

Abstract
Purpose
Progress in identification of effective therapies for diabetic nephropathy continues to be limited by the lack of ideal animal models. Here we review the current status of some leading murine models of this disorder.
Recent Findings
A consensus statement of the Animals Models of Diabetic Complications Consortium sets forth guidelines and standards for measuring renal function and structural parameters necessary for validating murine models of diabetic nephropathy. Two murine models exploiting eNOS deficiency as a major susceptibility factor for development of diabetic nephropathy are among the very few options for studying features of advanced diabetic nephropathy. Akita and OVE26 mice with mutations that result in Type I diabetes are also useful models of diabetic nephropathy. The recently described BTBR ob/ob (leptin deficient) mouse with Type II diabetes demonstrates key features of early podocyte loss and mesangiolysis characteristic of human diabetic nephropathy.
Summary
While there are many murine models of mesangial matrix expansion in the setting of diabetes, few progress to develop advanced diabetic lesions. Mice with eNOS deficiency, OVE26 mice, and the recently described BTBR ob/ob mouse currently appear to be the best murine models of advanced disease. A model that allows testing of interventions that modulate podocyte loss and regeneration, such as the BTBR ob/ob mouse, may be of particular benefit in developing therapeutics for diabetic nephropathy.
Keywords: diabetic nephropathy, animal models, podocytes, leptin, eNOS
Introduction
The value of animal models for studies of the pathogenesis and natural history of diabetic nephropathy (DN) is beyond question. An ideal animal model would reproduce most or all of the lesions of human DN as described below and would be susceptible to genetic analysis. For practical reasons such an animal model should be cheap, easy to maintain and widely available. These last requirements generally preclude consideration of non-human primates, dogs, and swine as widely utilizable animal species for model development. For these reasons, we focus on murine systems as the most useful and widely used models, although there are a few examples where investigators have used lower order model systems such as fruit flies and zebrafish to gain mechanistic insights into diabetic type renal injury. In this consideration of animal models of DN, we begin with this question: What are the features of human DN that should be present in an animal model?
Structural Alterations Characteristic of Diabetic Nephropathy in Humans
Diabetic nephropathy is in large part a glomerular disease. Abnormalities of glomerular structure are among the most striking and consistent changes identified in biopsy material from patients with DN [1]. A well-defined sequence of glomerular injury has been identified, based on experimental studies and observations in human patients. In the earliest stages of injury, which correlate at least in part with the hemodynamic changes of hyperfiltration, glomerular (and renal) hypertrophy occurs [1].
Mesangial injury
Increased accumulation of mesangial matrix then develops. This process is generally global – involving all segments of the glomeruli – and diffuse – involving all the glomeruli throughout the cortex. This process of diffuse mesangial sclerosis – or diffuse intercapillary glomerulosclerosis as termed by Kimmelstiel and Wilson – is the hallmark lesion of diabetic glomerulopathy.
The lesions of diffuse mesangial sclerosis are progressive, culminating in the distinctive nodular accumulations of mesangial matrix, so-called Kimmelstiel-Wilson nodules, generally 15 years or more after the onset of diabetes. Nodular mesangial sclerosis has been estimated to occur in approximately 25% of renal biopsies in patients with advanced DN, although the precise prevalence of this lesion is not known [1,2].
Related to, and likely central to, the process of nodular mesangial sclerosis are mesangiolysis and capillary microaneurysm formation. Mesangiolysis – the fraying and focal dissolution of the mesangial matrix, and capillary microaneurysms – the ballooning out into the urinary space of glomerular capillaries with frequent accumulations of hyaline in the enlarged capillary lumina–often occur in conjunction with mesangial nodules. Some observers have deduced a sequence of injury of repetitive mesangiolysis, microaneurysm formation, and subsequent capillary collapse that leads to the formation of mesangial nodules [3]. These alterations in mesangial cells and the mesangial matrix have long been considered central to the evolution of DN.
