

Abstract
Cancer is currently diagnosed and treated based on the results of a tissue biopsy of the primary tumor or a metastasis using invasive techniques such as surgical resection or needle biopsy. New technology for retrieving cancer cells from the circulation, developed in the last 5 years, has made it possible to obtain a ‘fluid biopsy’ from the bloodstream without the need for an invasive procedure. This technological development makes it possible to diagnose and manage cancer from a blood test rather than from a traditional biopsy. It also allows the repeated sampling of cancer cells from a patient, making it possible, in a practical manner, to interrogate the disease repeatedly in order to understand the mechanisms by which cancer cells evolve within a given individual. The ability to obtain cancer cells repeatedly also has the potential to substantially advance drug development by enabling early ex vivo validation of both targets and early-stage compounds, as well as creating new efficiencies in the drug development process during clinical trials.
Keywords: cancer diagnosis, circulating tumor cell, fluid biopsy, prognostic indicator
Rationale for fluid biopsy
The natural history of metastatic cancer within an individual
Epithelial malignancies, such as breast, colon, lung, prostate, liver and ovarian cancer, are the most common causes of cancer death [1]. For an individual cancer patient with metastatic disease, there is a repeating pattern to the treatment and progression of the disease that is seen in cancer centers all over high-resource environments. Following the diagnosis, and potentially surgical resection, the patient receives anticancer drugs, such as chemotherapy or biologically targeted treatments. However, regardless of which drug strategy is used and which metastatic tumor is the target of therapy, the outcome is quite predictable. The drug regimens work for a while – perhaps 4 months, perhaps 20 months – but, in the overwhelming majority of cases, the drug(s) stops working at some point, resistance to the agent(s) develops and the cancer grows. The patient’s physician will then select a new drug or combination, and the cycle will repeat itself, although with each subsequent cycle the probability of achieving a response becomes less and the duration of remission becomes shorter: 2–10 months for the second-line therapy, 0–5 months for the third-line and so forth. After receiving a therapy that is unsuccessful, the patient’s cancer grows, the performance status of the patient declines and they are deemed no longer fit enough to receive drug therapy, with death usually following within less than 1 month [2]. It is the goal of cancer physicians and scientists to change this natural history of cancer.
Identifying the drivers of cancer
Tumor cells themselves can possess a wealth of information to guide therapeutic decision-making; for example, knowledge of whether there is an EGFR-activating mutation, which can result in prolonged disease-free survival for lung cancer patients [3-5]. The list of clinically important molecular targets grows annually. Estrogen receptor and Her2/neu [6-10] in breast cancer, EGFR [3-5] and EML4-ALK [11,12] in lung cancer, KRAS in colon cancer [13-15] and BRAF in melanoma [16] are all critical for deciding on whether or not a particular drug is likely to provide a benefit. It should be noted, however, that during the course of a patient’s illness, the decision-making regarding the optimal drug therapy will be based almost exclusively on the results from the biopsy taken at the time of the original cancer diagnosis. Once metastatic disease has been established, most cancer patients never have a second biopsy to look for changes in drug-resistance genes, although discordance between metastasis and the primary tumor has been described [17]. With some recent notable exceptions, the scientific and drug-development community has failed to find the essential cellular changes that confer drug resistance after a period of drug therapy [18-22]. Second biopsies of tumor tissue are avoided both because they typically confer risk to the patient associated with an invasive procedure, but also because there is very little seen under conventional pathology techniques that results in useful information from the second biopsy. In a functional personalized cancer care environment, each drug decision would be made using a real-time biopsy with multiple re-evaluations possible anytime the disease converted from drug sensitive to drug resistant.
Understanding cancer evolution
If a technology were available that would permit patients to undergo multiple repeated biopsies of their tumor, it would permit scientists to recognize the changes in the cells that drive chemotherapy-induced evolution of resistance, and also permit physicians to recognize the changes in phenotype that result in resistance. There is no question that cancers evolve resistance to chemotherapy; were it otherwise, no patient with a successful response to first-line chemotherapy would ever die. The important and, as yet, unanswered problem is to understand the fundamental changes in human cancer cells that drive resistance.
It is the topic of understanding drug resistance for which fluid biopsy of tumor material is especially important. The last 50 years have seen an explosion in the understanding of fundamental cell processes within tissue culture. From experiments with cells in culture, we have found a greater understanding of cellular signaling pathways and fundamental cellular processes. What we have not gained is an understanding of how these processes translate into the mechanisms of resistance in humans. Cells in culture, after all, bear very little resemblance to their counterparts in human disease. The gene expression patterns, for example, are entirely different [23]. The same can be said for cell–cell interactions, the relationship with the microenvironment and signaling milieu. For these reasons, among others, we have failed to predict and understand how cancer evolves in humans as opposed to cell culture systems. Having ready access to repeated samples of cancer cells from humans with disease should permit the necessary experiments required to bridge this gap and create the kind of predictive mathematical models that might offer insights. Such a model cannot be predictive without the necessary data on cellular changes in cancer cells retrieved directly from the patient over time.
In describing the fluid biopsy within this perspective, we will refrain from calling it a circulating tumor cell (CTC) assay. There are, after all, a myriad of methods that can be used to count cancer cells in the circulation and these will be briefly summarized herein. But mere counting and other bulk analyses of cells is insufficient to serve as a biopsy in which additional data, such as cell morphology and cell phenotype, are reported. Furthermore, to be regarded as a fluid biopsy, a CTC technology must provide information at the macro level about tumor–microenvironment interactions with blood being considered the third microenvironment. Similar to our understanding of the primary tumor, the other cells in the micro-environment are likely to play a significant role in the disease. At the micro level, information about cell size, nuclear shape and nuclear:cytoplasmic ratio must be accessible for studies of protein and gene expression, as are carried out with present-day biopsy specimens. In essence, the same requirements that a pathologist has of biopsy specimens from human tumors must be fulfilled by a fluid biopsy if the material is to be of diagnostic quality.
