cardinale A, Filesi I, Vetrugno V, Pocchiari M, Sy MS, Biocca S. Trapping prion protein in the endoplasmic reticulum impairs PrPC maturation and prevents PrPSc accumulation. Journal of Biological Chemistry. 2005;280(1):685–694. doi: 10.1074/jbc.M407360200. [DOI] [PubMed] [Google Scholar]
- 183.Vetrugno V, Cardinale A, Filesi I, et al. KDEL-tagged anti-prion intrabodies impair PrP lysosomal degradation and inhibit scrapie infectivity. Biochemical and Biophysical Research Communications. 2005;338(4):1791–1797. doi: 10.1016/j.bbrc.2005.10.146. [DOI] [PubMed] [Google Scholar]
- 184.White AR, Enever P, Tayebi M, et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422(6927):80–83. doi: 10.1038/nature01457. [DOI] [PubMed] [Google Scholar]
- 185.Sigurdsson EM, Sy MS, Li R, et al. Anti-prion antibodies for prophylaxis following prion exposure in mice. Neuroscience Letters. 2003;336(3):185–187. doi: 10.1016/s0304-3940(02)01192-8. [DOI] [PubMed] [Google Scholar]
- 186.Heppner FL, Musahl C, Arrighi I, et al. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science. 2001;294(5540):178–182. doi: 10.1126/science.1063093. [DOI] [PubMed] [Google Scholar]
- 187.Weurtzer CA, Sulivan MA, Qiu X, Federoff HJ. CNS delivery of vectored prion-specific single-chain antibodies delays disease onset. Molecular Therapy. 2008;16(3):481–486. doi: 10.1038/sj.mt.6300387. [DOI] [PubMed] [Google Scholar]
- 188.Sigurdsson EM, Brown DR, Daniels M, et al. Immunization delays the onset of prion disease in mice. American Journal of Pathology. 2002;161(1):13–17. doi: 10.1016/S0002-9440(10)64151-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Schwarz A, Krätke O, Burwinkel M, et al. Immunisation with a synthetic prion protein-derived peptide prolongs survival times of mice orally exposed to the scrapie agent. Neuroscience Letters. 2003;350(3):187–189. doi: 10.1016/s0304-3940(03)00907-8. [DOI] [PubMed] [Google Scholar]
- 190.Rosset MB, Sacquin A, Lecollinet S, et al. Dendritic cell-mediated-immunization with xenogenic PrP and adenoviral vectors breaks tolerance and prolongs mice survival against experimental scrapie. PLoS ONE. 2009;4(3) doi: 10.1371/journal.pone.0004917. Article ID e4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Goñi F, Prelli F, Schreiber F, et al. High titers of mucosal and systemic anti-PrP antibodies abrogate oral prion infection in mucosal-vaccinated mice. Neuroscience. 2008;153(3):679–686. doi: 10.1016/j.neuroscience.2008.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hundt C, Peyrin JM, Haïk S, et al. Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. EMBO Journal. 2001;20(21):5876–5886. doi: 10.1093/emboj/20.21.5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kim CL, Karino A, Ishiguro N, Shinagawa M, Sato M, Horiuchi M. Cell-surface retention of PrPC by anti-PrP antibody prevents protease-resistant PrP formation. Journal of General Virology. 2004;85(11):3473–3482. doi: 10.1099/vir.0.80113-0. [DOI] [PubMed] [Google Scholar]
- 194.Peters PJ, Mironov A, Jr., Peretz D, et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. Journal of Cell Biology. 2003;162(4):703–717. doi: 10.1083/jcb.200304140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Shyng SL, Heuser JE, Harris DA. A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. Journal of Cell Biology. 1994;125(6):1239–1250. doi: 10.1083/jcb.125.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. Journal of Biological Chemistry. 1992;267(23):16188–16199. [PubMed] [Google Scholar]
- 197.Adelstein S, Pritchard-Briscoe H, Anderson TA, et al. Induction of self-tolerance in T cells but not B cells of transgenic mice expressing little self antigen. Science. 1991;251(4998):1223–1225. doi: 10.1126/science.1900950. [DOI] [PubMed] [Google Scholar]
- 198.Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow B cells. Journal of Experimental Medicine. 1993;177(4):1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Solforosi L, Criado JR, McGavern DB, et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303(5663):1514–1516. doi: 10.1126/science.1094273. [DOI] [PubMed] [Google Scholar]
- 200.Hanan E, Goren O, Eshkenazy M, Solomon B. Immunomodulation of the human prion peptide 106-126 aggregation. Biochemical and Biophysical Research Communications. 2001;280(1):115–120. doi: 10.1006/bbrc.2000.4097. [DOI] [PubMed] [Google Scholar]
- 201.Koller MF, Grau T, Christen P. Induction of antibodies against murine full-length prion protein in wild-type mice. Journal of Neuroimmunology. 2002;132(1-2):113–116. doi: 10.1016/s0165-5728(02)00316-8. [DOI] [PubMed] [Google Scholar]
- 202.Rosset MB, Ballerini C, Gregoire S, Metharom P, Carnaud C, Aucouturier P. Breaking immune tolerance to the prion protein using prion protein peptides plus oligodeoxynucleotide-CpG in mice. Journal of Immunology. 2004;172(9):5168–5174. doi: 10.4049/jimmunol.172.9.5168. [DOI] [PubMed] [Google Scholar]
- 203.Polymenidou M, Heppner FL, Pellicioli EC, et al. Humoral immune response to native eukaryotic prion protein correlates with anti-prion protection. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(supplement 2):14670–14676. doi: 10.1073/pnas.0404772101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hanan E, Priola SA, Solomon B. Antiaggregating antibody raised against human PrP 106-126 recognizes pathological and normal isoforms of the whole prion protein. Cellular and Molecular Neurobiology. 2001;21(6):693–703. doi: 10.1023/a:1015199904354. [DOI] [PubMed] [Google Scholar]