While mesangial alterations similar to those found in human DN are the most characteristic and easily recognized abnormalities of DN, there has been a recent focus on podocyte alterations, since podocytes are reduced in number in both Type I and Type II diabetics with DN, and this reduction in podocytes may precede and even predict the onset of clinically detectable proteinuria [4]. Other lesions that characterize the pathology of diabetic glomerular injury include diffuse thickening of glomerular basement membranes and loss of charge and permselectivity of the glomerular filtration barrier. The pathophysiologic basis for basement membrane thickening, mesangial expansion, mesangiolysis, microaneurysm formation, and podocyte loss largely remain unknown. An ideal animal model of DN would recapitulate each of these changes.
Mouse Models of Diabetic Nephropathy
There are numerous rodent and rabbit models of spontaneous and acquired (e.g. induction with streptozotocin) diabetes, which have been the subject of recent reviews [5–7]**. Most of these models, including NOD mice and mice administered streptozotocin as models of Type I diabetes, and db/db mice as the most widely used model of Type II diabetes, develop functional and morphologic renal abnormalities in conjunction with hyperglycemia. The functional abnormalities include hyperfiltration and proteinuria; morphologic abnormalities include variable degrees of mesangial matrix expansion and glomerular capillary basement membrane thickening. These features replicate the changes of morphologically early DN, but very few of these models develop the nodular glomerulosclerosis characteristic of advanced human DN and certainly do not do it quickly or reliably.
The NIH-supported Animal Models of Diabetic Complications Consortium (AMDCC) was created in part to address these deficiencies, and remains engaged in efforts to identify and characterize more comprehensive mouse models of DN. A consensus publication from this group in 2005 [5], and updated in 2009 [7],** provides explicit standards for phenotyping murine models of DN and indicates major methodological pitfalls that have made the results from many studies of various models problematic to interpret. One premise of this group is that the standardization of phenotyping methodology will allow better comparisons of results obtained by different groups using different animal models, and ensure that future studies will increasingly use murine models that closely and reliably model human disease. The AMDCC initially proposed the following three criteria for an ideal mouse model: 1) Progressive renal insufficiency in the setting of hyperglycemia; 2) Albuminuria, and 3) Characteristic pathologic changes including basement membrane thickening by electron microscopy, mesangial matrix expansion ± nodular mesangial sclerosis, interstitial fibrosis and arteriolar hyalinosis [5].
These criteria have been updated to include:
Greater than 50% decline in GFR over the lifetime of the animal.
Greater than 10-fold increase in albuminuria compared with controls for that strain at the same age and gender.
Pathologic changes in kidneys, including advanced mesangial matrix expansion +/− nodular sclerosis and mesangiolysis, any degree of arteriolar hyalinosis, glomerular basement membrane thickening by >50% over baseline, and tubulointerstitial fibrosis [7].
Additional histologic phenotyping for diabetic nephropathy models with advanced disease should include:
Quantification of mesangial matrix expression, ideally with morphometric analyses, and with mesangiolysis and microaneurysms detected with appropriate tissue stains.
Exclusion of immune complex disease with immunohistochemistry on frozen sections for IgG, IgM, and IgA.
Demonstration of glomerular basement membrane thickening by electron microscopy.
Demonstration of podocyte loss by a reasonable morphometric system [7].
Demonstration of podocyte loss is critically important in view of the extensive data that podocyte loss is an early and likely essential event in progressive human DN, yet this is infrequently determined in animal models. Podocyte loss cannot be recognized in routine histologic preparations. Immunohistochemical stains are necessary to identify podocytes and podocyte loss, and the methodology used must account for differences in podocyte density as glomerular volume is altered (e.g., by the hypertrophy characteristic of DN) [8,9]. These standards also emphasize the importance of going beyond measures of altered mesangial matrix volume alone to characterize the structural abnormalities in models of DN.
Commonly overlooked by investigators and reviewers of studies is that these criteria were established by consensus, and represent goals and not absolute standards. Failure to achieve any one specific standard should not doom a promising mouse model to oblivion, but in aggregate a model of DN that meets most of these criteria is likely to provide useful as a research tool. As long as this caveat is kept in mind, these standards have begun to gain wide acceptance for being both unambiguous and rigorous [10].