Past history of CTC technology
Cancer cells can be biopsied from a liquid media and this is frequently carried out in current clinical practice. Cytologic analysis of body fluids, such as urine and sputum as well as pleural, peritoneal and cerebrospinal fluid, will often reveal shed epithelial cancer cells that can be analyzed using conventional cytology techniques and from which a diagnosis of cancer can be made using light microscopy alone. The analysis of cells using conventional cytology techniques involves, among other things, cell size, nuclear appearance and, for epithelial tumors, the presence of clustering behavior.
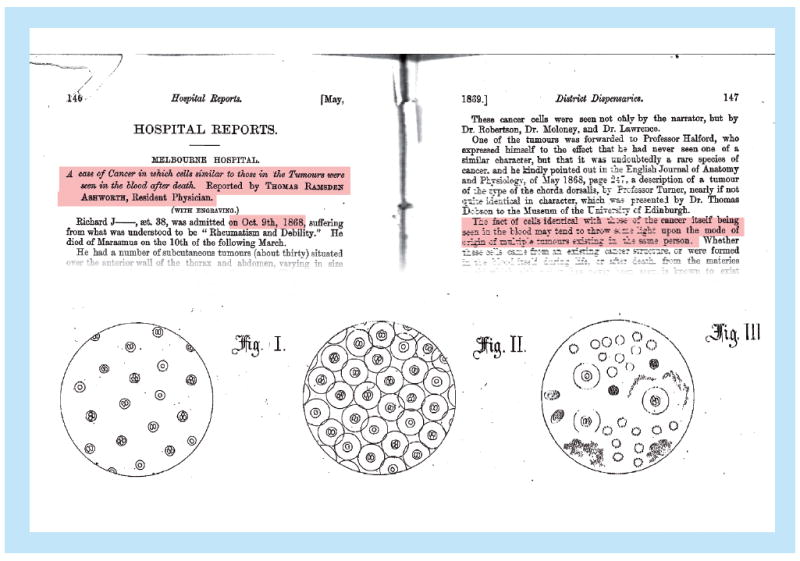
Thomas Ashworth first documented the presence of epithelial cells in the blood compartment in 1869 in a patient dying of cancer and raised the possibility that those cells were tumor derived, which potentially explained the presence of multiple tumor metastases in various anatomic locations in the patient (Figure 1) [24]. As the identification of epithelial cells in the circulation in such large numbers that they can be seen in nonenriched blood samples occurs only immediately before death, very little work was done with these cells over the next 120 years.
Figure 1. Historical medical journal article by Thomas Ashworth from 1869 describing circulating tumor cells.
The blood is not, however, the only hematopoetic compartment and in the 1990s significant work was carried out on what were then called occult tumor cells in bone marrow [25-27]. Unlike the circulation, which has a constant turnover of cells, the bone marrow has the ability to filter and retain epithelial cancer cells, leading to their detection in reasonably high numbers using conventional immunodetection techniques without enrichment of the sample for these cells. The pioneering work of Braun and Pantel in this field led to the recognition that cancer cells in this space could be detected and were associated with an inferior prognosis [26]. However, because sampling of bone marrow is painful, the practice of using occult tumor cells as a prognostic tool in cancer patients did not gain widespread adoption in the clinic despite excellent data showing prognostic significance and the availability of standardized methods for detection [28,29]. Following the recognition that tumor cells could be detected in the hematopoetic compartment, a number of investigators attempted to enumerate these cells from the bloodstream using a variety of enrichment techniques [30-33].
As the presence of cancer cells in the bloodstream presumably occurs at very low concentrations, in the order of 1–1000 cells per 10 ml, there was a need to develop new and specialized technologies to make the identification of these cells clinically feasible. These cells are 100–1000-fold more rare than others of the less-common hematopoetic cells, such as basophils and eosinophils, and are not identified by hematopathologists in routine sample processing except in rare instances when the burden of cells is overwhelming, as was seen by Ashworth. The specialized techniques used range widely, from the detection of epithelial cell mRNA [34-36] to flow cytometry with or without cell sorting [37] and differential density gradient centrifugation [38] as well as immunomagnetic enrichment, which was by far the most successful technology to accomplish this goal [39,40]. For all of these methodologies, the goal was to successfully enumerate CTCs in order to stratify patients into low- and high-risk groups based on CTC enumeration. It was hypothesized that knowledge of a patient’s CTC status could be used to drive decision-making to more appropriate uses of chemotherapy.
Molecular techniques
The detection of epithelial cell mRNA was an obvious choice of technology for molecular biologists. After all, tumor cells express cytokeratins as well as a host of other unique proteins that are not normally expressed by blood cells, and PCR techniques for detecting these cells have superb sensitivity [34,41]. There were, however, two challenges to the widespread adoption of this technology. One problem was that of illegitimate transcription [42]; on occasion, nonepithelial cells could produce a transcript of an epithelial protein. Because the ratio in blood of heme-derived cells:tumor cells is as high as 50 million:1, relatively low levels of illegitimate transcription produced noise that limited the utility of the assay. The more problematic challenge with an mRNA-detecting system with a prognostic assay is that well-differentiated tumors would produce higher amounts of the epithelial transcript of interest than poorly differentiated tumors [43]. Hence, those tumors with the worst prognosis (poorly differentiated) would have lower levels of epithelial cell transcripts than the well-differentiated, good-prognosis tumors, making the total transcript number an unreliable indicator of prognosis.
Circulating DNA assays have recently been developed to identify changes in cancer cell genomes and specific mutations. However, because these techniques do not permit whole-cell analysis, they will not be described further. Additionally, various attempts at identifying cells in epithelial–mesenchymal transition have been made using both cell-based and molecular technologies to identify the expression of mesenchymal antigens such as TWIST and vimentin. This review is limited to cellular detection techniques of epithelial cancer cells.