- 205.Li L, Napper S, Cashman NR. Immunotherapy for prion diseases: opportunities and obstacles. Immunotherapy. 2010;2(2):269–282. doi: 10.2217/imt.10.3. [DOI] [PubMed] [Google Scholar]
- 206.Goñi F, Knudsen E, Schreiber F, et al. Mucosal vaccination delays or prevents prion infection via an oral route. Neuroscience. 2005;133(2):413–421. doi: 10.1016/j.neuroscience.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 207.Nicolll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nature Medicine. 2003;9(4):448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 208.Solomon B. Alzheimer's disease and immunotherapy. Current Alzheimer Research. 2004;1(3):149–163. doi: 10.2174/1567205043332126. [DOI] [PubMed] [Google Scholar]
- 209.Gregoire S, Bergot AS, Feraudet C, Carnaud C, Aucouturier P, Rosset MB. The murine B cell repertoire is severely selected against endogenous cellular prion protein. Journal of Immunology. 2005;175(10):6443–6449. doi: 10.4049/jimmunol.175.10.6443. [DOI] [PubMed] [Google Scholar]
- 210.Nikles D, Bach P, Boller K, et al. Circumventing tolerance to the prion protein (PrP): vaccination with PrP-displaying retrovirus particles induces humoral immune responses against the native form of cellular PrP. Journal of Virology. 2005;79(7):4033–4042. doi: 10.1128/JVI.79.7.4033-4042.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Handisurya A, Gilch S, Winter D, et al. Vaccination with prion peptide-displaying papillomavirus-like particles induces autoantibodies to normal prion protein that interfere with pathologic prion protein production in infected cells. FEBS Journal. 2007;274(7):1747–1758. doi: 10.1111/j.1742-4658.2007.05721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(24):12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Krug A, Towarowski A, Britsch S, et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with Cd40 ligand to induce high amounts of IL-12. European Journal of Immunology. 2001;31(10):3026–3037. doi: 10.1002/1521-4141(2001010)31:10<3026::aid-immu3026>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 214.Krieg AM, Yi AK, Matson S, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374(6522):546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 215.Ishibashi D, Yamanaka H, Yamaguchi N, et al. Immunization with recombinant bovine but not mouse prion protein delays the onset of disease in mice inoculated with a mouse-adapted prion. Vaccine. 2007;25(6):985–992. doi: 10.1016/j.vaccine.2006.09.078. [DOI] [PubMed] [Google Scholar]
- 216.Tang Y, Zhang L, Yuan J, et al. Multistep process through which adenoviral vector vaccine overcomes anergy to tumor-associated antigens. Blood. 2004;104(9):2704–2713. doi: 10.1182/blood-2003-12-4319. [DOI] [PubMed] [Google Scholar]
- 217.Paramithiotis E, Pinard M, Lawton T, et al. A prion protein epitope selective for the pathologically misfolded conformation. Nature Medicine. 2003;9(7):893–899. doi: 10.1038/nm883. [DOI] [PubMed] [Google Scholar]
- 218.Pilon J, Loiacono C, Okeson D, et al. Anti-prion activity generated by a novel vaccine formulation. Neuroscience Letters. 2007;429(2-3):161–164. doi: 10.1016/j.neulet.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 219.Hedlin PD, Cashman NR, Li L, et al. Design and delivery of a cryptic PrPc epitope for induction of PrPSc-specific antibody responses. Vaccine. 2010;28(4):981–988. doi: 10.1016/j.vaccine.2009.10.134. [DOI] [PubMed] [Google Scholar]
- 220.Guest W, Plotkin S, Cashman N. An estimate of the PrPC beta sheet dissociation Gibbs free energy: implications for prion conversion. NeuroPrion. In press. [Google Scholar]
- 221.Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nature Reviews Drug Discovery. 2007;6(5):404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 222.Purcell AW, Zeng W, Mifsud NA, Ely LK, Macdonald WA, Jackson DC. Dissecting the role of peptides in the immune response: theory, practice and the application to vaccine design. Journal of Peptide Science. 2003;9(5):255–281. doi: 10.1002/psc.456. [DOI] [PubMed] [Google Scholar]
- 223.Conner MM, Miller MW, Ebinger MR, Burnham KP. A meta-baci approach for evaluating management intervention on chronic wasting disease in mule deer. Ecological Applications. 2007;17(1):140–153. doi: 10.1890/1051-0761(2007)017[0140:amafem]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 224.Blancou J, Kieny MP, Lathe R, et al. Oral vaccination of the fox against rabies using a live recombinant vaccinia virus. Nature. 1986;322(6077):373–375. doi: 10.1038/322373a0. [DOI] [PubMed] [Google Scholar]
- 225.Cross ML, Buddle BM, Aldwell FE. The potential of oral vaccines for disease control in wildlife species. Veterinary Journal. 2007;174(3):472–480. doi: 10.1016/j.tvjl.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 226.Gross JE, Miller MMW. Chronic wasting disease in mule deer: disease dynamics and control. Journal of Wildlife Management. 2001;65(2):205–215. [Google Scholar]