In addition to defining goals for phenotyping mouse models of DN and reviewing strengths of several models under active development, the AMDCC consensus paper provides a perspective on where the field is moving. One trend followed by both the consortium and other investigators to overcome the limitations of current models is a strategy of breeding critical diabetogenic mutations into mouse strains found to be susceptible to the development of specific complications such as nephropathy (e.g. the DBA/2 strain) versus strains that are resistant (e.g. C57BL/6). To date a few monogenic mutations (eNOS [11] and bradykinin receptor [12] deficiencies are examples) have been shown to impart significant features of DN when introduced into susceptible mice, but there have been few such mutations identified to date and none as yet fully model human DN.
db/db
The db/db mouse model of leptin deficiency is currently the most widely used mouse for modeling DN in settings of Type II diabetes. Leptin deficiency confers susceptibility to obesity, insulin resistance, and Type II diabetes. db/db mice have a mutation deletion of the leptin receptor and an underlying genetic background that is susceptible to diabetic complications such as nephropathy [13]. DN in these mice is manifest by albuminuria, podocyte loss, and mesangial matrix expansion [5,13,14]. The mesangial expansion develops relatively slowly, often needing a half year or more to fully evolve. In general, these mice do not develop mesangiolysis or nodular mesangial sclerosis, nor do they develop progressive renal insufficiency. Hence, these mice are a good model of the early changes of human DN, but fail to recapitulate later and morphologically advanced features of DN.
The importance of strain as a major co-factor for susceptibility to nephropathy is particularly evidenced by the observation that mice of the C57BL/6 KS strain (a strain that has been modified to contain part of the DBA/2 strain genetic endowment) that carry the db/db mutation can develop features of DN, while the identical mutation in mice of C57BL/6 strain, without the contribution of DBA/2, fail to develop significant nephropathy [5].
OVE26 mice
The OVE26 mouse is characterized by transgenic overexpression of calmodulin in pancreatic β cells leading to deficient production of insulin and Type I diabetes [15]*. This overexpression of calmodulin, in mice of FVB background, results in significant nephropathy with marked albuminuria far in excess of the standard recommended by the AMDCC. The nephropathy in these mice is characterized by increased mesangial matrix, which can be severe and extends to more global glomerulosclerosis, diminished podocyte number, and mild degrees of renal fibrosis. The diminished podocyte number, albuminuria, and mesangial matrix increase all are significant strengths for using these mice for studies of DN. A significant limitation of these mice is the requirement for the mutation to be expressed in the FVB mouse strain to obtain the desired phenotypic features. Albuminuria, mesangial matrix expansion, and fibrosis are all significantly diminished when the transgene is introduced into the C57BL/6 or DBA/2 mouse strains (despite the enhanced susceptibility of DBA/2 mice to develop DN) [15]. This particular strain dependence makes it challenging to introduce other genetic mutations from other mouse strains into this model. Although OVE26 mice on an FVB background develop mesangial sclerosing lesions of advanced diabetic nephropathy including nodular mesangial sclerosis, it has been difficult to glean from published studies how this has been quantitated and therefore how uniform and widespread these changes may be. These mice develop reduction in podocyte number and density, but most of this loss appears to develop late in the disease course (as assessed at days 60, 150, and 450) and hence follows, rather than precedes, other manifestations of DN [16]. Finally, based on the initial promise of the model, a number of laboratories have investigated its use. At least one group other than that of Epstein et al has found that these mice develop features of advanced diabetic nephropathy [17], but difficulties with the viability of these mice also have been reported [7].