Physical separation methods
Flow cytometry has also been used as a CTC enumeration device [37]; however, the very low concentration of CTCs has limited the use of this technology. Most flow cytometric assays are designed to characterize 10,000–100,000 nucleated cells, a far larger number than is appropriate for CTC measurements. The strength of the technology is in its identification of unique populations of cells, but not populations that make up less than 0.0001% of analyzed events. Attempts have been made to pair a flow cytometer with a cell sorter in order to capture cells for downstream analysis and confirm the finding that the identified events are bona fide CTCs; however, this has not gained widespread use.
Density gradient methods have been used as a research tool to enrich the population of CTCs in any CTC-containing sample [38]; however, unfavorable comparisons to the current gold standard of immunomagnetic enrichment resulted in the technology falling out of favor as a clinical tool [44].
Immunomagnetic enrichment is currently the only US FDA-approved technology for the enumeration of CTCs. The technique relies on the separation of CTCs from nucleated white blood cells using, first, a single parameter, that being antibodies against EpCAM (also known as CD326 or CA17-1). After antibodies are bound to the cells they are placed in a magnetic field that isolates the cells from surrounding white blood cells. The cells are counterstained using a multiparameter fluorescent immunoassay technique in order to identify a cytokeratin-positive and CD45-negative population of nucleated cells that are deemed the white blood cells.
The immunomagnetic enrichment technique has correlated reasonably well with prognosis in breast, colorectal, prostate and lung cancer to separate populations that have a shorter survival from populations that have longer survival [45-49]. Those patients with unfavorable results on the assay appear to have a worse prognosis than those patients with a favorable result. For example, in breast cancer patients with metastatic disease, those patients with five or more CTCs have a hazard ratio for death that is 4.26 relative to those with a lower CTC count. Unfortunately, this level of risk stratification was not clearly superior to currently available protein-based tumor marker assays [50] and did not gain universal adoption in the medical oncology community. The company that developed the technology, Immunicon, filed for bankruptcy in 2008. While the platform continues to be marketed by Veridex Corporation (NJ, USA) for clinical use, it has not become popular among oncologists because of its high cost and lack of predictive ability to guide decision-making despite its prognostic value.
The limitations of the Immunicon platform to serve as a successful fluid biopsy were in part driven by technology and in other ways driven by biology. The Immunicon platform was never designed to be a fluid biopsy, but rather a prognostic test. The technical quality of the cell images was insufficiently detailed for a diagnostic quality assay.
The numbers of cells collected were significantly lower than those seen with present-day technology; this is, in part, because the target of enrichment, EpCAM, is downregulated when cancer cells leave the primary tumor and enter the circulation [51]. The other failure of the technology may be the result of the mathematical limitations of an enrichment-based technique for rare cells. As highlighted in Figure 2, when analyzing two populations of equal size on the basis of a single parameter, such as cell size or nuclear complexity, it is possible to separate the populations quite easily. However, when one population is 5 million-times larger than the other, the signal intensity from the rare population is no larger than the background noise in the larger one. For this reason, future technologies that use the fluid biopsy successfully will not rely on single-parameter measurements to separate the rare CTC population from the background of hematopoetic cells.
Figure 2. The central challenge to using single-parameter enrichment techniques in identifying rare cells.
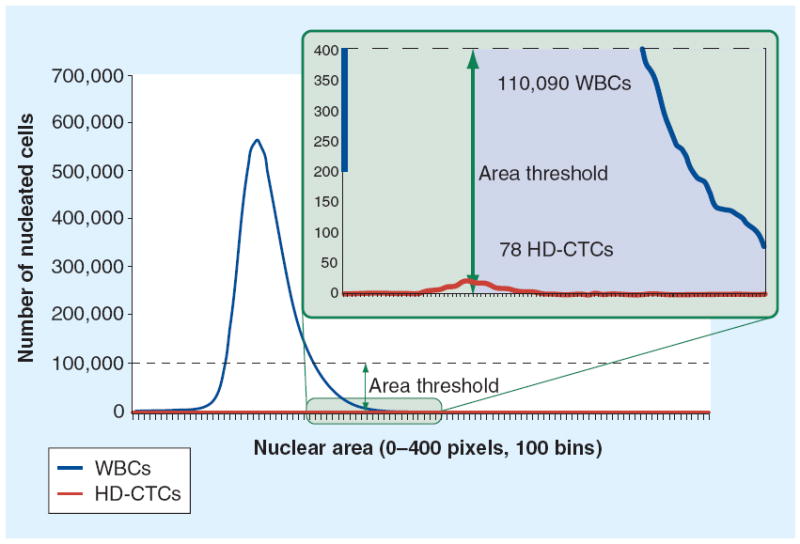
This example of computational size filtering of a single patient sample demonstrates the limitations of single-parameter enrichment. Assuming an area threshold at the 50% level (allowing for 50% false-negative rates) would result in an overwhelming number of 110,090 false-positive events.
CTC: Circulating tumor cell; HD: High definition; WBC: White blood cell.
Present fluid biopsy technologies
There are presently two technologies with the potential to evolve as fluid biopsies. These are the CTC-chip and the high-definition (HD)-CTC assay. Both assays have many of the fundamental features necessary for a fluid biopsy including multiparameter measurements, the ability for gene expression analysis and the ability to capture cells for conventional histologic analysis.
CTC-chip
First described in 2007, and developed at the laboratories of Mehmet Toner and Daniel Haber at Massachusetts General Hospital (MA, USA), the CTC-chip is a microfluidic device that uses silicone posts coated with anti-EpCAM antibodies to arrest CTCs and allow other hematopoetic cells to pass through the filtering device (Figure 3) [52]. Once captured in the device, the cells can be released for further analysis. CTCs were identified in 99% of patient specimens tested with mean CTC counts of 79–155 CTCs/ml from patient samples. These results compare quite favorably with results seen using the established immunomagnetic enrichment techniques. Having higher numbers of CTCs detected from each patient is important because the statistics of ‘small numbers’ result in unacceptable variability of interpretation when, for example, a CTC count changes from three to six, both of these numbers being within the intrinsic Poisson noise of the numbers four and five [53]. It is difficult to make the case that these integer differences reflect true changes in disease biology rather than sampling variability. Detecting larger numbers of cells in each patient mitigates against the risk imposed by Poisson statistics and creates two populations, one more likely and the other less likely to have CTCs – a far cry from personalized cancer diagnostics.