Akita Mice
Akita mice carry the Ins2+/C96Y mutation, a single nucleotide substitution in the Ins2 gene. This mutation causes abnormal folding of the insulin protein, toxic injury to pancreatic β cells, and diminished capacity to secrete insulin, resulting in Type I diabetes [5,18]*. As originally described in mice of the C57BL/6 strain, diabetic mice with the Akita mutation develop modest levels of albuminuria and modest structural changes of increased mesangial matrix and basement membrane thickening. Depletion of podocytes, due at least in part to increased apoptosis, manifests early in development of DN [14]. Gurley et al. have studied the effect of breeding the Ins2+/C96Y mutation into strains more susceptible to nephropathy, i.e. DBA/2 and 129/SvEv [18]. They found that C57BL/6, DBA/2, and 129/SvEv mice with the Akita mutation develop similar degrees of hyperglycemia, but differed in their manifestation of nephropathy. DBA/2 mice had more albuminuria than the others, but only C57BL/6 and 129/SvEv strain mice demonstrated increased mesangial matrix.
These and other previously published studies show that Akita mice have potential to model some features of diabetic nephropathy, but as is the case with OVE26 mice, these features are heavily dependent on the mouse strain in which the mutation is introduced. To date, none of the published studies of Akita mice, regardless of strain, have demonstrated structural alterations that model advanced human diabetic nephropathy such as mesangiolysis or widespread marked or nodular mesangial sclerosis. A particular caveat to be considered when using these mice is the propensity for Akita mice, for unknown reasons, to develop mesangial deposits of IgA [19]. Since mesangial deposition of IgA is a major cause of mesangial proliferative glomerulopathy in humans, understanding the basis for mesangial matrix increase in Akita mice (diabetes, mesangial immune complexes, or both?) can be problematic and limit the value of studies in which alterations of mesangial volume and matrix accumulation are a key endpoint.
The number of background strains studied with the Ins2+/C96Y mutation is small. It can be expected that studies of this mutation in mice of other genetic backgrounds may result in a more robust model of Type I diabetes and nephropathy, and it is possible the confounding issue of IgA deposition may not arise when the mutation is expressed in a different mouse strain.
Podocyte Specific Insulin Receptor Knockout Mouse
An exciting recent development has been the report of a mixed mouse strain (with contributions of C57BL/6, 129/Sv, and FVB backgrounds) with podocyte specific deletion of insulin receptors using both podocin and nephrin promoters to drive cre-recombinase directed excision of the receptor [20]. These mice develop proteinuria and were reported to develop histologic features of DN despite maintaining normal insulin levels and normoglycemia. Features of DN exhibited by these mice included increased podocyte apoptosis at 8 weeks of age, detected at a time when significant proteinuria was becoming established, podocyte foot process effacement, and increased mesangial matrix. However, the published histologic lesions in these mice suggests a non-specific process of progressive glomerulosclerosis rather than development of classic features of DN. Features of mesangiolysis and/or nodular glomerulosclerosis were not characterized, nor was podocyte number. Nonetheless, this first publication showed considerable promise for this model as a tool for dissecting important podocyte specific activities that may mediate some of the structural changes of DN, and particularly identifies podocyte insulin sensitivity as an important regulatory element in the maintenance of normal glomerular structure and function.
eNOS Deficient Mice
There are currently two models of DN dependent on genetic deletion of endothelial nitric oxide synthase (eNOS−/−). eNOS provides the principal means by which nitric oxide is generated in the vasculature; NO is in turn a major regulator of vascular tone. Diminution of NO availability has been considered a major mechanism underlying development of diabetic complications involving the vasculature, including DN.
A major advance in DN model development was the introduction of eNOS deficiency into db/db mice. eNOS−/−/db/db mice develop Type II diabetes, obesity, hypertension, albuminuria, marked mesangial expansion, and mesangiolysis [11,21]. These features establish this mouse as one of the very few to develop features of advanced DN, and hence offering great potential for studying mechanisms underlying progression. However, there have been no subsequent publications on this model following its initial description; we speculate this may be attributable to difficulty in breeding mice with these combined mutations.
An alternate approach to exploit eNOS deficiency has been to use streptozotocin induced Type I diabetes in eNOS−/− mice of C57BL/6 background. Though C57BL/6 mice are resistant to development of DN, Nakagawa et al. have shown eNOS deficiency is sufficient to permit development of features of advanced DN including hypertension, albuminuria, mesangial matrix expansion, mesangiolysis and mesangial nodule formation, and renal insufficiency in these mice [22]. Like eNOS−/−/db/db mice, this model also offers a means to study mechanisms underlying advanced DN, but several important caveats need to be considered. Use of high streptozotocin doses in the initial study by Nakagawa also led to high mortality. When a lower dose streptozotocin protocol was used to evaluate the effects of diabetes in eNOS−/− C57BL/6 mice, Kanetsuna et al. found a milder phenotype with no significant renal insufficiency and only mild mesangial expansion despite the presence of mesangiolysis and albuminuria [23]. eNOS−/− mice without diabetes also have a low but baseline background of mesangiolysis and microthrombosis, complicating interpretations of whether the mesangiolysis in diabetic eNOS−/− mice models the mesangiolysis of human DN or is an exacerbation of an alternate injury process. Some support for this scenario comes both from the Kanetsuna study and from a subsequent study of this model by Nakagawa’s group which demonstrated the phenotypic features of diabetic glomerulopathy (mesangiolysis and mesangial nodule formation) were the result of hypertension and not diabetes and could be prevented, despite persistent hyperglycemia, solely by administration of hydralazine to lower blood pressure [24]*.
In spite of these limitations, the advanced renal morphologic changes encountered in a diabetic milieu when eNOS is deficient, especially in mice of susceptible genetic background, are rarely encountered in most other DN models. This makes such mice extremely promising for further model development. Perhaps when this mutation is shifted to an alternate genetic background, or a model is developed that allows regulatable and/or anatomic site-specific deletion of eNOS (e.g., in the glomerular capillaries after an extended period of diabetes), an even more robust model of advanced DN may emerge.
BTBR ob/ob mice
A mouse model of insulin resistance that develops in the BTBR (black and tan brachyuric, i.e. short tailed) mouse strain in which the ob/ob mutation has been introduced has been created by Attie et. al.[25]. In brief, the characteristics of the BTBR ob/ob mice are insulin resistance (measured as early as four weeks of age) with elevated serum insulin levels and pancreatic islet hypertrophy and subsequent sustained hyperglycemia at 350–400 mg/dl. They are largely resistant to significant lowering of blood glucose by insulin administration. The kidney phenotype of these mice includes early loss of podocytes, chronologically coinciding with onset of proteinuria, both of which are detectable by 8 weeks of age [26]**. These changes precede mesangial expansion, identifiable by 10 weeks of age. These features model morphologically early human DN, and by 18 weeks of age, these mice model progressive, advanced DN with features of marked proteinuria, more extensive mesangial expansion, mesangiolysis, persistent podocyte loss, basement membrane thickening, and modest but detectable interstitial fibrosis (Figure 1).
Figure 1.
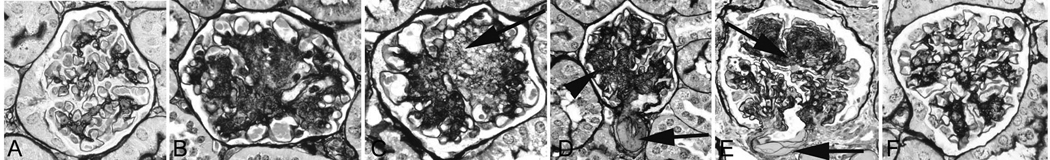
BTBR ob/ob mice show distinct mesangial expansion at 14 wks (B) compared to BTBR wild type (A). These mesangial lesions are progressive as shown at 21 wks (C, D) and result in part from mesangiolysis, with lucency and dissolution of the mesangial (C, arrow). The glomerulus in D shows diffuse and focally nodular mesangial sclerosis (arrowhead) and hyalinosis of an arteriole (D, arrow). Compare to human DN in E with characteristic hyalinosis of arteriole (arrow) and nodular sclerosis (arrowhead), and to the limited mesangial change in leptin receptor deficient db/db mice (F), currently the most widely used murine model of DN in Type II diabetes, at 22 weeks. A–F, silver methenamine stain. Modified from Hudkins KL et al. J Am Soc Nephrol. 2010; 21:1533–1542, reprinted with permission.