Figure 3. The CTC-chip developed at the Massachusetts General Hospital (MA, USA) uses silicone posts coated with anti-EpCAM antibodies to capture circulating tumor cells.
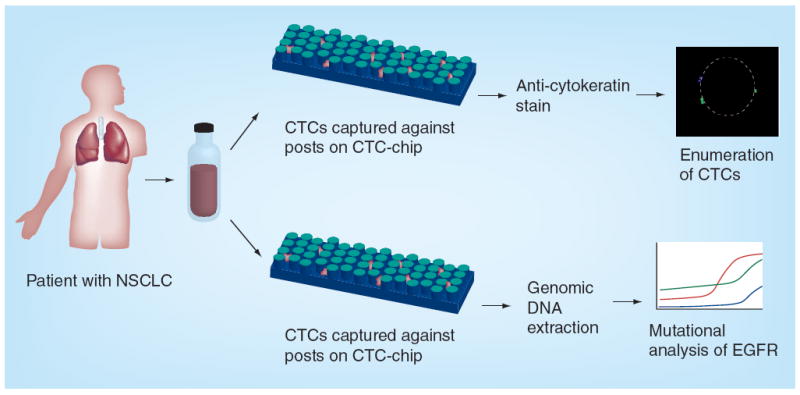
CTC: Circulating tumor cell; NSCLC: Non-small-cell lung cancer.
Reproduced with permission from [52].
Using the CTC-chip device, activating mutations in the EGF receptor were detected in CTCs and correlated with response to therapy directed against this target [54]. In addition to the original design, the same group has advanced the technology with multiple iterations and refinement to enable the move beyond the initial proof of concept, and into both clinical validation and clinical utility.
HD-CTC fluid biopsy
Another promising technology comes from Scripps Physics Oncology Center (CA, USA) and uses mathematically derived data collection in automated digital microscopy-based techniques to scan entire blood specimens in order to detect CTCs without the need for EpCAM-based enrichment [55-60]. While the CTC-chip has the advantage of being a point-of-care test, the HD-CTC technology is optimally run at a central laboratory from previously collected specimens. The HD-CTC assay has been successfully commercialized and is presently in use not only as a clinical research diagnostic, but also as a drug-development tool used to evaluate the effects of pharmaceutical products on cell targets within CTCs of clinical trial participants (Epic Sciences, La Jolla, CA, USA).
Using the HD-CTC assay, the Scripps group has focused on evaluating CTC morphology and has shown that the variability in CTC morphology resembles that seen in the primary tumor [61,62], suggesting that the release of CTCs is, in part, a stochastic phenomenon of tumor cell release. Large clusters of CTCs are frequently seen using the HD-CTC assay [59], permitting pathologists to view cell–cell interactions in a manner analogous to that seen in cytology preparations from body fluids such as pleural and peritoneal fluid (Figure 4) [57]. The primary limitation of the HD-CTC assay is that the cells are fixed and permeabilized on the glass slide, precluding analysis requiring live cells.
Figure 4. The high-definition circulating tumor cell assay and identification process.
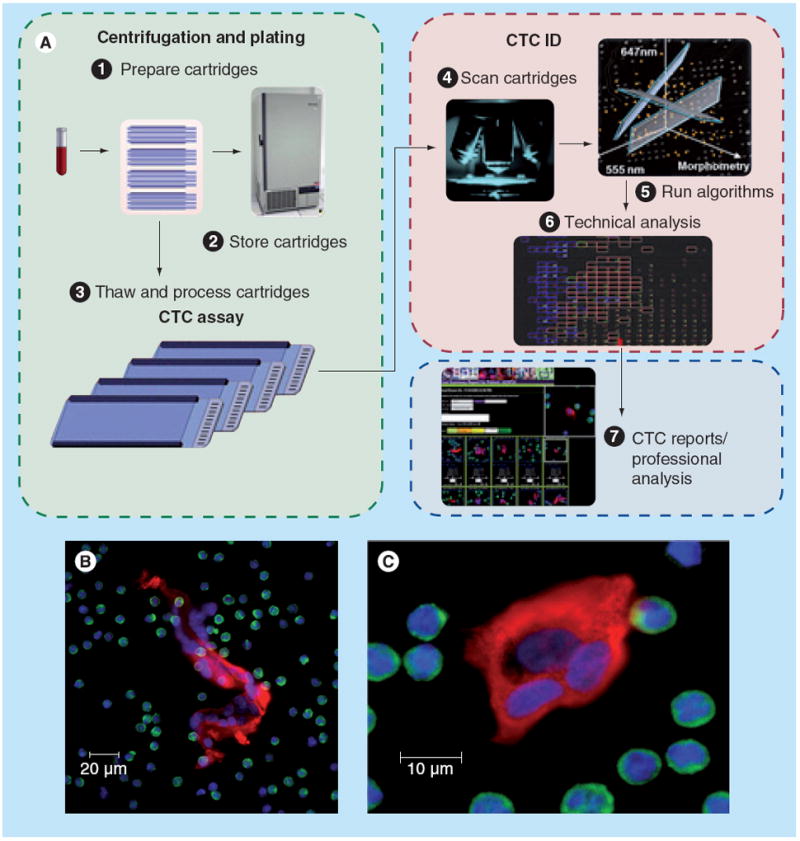
(A) The sequence of events in a high-definition CTC assay. (B) and (C) show two high-definition CTC clusters displaying the typical high-definition CTC fluorescence patterns.