These mice offer a unique opportunity to study mechanisms that may lead to new therapies aimed at regression of DN, and not just its stabilization. Reversibility of morphologically advanced DN has not been previously demonstrable in other animal models or in human disease, except in the case of longstanding (10 years) pancreatic transplantation in humans. One particular advantage of BTBR ob/ob mice as a model of DN is the relatively rapid time course for development of advanced disease, which reduces the time needed to conduct interventional studies that may ameliorate advanced DN after it is already established. The mutation of leptin deficiency (versus deficiency of the leptin receptor in db/db mice), allows for studies of mechanisms underlying potential reversibility of DN by utilizing leptin replacement as a strategy. Demonstration that this potential can be realized in these mice has been recently reported (in abstract form) by Pichaiwong et al. [27], who demonstrated full reversal of the lesions of advanced DN by continuous infusion of leptin via osmotic minipump. This model may be a model for other models of DN; i.e., by setting a goal for new models that they should be suitable for studies of interventions that ameliorate or even reverse established and advanced DN, as commonly encountered in patients, and not just prevent or ameliorate DN in its earliest stages.
The BTBR ob/ob mouse, as other models, has significant limitations. Like other mice with leptin deficiency, these mice are infertile and breeding mice in sufficient numbers for interventional studies is labor intensive and expensive. The BTBR strain is not commonly studied, and so introduction of genetic mutations into these mice to test specific pathophysiologic concepts requires extensive and time consuming backcrossing strategies. However, given the unique possibilities to study reversibility of DN, and the limitations of alternative models, these mice are likely to be a worthwhile research tool for the foreseeable future.
Conclusions
There are multiple murine models, modified by strain and superimposed genetic mutations, of DN. We have considered selected currently used animal models. Some mutations that have been important for recent pathophysiologic studies of mouse models of DN, but not considered here, include genetic deletions of decorin [28], bradykin receptors [12], the receptor for advanced glycation end products (RAGE) [17], and osteopontin [29]. Mouse models of diabetes that reliably recapitulate early morphological changes of human diabetic nephropathy, principally mesangial matrix expansion and in some cases, podocyte loss, include db/db and Akita mice. There are few models that recapitulate features of morphologically early and late diabetic nephropathy; of these, mice with deficiency of eNOS, OVE26 FVB mice, and BTBR ob/ob mice appear to be the most robust. The 2009 review of the AMDCC suggested that eNOS−/− db/db mice may be the best current mouse model for recapitulating the functional and structural changes of morphologically advanced human diabetic nephropathy [7]. The recently described BTBR ob/ob mouse model rivals the eNOS−/− db/db mouse in this regard. The relative rapidity in which lesions develop in this model make it well suited for interventional studies. Despite these advances, all of the mouse models available to date possess important limitations. Designing better models of DN that will allow identification of underlying mechanisms, or testing of therapeutic interventions, must continue.
Key Points.
-
-
Phenotyping of pathologic changes in mouse models of diabetic nephropathy should be comprehensive and include, whenever possible, measures of podocyte number or density, characterization of mesangiolysis and mesangial expansion, and exclusion of concurrent immune complex deposition processes, in addition to extraglomerular alterations in the tubulointerstitium and vasculature.
-
-
There are few murine models of morphologically advanced diabetic nephropathy; currently these include OVE26 mice, diabetic eNOS deficient mice, and BTBR ob/ob mice.
-
-
BTBR ob/ob mice with Type II diabetes due to leptin deficiency offer the potential to study reversibility of nephropathy with leptin restoration.
Acknowledgments
This work was supported in part by a grant (DK 76126) from the National Institutes of Health
Abbreviations
- DN
diabetic nephropathy
- eNOS
endothelial nitric oxide synthase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, Ferrario F, Fogo AB, Haas M, de Heer E, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 2.Remuzzi G, Schieppati A, Ruggenenti P. Clinical practice. Nephropathy in patients with type 2 diabetes. N Engl J Med. 2002;346:1145–1151. doi: 10.1056/NEJMcp011773. [DOI] [PubMed] [Google Scholar]
- 3.Stout LC, Kumar S, Whorton EB. Focal mesangiolysis and the pathogenesis of the Kimmelstiel-Wilson nodule. Hum Pathol. 1993;24:77–89. doi: 10.1016/0046-8177(93)90066-p. [DOI] [PubMed] [Google Scholar]
- 4.Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int. 2008;74:22–36. doi: 10.1038/ki.2008.128. [DOI] [PubMed] [Google Scholar]
- 5.Breyer MD, Bottinger E, Brosius FC, 3rd, Coffman TM, Harris RC, Heilig CW, Sharma K Amdcc. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 6.Breyer MD, Qi Z, Tchekneva EE, Harris RC. Insight into the genetics of diabetic nephropathy through the study of mice. Curr Opin Nephrol Hypertens. 2008;17:82–86. doi: 10.1097/MNH.0b013e3282f49cc9. [DOI] [PubMed] [Google Scholar]
- 7. Brosius FC, 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. doi: 10.1681/ASN.2009070721.. An essential review of the status of available diabetic nephropathy models through 2009 . This paper from a consortium of investigators focused on diabetic complications sets forth clearly defined standards for phenotyping mouse models of diabetic nephropathy.
- 8.Sanden SK, Wiggins JE, Goyal M, Riggs LK, Wiggins RC. Evaluation of a thick and thin section method for estimation of podocyte number, glomerular volume, and glomerular volume per podocyte in rat kidney with Wilms' tumor-1 protein used as a podocyte nuclear marker. J Am Soc Nephrol. 2003;14:2484–2493. doi: 10.1097/01.asn.0000089829.45296.7c. [DOI] [PubMed] [Google Scholar]
- 9.White KE, Bilous RW. Estimation of podocyte number: a comparison of methods. Kidney Int. 2004;66:663–667. doi: 10.1111/j.1523-1755.2004.00787.x. [DOI] [PubMed] [Google Scholar]
- 10.Schlondorff D. Choosing the right mouse model for diabetic nephropathy. Kidney Int. 2010;77:749–750. doi: 10.1038/ki.2009.545. [DOI] [PubMed] [Google Scholar]
- 11.Zhao HJ, Wang S, Cheng H, Zhang MZ, Takahashi T, Fogo AB, Breyer MD, Harris RC. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17:2664–2669. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kakoki M, Sullivan KA, Backus C, Hayes JM, Oh SS, Hua K, Gasim AM, Tomita H, Grant R, Nossov SB, et al. Lack of both bradykinin B1 and B2 receptors enhances nephropathy, neuropathy, and bone mineral loss in Akita diabetic mice. Proc Natl Acad Sci U S A. 2010;107:10190–10195. doi: 10.1073/pnas.1005144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol. 2003;284:F1138–F1144. doi: 10.1152/ajprenal.00315.2002. [DOI] [PubMed] [Google Scholar]
- 14.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 15. Xu J, Huang Y, Li F, Zheng S, Epstein PN. FVB mouse genotype confers susceptibility to OVE26 diabetic albuminuria. Am J Physiol Renal Physiol. 2010;299:F487–F494. doi: 10.1152/ajprenal.00018.2010.. A recent consideration of the potential of the OVE26 mutation to induce diabetic nephropathy in mice of differing strains.
- 16.Teiken JM, Audettey JL, Laturnus DI, Zheng S, Epstein PN, Carlson EC. Podocyte loss in aging OVE26 diabetic mice. Anat Rec (Hoboken) 2008;291:114–121. doi: 10.1002/ar.20625. [DOI] [PubMed] [Google Scholar]
- 17.Reiniger N, Lau K, McCalla D, Eby B, Cheng B, Lu Y, Qu W, Quadri N, Ananthakrishnan R, Furmansky M, et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes. 2010;59:2043–2054. doi: 10.2337/db09-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gurley SB, Mach CL, Stegbauer J, Yang J, Snow KP, Hu A, Meyer TW, Coffman TM. Influence of genetic background on albuminuria and kidney injury in Ins2(+/C96Y) (Akita) mice. Am J Physiol Renal Physiol. 2010;298:F788–F795. doi: 10.1152/ajprenal.90515.2008.. The most current assessment of phenotypic features of the Akia mouse model.