CTC: Circulating tumor cell.
Future of the fluid biopsy
The future is expected to bring uses for the fluid biopsy both as a drug-development tool as well as a clinical tool. It is now increasingly understood that cell culture systems do not fully replicate the effects of drugs when viewed in the context of the heterogeneity of a cancer type within a population. Similarly, animal models that rely on the same type of cell-cultured cell line to determine the effects of drugs only replicate the effects of physiologic and metabolic heterogeneity, and do not reflect the disease variability. Success in the war on cancer will require an increasing shift away from animal models and cell culture systems, and requires increased analysis of cancer cells in affected humans.
Understanding cancer evolution
It will be important for pharmaceutical companies to understand what happens to cancer cells at the moments when their drug is first effective and then no longer works. That is, to take clinical trial patients who have experienced a response to drug therapy and then analyze their tumor cells at the moment that the tumor begins to regrow and display resistance. Cells in culture undergo all sorts of phenomena to make this happen, such as upregulation of multidrug resistance channels and downregulation of apoptosis pathways. However, despite this knowledge of how cells in culture develop resistance, targeting these pathways has not been successful as drug targets in patients. Perhaps what happens in humans and what happens in culture are not the same, and the focus should be on the former rather than the latter in understanding drug resistance. It will be from studies in patients, and not those in culture systems, that we will develop an understanding of how tumors evolve within individuals.
As a drug-development tool, there is increasing emphasis on understanding the downstream cell signaling effects of a drug after it is administered to actual patients. Pharmaceutical companies must understand whether their products successfully bind the target and mediate the expected downstream effects. This is especially important in early Phase I and II drug development when efficacy against the target must be weighed against toxicity in the decision to move a drug forward to the next phase of development. The successful pharmaceutical drug developers of the future will use the fluid biopsy to gain early insights into their drug’s effect on tumor cells by developing custom-built secondary assays on captured tumor cells that provide this kind of information. In the present day, this approach to drug development requires additional biopsies of tumor cells, an expensive process and one that requires additional human subject risk that may not be viewed favorably by institutional review boards. One might anticipate that, once the fluid biopsy becomes readily available, drug developers seeking to ask human subjects to undergo the risk of a second biopsy will need to justify to institutional review boards why they are choosing such an approach when a fluid biopsy could garner the same information without additional risk to human subjects.
Clinical applications
As a clinical tool, the fluid biopsy is expected to reach a stage where this technology is used both for diagnosis as well as for the monitoring of cancer cells. There is no a priori reason why cells shed into the circulation should be treated differently than cells shed into other body fluids such as sputum or urine. However, in order for this to happen, the technology developers will need to be able to present images to pathologists from light rather than darkfield microscopes. The technological tools need to give pathologists what they want rather than ask pathologists to change the way they presently practice if they are to make an impact. Fluid biopsy technologies of the future should be able to present cell images to pathologists using the conventional hematoxylin and eosin, Wright–Giemsa and Papanicolaou staining, which is presently used by these professionals to make a cancer diagnosis.
In the future, patients who are at high risk of complications from needle biopsy to establish a diagnosis such as coagulopathy, emphysema or tumor at an anatomically high-risk site should be able to receive a diagnosis without the need for unnecessary procedure-related risk. Primary care clinicians should be able to see a tumor in a patient’s x-ray and send a fluid biopsy for evaluation before the patient sees a procedural specialist solely for the purpose of obtaining diagnostic tissue. This is expected to lead to better utilization of the increasingly limited supply of surgical specialists.
The future should also bring the entire arsenal of pathology-related gene expression assays to the fluid biopsy field. FISH testing for gene rearrangements and expression analysis has already been successfully accomplished and will, in the future, become clinical routine as a companion diagnostic test (commercialized by Epic Sciences, Inc., CA, USA). The first HD-CTC FISH assay in routine use is the PTEN loss test (Epic Sciences). Mutation-specific antibodies should make the detection of DNA mutations possible in cells that have cancer-driving mutations and PCR testing should become feasible on small numbers of cells, even those that have been permeabilized to permit cytokeratin detection.
The monitoring of metastatic disease, the current use of immunomagnetic enrichment technologies, may or may not continue to provide a role for fluid biopsy in the future. It is not clear that enumeration of CTCs using today’s technology provides benefits beyond those that are seen using cheaper protein-based biomarkers. For monitoring purposes, CTC enumeration technologies may have value in their universality, that is, the same assay can be used for patients with many different epithelial tumor types. For this application, the technology will need to undergo significant cost reductions and be readily available as a point-of-care device within physician’s offices. It is in this area that the present day CTC-chip technology may be expected to have the largest impact given the simplicity and scalability of the platform design.
Conclusion
There is no question that the fluid biopsy will have a future in cancer medicine. Indeed, there are already several pharmaceutical companies that have recognized the importance of real-time information on cancer cells from patients who are undergoing novel therapies. Both HD-CTC technologies and the CTC-chip continue to be developed and refined to the point where they will become everyday tools in the war on cancer, deployed by clinicians and pharmaceutical scientists alike.
One can imagine a future where a patient comes to his primary care doctor’s office with a cough and imaging studies reveal multiple tumors in the lung, liver, adrenal gland and bone. A fluid biopsy is ordered, and it is able to report that the patient not only has cancer, but has lung cancer, non-small-cell type, with an activating mutation in EGFR. The patient is started on erlotinib and continues on the agent until it is no longer effective. At that point, he undergoes another fluid biopsy that identifies the molecular cause for the erlotinib resistance and a second, as-yet undeveloped, pharmaceutical is added and remission is regained.
There remains much more work to be done, by both the drug-development community and physicians alike.