- 19.Haseyama T, Fujita T, Hirasawa F, Tsukada M, Wakui H, Komatsuda A, Ohtani H, Miura AB, Imai H, Koizumi A. Complications of IgA nephropathy in a non-insulin-dependent diabetes model, the Akita mouse. Tohoku J Exp Med. 2002;198:233–244. doi: 10.1620/tjem.198.233. [DOI] [PubMed] [Google Scholar]
- 20.Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, Pons DA, Owen RJ, Satchell SC, Miles MJ, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12:329–340. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohan S, Reddick RL, Musi N, Horn DA, Yan B, Prihoda TJ, Natarajan M, Abboud-Werner SL. Diabetic eNOS knockout mice develop distinct macro- and microvascular complications. Lab Invest. 2008;88:515–528. doi: 10.1038/labinvest.2008.23. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa T, Sato W, Glushakova O, Heinig M, Clarke T, Campbell-Thompson M, Yuzawa Y, Atkinson MA, Johnson RJ, Croker B. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18:539–550. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- 23.Kanetsuna Y, Takahashi K, Nagata M, Gannon MA, Breyer MD, Harris RC, Takahashi T. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol. 2007;170:1473–1484. doi: 10.2353/ajpath.2007.060481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kosugi T, Heinig M, Nakayama T, Connor T, Yuzawa Y, Li Q, Hauswirth WW, Grant MB, Croker BP, Campbell-Thompson M, et al. Lowering blood pressure blocks mesangiolysis and mesangial nodules, but not tubulointerstitial injury, in diabetic eNOS knockout mice. Am J Pathol. 2009;174:1221–1229. doi: 10.2353/ajpath.2009.080605.. A key paper demonstrating the role of hypertension in mediating some features of diabetic glomerulopathy in an eNOS deficient model of diabetic nephropathy.
- 25.Clee SM, Attie AD. The genetic landscape of type 2 diabetes in mice. Endocr Rev. 2007;28:48–83. doi: 10.1210/er.2006-0035. [DOI] [PubMed] [Google Scholar]
- 26. Hudkins KL, Pichaiwong W, Wietecha T, Kowalewska J, Banas MC, Spencer MW, Muhlfeld A, Koelling M, Pippin JW, Shankland SJ, et al. BTBR ob/ob mutant mice model progressive diabetic nephropathy. J Am Soc Nephrol. 2010;21:1533–1542. doi: 10.1681/ASN.2009121290.. A new murine model of diabetic nephropathy noteworthy for its relatively rapid development, documentation of early podocyte loss, and robust development of morphologically advanced lesions including mesangiolysis and mesangial matrix accumulation.
- 27.Pichaiwong W, Hudkins KL, Wietecha TA, Shankland SJ, Alpers CE. Reversibility of Diabetic Nephropathy and Podocyte Loss in the BTBR ob/ob Mouse. Presented at the American Society of Nephrology Annual Meeting November 2010; Denver, CO. 2010. Nov, [Google Scholar]
- 28.Williams KJ, Qiu G, Usui HK, Dunn SR, McCue P, Bottinger E, Iozzo RV, Sharma K. Decorin deficiency enhances progressive nephropathy in diabetic mice. Am J Pathol. 2007;171:1441–1450. doi: 10.2353/ajpath.2007.070079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholas SB, Liu J, Kim J, Ren Y, Collins AR, Nguyen L, Hsueh WA. Critical role for osteopontin in diabetic nephropathy. Kidney Int. 2010;77:588–600. doi: 10.1038/ki.2009.518. [DOI] [PubMed] [Google Scholar]