Future perspective
In the coming decade, many, if not most, early-phase clinical trials will include fluid biopsy as a routine method for determining the on-target effects of new medications before those drugs enter randomized trials. This will lead to a paradigm shift in the goals of early-phase clinical trials by allowing for efficacy end points, as measured on a cellular level, to be obtained simultaneously with toxicity end points in early drug development. With regard to clinical cancer care, the fluid biopsy will serve as a substitute for a tissue biopsy in those patients who are deemed too frail or too ill to undergo an invasive tissue biopsy and will gain growing acceptance in the pathology community as an equivalent to the cytologic examination of other fluid compartments, but with improved convenience of sample procurement.
Executive summary.
-
▪
Circulating tumor cells can be detected in the bloodstream of cancer patients with epithelial malignancies.
-
▪
The presence of circulating tumor cells correlates with poor prognosis.
-
▪
A variety of technological approaches to circulating tumor cell detection exist.
-
▪
Technologies that permit phenotypic characterization of whole cells represent the future of the field and serve as a ‘fluid biopsy’.
-
▪
The fluid biopsy has potential application in early-phase drug development for the resampling of tumor cells after drug administration to determine whether a drug target has been affected.
-
▪
The fluid biopsy has potential application as a diagnostic assay for cancer in patients in whom an invasive biopsy would confer unacceptable risk.
Acknowledgments
This manuscript was supported by NIH grants R01CA125653 and U54CA143906 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the NIH. This is manuscript #21739 from The Scripps Research Institute. The HD-CTC assay technology described here has been licensed to Epic Sciences. JJ Nieva and P Kuhn have ownership in Epic Sciences.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
- 1.Cancer Facts & Figures 2011. American Cancer Society; GA, USA: 2011. [Google Scholar]
- 2.Hardy D, Chan W, Liu CC, et al. Racial disparities in length of stay in hospice care by tumor stage in a large elderly cohort with non-small cell lung cancer. Palliat Med. 2012;26(1):61–71. doi: 10.1177/0269216311407693. [DOI] [PubMed] [Google Scholar]
- 3.Gazdar AF. Personalized medicine and inhibition of EGFR signaling in lung cancer. N Engl J Med. 2009;361(10):1018–1020. doi: 10.1056/NEJMe0905763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doroshow JH. Targeting EGFR in non-small-cell lung cancer. N Engl J Med. 2005;353(2):200–202. doi: 10.1056/NEJMe058113. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 6.Bonadonna G, Valagussa P, Tancini G, Di Fronzo G. Estrogen-receptor status and response to chemotherapy in early and advanced breast cancer. Cancer Chemother Pharmacol. 1980;4(1):37–41. doi: 10.1007/BF00255456. [DOI] [PubMed] [Google Scholar]
- 7.Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20(3):719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 8.Eiermann W. Trastuzumab combined with chemotherapy for the treatment of HER2-positive metastatic breast cancer: pivotal trial data. Ann Oncol. 2001;12(Suppl. 1):S57–S62. [PubMed] [Google Scholar]
- 9.Burstein HJ, Kuter I, Campos SM, et al. Clinical activity of trastuzumab and vinorelbine in women with HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2001;19(10):2722–2730. doi: 10.1200/JCO.2001.19.10.2722. [DOI] [PubMed] [Google Scholar]
- 10.Seidman AD, Fornier MN, Esteva FJ, et al. Weekly trastuzumab and paclitaxel therapy for metastatic breast cancer with analysis of efficacy by HER2 immunophenotype and gene amplification. J Clin Oncol. 2001;19(10):2587–2595. doi: 10.1200/JCO.2001.19.10.2587. [DOI] [PubMed] [Google Scholar]
- 11.Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12(11):1004–1012. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ou SH, Bazhenova L, Camidge DR, et al. Rapid and dramatic radiographic and clinical response to an ALK inhibitor (crizotinib, PF02341066) in an ALK translocation-positive patient with non-small cell lung cancer. J Thorac Oncol. 2010;5(12):2044–2046. doi: 10.1097/JTO.0b013e318200f9ff. [DOI] [PubMed] [Google Scholar]
- 13.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 14.Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 15.Nagasaka T, Sasamoto H, Notohara K, et al. Colorectal cancer with mutation in BRAF, KRAS, and wild-type with respect to both oncogenes showing different patterns of DNA methylation. J Clin Oncol. 2004;22(22):4584–4594. doi: 10.1200/JCO.2004.02.154. [DOI] [PubMed] [Google Scholar]
- 16.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17▪.Pusztai L, Viale G, Kelly CM, Hudis CA. Estrogen and HER-2 receptor discordance between primary breast cancer and metastasis. Oncologist. 2010;15(11):1164–1168. doi: 10.1634/theoncologist.2010-0059. Observed that there is heterogeneity between primary tumors and metastasis, laying the foundation for assays that can reassess tumors as their phenotype evolves. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell LJ, Patsouris C, Rayeroux KC, Somana K, Januszewicz EH, Szer J. BCR/ABL amplification in chronic myelocytic leukemia blast crisis following imatinib mesylate administration. Cancer Genet Cytogenet. 2002;139(1):30–33. doi: 10.1016/s0165-4608(02)00615-5. [DOI] [PubMed] [Google Scholar]
- 19.Gorre ME, Ellwood-Yen K, Chiosis G, Rosen N, Sawyers CL. BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL chaperone heat shock protein 90. Blood. 2002;100(8):3041–3044. doi: 10.1182/blood-2002-05-1361. [DOI] [PubMed] [Google Scholar]
- 20.Luzzatto L, Melo JV. Acquired resistance to imatinib mesylate: selection for pre-existing mutant cells. Blood. 2002;100(3):1105. doi: 10.1182/blood-2002-05-1578. [DOI] [PubMed] [Google Scholar]
- 21.Kosaka T, Yatabe Y, Endoh H, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12(19):5764–5769. doi: 10.1158/1078-0432.CCR-06-0714. [DOI] [PubMed] [Google Scholar]
- 22.Gow CH, Shih JY, Chang YL, Yu CJ. Acquired gefitinib-resistant mutation of EGFR in a chemonaive lung adenocarcinoma harboring gefitinib-sensitive mutation L858R. PLoS Med. 2005;2(9):e269. doi: 10.1371/journal.pmed.0020269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniel VC, Marchionni L, Hierman JS, et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009;69(8):3364–3373. doi: 10.1158/0008-5472.CAN-08-4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashworth T. A case of cancer in which cells similar to those in the tumors were seen in blood after death. Aust Med J. 1869;14:146–147. [Google Scholar]
- 25.Braun S, Muller M, Hepp F, Schlimok G, Riethmuller G, Pantel K. Re: Micrometastatic breast cancer cells in bone marrow at primary surgery: prognostic value in comparison with nodal status. J Natl Cancer Inst. 1998;90(14):1099–1101. doi: 10.1093/jnci/90.14.1099. [DOI] [PubMed] [Google Scholar]
- 26.Pantel K, Izbicki JR, Angstwurm M, et al. Immunocytological detection of bone marrow micrometastasis in operable non-small cell lung cancer. Cancer Res. 1993;53(5):1027–1031. [PubMed] [Google Scholar]
- 27.Pantel K, Braun S, Schlimok G, Riethmuller G. Micrometastatic tumour cells in bone marrow in colorectal cancer. Lancet. 1993;341(8843):501. doi: 10.1016/0140-6736(93)90262-f. [DOI] [PubMed] [Google Scholar]
- 28.Fehm T, Braun S, Muller V, et al. A concept for the standardized detection of disseminated tumor cells in bone marrow from patients with primary breast cancer and its clinical implementation. Cancer. 2006;107(5):885–892. doi: 10.1002/cncr.22076. [DOI] [PubMed] [Google Scholar]
- 29▪.Braun S, Pantel K, Muller P, et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N Engl J Med. 2000;342(8):525–533. doi: 10.1056/NEJM200002243420801. Identified the important prognostic implications of hematogenously seeded micrometastasis in the early stages of cancer. [DOI] [PubMed] [Google Scholar]
- 30.Gertler R, Rosenberg R, Fuehrer K, Dahm M, Nekarda H, Siewert JR. Detection of circulating tumor cells in blood using an optimized density gradient centrifugation. Recent Results Cancer Res. 2003;162:149–155. doi: 10.1007/978-3-642-59349-9_13. [DOI] [PubMed] [Google Scholar]
- 31.Ma PC, Blaszkowsky L, Bharti A, et al. Circulating tumor cells and serum tumor biomarkers in small cell lung cancer. Anticancer Res. 2003;23(1A):49–62. [PubMed] [Google Scholar]
- 32.Sabbatini R, Federico M, Morselli M, et al. Detection of circulating tumor cells by reverse transcriptase polymerase chain reaction of maspin in patients with breast cancer undergoing conventional-dose chemotherapy. J Clin Oncol. 2000;18(9):1914–1920. doi: 10.1200/JCO.2000.18.9.1914. [DOI] [PubMed] [Google Scholar]
- 33.Yamashita JI, Kurusu Y, Fujino N, Saisyoji T, Ogawa M. Detection of circulating tumor cells in patients with non-small cell lung cancer undergoing lobectomy by video-assisted thoracic surgery: a potential hazard for intraoperative hematogenous tumor cell dissemination. J Thorac Cardiovasc Surg. 2000;119(5):899–905. doi: 10.1016/S0022-5223(00)70084-5. [DOI] [PubMed] [Google Scholar]
- 34.Ignatiadis M, Xenidis N, Perraki M, et al. Different prognostic value of cytokeratin-19 mRNA positive circulating tumor cells according to estrogen receptor and HER2 status in early-stage breast cancer. J Clin Oncol. 2007;25(33):5194–5202. doi: 10.1200/JCO.2007.11.7762. [DOI] [PubMed] [Google Scholar]
- 35.Xenidis N, Perraki M, Kafousi M, et al. Predictive and prognostic value of peripheral blood cytokeratin-19 mRNA-positive cells detected by real-time polymerase chain reaction in node-negative breast cancer patients. J Clin Oncol. 2006;24(23):3756–3762. doi: 10.1200/JCO.2005.04.5948. [DOI] [PubMed] [Google Scholar]
- 36.Stathopoulou A, Ntoulia M, Perraki M, et al. A highly specific real-time RT-PCR method for the quantitative determination of CK-19 mRNA positive cells in peripheral blood of patients with operable breast cancer. Int J Cancer. 2006;119(7):1654–1659. doi: 10.1002/ijc.22017. [DOI] [PubMed] [Google Scholar]
- 37.Racila E, Euhus D, Weiss AJ, et al. Detection and characterization of carcinoma cells in the blood. Proc Natl Acad Sci USA. 1998;95(8):4589–4594. doi: 10.1073/pnas.95.8.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberg R, Gertler R, Friederichs J, et al. Comparison of two density gradient centrifugation systems for the enrichment of disseminated tumor cells in blood. Cytometry. 2002;49(4):150–158. doi: 10.1002/cyto.10161. [DOI] [PubMed] [Google Scholar]
- 39▪.Cristofanilli M, Budd GT, Ellis MJ, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351(8):781–791. doi: 10.1056/NEJMoa040766. Describes the use of circulating tumor cells as a prognostic test. [DOI] [PubMed] [Google Scholar]
- 40.Tibbe AG, de Grooth BG, Greve J, Liberti PA, Dolan GJ, Terstappen LW. Cell analysis system based on immunomagnetic cell selection and alignment followed by immunofluorescent analysis using compact disk technologies. Cytometry. 2001;43(1):31–37. [PubMed] [Google Scholar]
- 41.Lianidou ES, Markou A. Circulating tumor cells in breast cancer: detection systems, molecular characterization, and future challenges. Clin Chem. 2011;57(9):1242–1255. doi: 10.1373/clinchem.2011.165068. [DOI] [PubMed] [Google Scholar]
- 42.Chelly J, Concordet JP, Kaplan JC, Kahn A. Illegitimate transcription: transcription of any gene in any cell type. Proc Natl Acad Sci USA. 1989;86(8):2617–2621. doi: 10.1073/pnas.86.8.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zach O, Kasparu H, Krieger O, Hehenwarter W, Girschikofsky M, Lutz D. Detection of circulating mammary carcinoma cells in the peripheral blood of breast cancer patients via a nested reverse transcriptase polymerase chain reaction assay for mammaglobin mRNA. J Clin Oncol. 1999;17(7):2015–2019. doi: 10.1200/JCO.1999.17.7.2015. [DOI] [PubMed] [Google Scholar]
- 44.Balic M, Dandachi N, Hofmann G, et al. Comparison of two methods for enumerating circulating tumor cells in carcinoma patients. Cytometry B Clin Cytom. 2005;68(1):25–30. doi: 10.1002/cyto.b.20065. [DOI] [PubMed] [Google Scholar]
- 45.Shaffer DR, Leversha MA, Danila DC, et al. Circulating tumor cell analysis in patients with progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13(7):2023–2029. doi: 10.1158/1078-0432.CCR-06-2701. [DOI] [PubMed] [Google Scholar]
- 46.Cohen SJ, Alpaugh RK, Gross S, et al. Isolation and characterization of circulating tumor cells in patients with metastatic colorectal cancer. Clin Colorectal Cancer. 2006;6(2):125–132. doi: 10.3816/CCC.2006.n.029. [DOI] [PubMed] [Google Scholar]
- 47.Moreno JG, Miller MC, Gross S, Allard WJ, Gomella LG, Terstappen LW. Circulating tumor cells predict survival in patients with metastatic prostate cancer. Urology. 2005;65(4):713–718. doi: 10.1016/j.urology.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 48.Cristofanilli M, Hayes DF, Budd GT, et al. Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J Clin Oncol. 2005;23(7):1420–1430. doi: 10.1200/JCO.2005.08.140. [DOI] [PubMed] [Google Scholar]
- 49.Krebs MG, Hou JM, Sloane R, et al. Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and -independent approaches. J Thorac Oncol. 2012;7(2):306–315. doi: 10.1097/JTO.0b013e31823c5c16. [DOI] [PubMed] [Google Scholar]
- 50.Hepp PGM, Rack BK, Tesch H, et al. Correlation of CA 27.29 and circulating tumor cells before, at the end, and 2 years after adjuvant chemotherapy in patients with primary breast cancer: the SUCCESS trial. J Clin Oncol. 2011;29(Suppl) Abstract 10626. [Google Scholar]
- 51.Rao CG, Chianese D, Doyle GV, et al. Expression of epithelial cell adhesion molecule in carcinoma cells present in blood and primary and metastatic tumors. Int J Oncol. 2005;27(1):49–57. [PubMed] [Google Scholar]
- 52▪.Sequist LV, Nagrath S, Toner M, Haber DA, Lynch TJ. The CTC-chip: an exciting new tool to detect circulating tumor cells in lung cancer patients. J Thorac Oncol. 2009;4(3):281–283. doi: 10.1097/JTO.0b013e3181989565. Description of a microfluidic device that is capable of a fluid biopsy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tibbe AG, Miller MC, Terstappen LW. Statistical considerations for enumeration of circulating tumor cells. Cytometry A. 2007;71(3):154–162. doi: 10.1002/cyto.a.20369. [DOI] [PubMed] [Google Scholar]
- 54▪.Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359(4):366–377. doi: 10.1056/NEJMoa0800668. Describes the molecular characterization of individual circulating tumor cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wendel M, Bazhenova L, Boshuizen R, et al. Fluid biopsy for circulating tumor cell identification in patients with early-and late-stage non-small cell lung cancer: a glimpse into lung cancer biology. Phys Biol. 2012;9(1):016005. doi: 10.1088/1478-3967/9/1/016005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nieva J, Wendel M, Luttgen MS, et al. High-definition imaging of circulating tumor cells and associated cellular events in non-small cell lung cancer patients: a longitudinal analysis. Phys Biol. 2012;9(1):016004. doi: 10.1088/1478-3975/9/1/016004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57▪.Marrinucci D, Bethel K, Kolatkar A, et al. Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys Biol. 2012;9(1):016003. doi: 10.1088/1478-3975/9/1/016003. Description of a whole-cell-based approach to the fluid biopsy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lazar DC, Cho EH, Luttgen MS, et al. Cytometric comparisons between circulating tumor cells from prostate cancer patients and the prostate-tumor-derived LNCaP cell line. Phys Biol. 2012;9(1):016002. doi: 10.1088/1478-3975/9/1/016002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59▪.Cho EH, Wendel M, Luttgen M, et al. Characterization of circulating tumor cell aggregates identified in patients with epithelial tumors. Phys Biol. 2012;9(1):016001. doi: 10.1088/1478-3975/9/1/016001. Describes circulating tumor cell aggregates, laying the foundation for a fluid biopsy to evaluate cell–cell interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marrinucci D, Bethel K, Bruce RH, et al. Case study of the morphologic variation of circulating tumor cells. Hum Pathol. 2007;38(3):514–519. doi: 10.1016/j.humpath.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 61.Marrinucci D, Bethel K, Lazar D, et al. Cytomorphology of circulating colorectal tumor cells: a small case series. J Oncol. 2010;2010:861341. doi: 10.1155/2010/861341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62▪.Marrinucci D, Bethel K, Luttgen M, Bruce RH, Nieva J, Kuhn P. Circulating tumor cells from well-differentiated lung adenocarcinoma retain cytomorphologic features of primary tumor type. Arch Pathol Lab Med. 2009;133(9):1468–1471. doi: 10.1043/1543-2165-133.9.1468. Observes that primary tumor and circulating tumor cells share similar morphology. [DOI] [PMC free article] [PubMed] [Google Scholar]