

Abstract
Background
Non-negative matrix factorization (NMF) has been shown to be a powerful tool for clustering gene expression data, which are widely used to classify cancers. NMF aims to find two non-negative matrices whose product closely approximates the original matrix. Traditional NMF methods minimize either the l2 norm or the Kullback-Leibler distance between the product of the two matrices and the original matrix. Correntropy was recently shown to be an effective similarity measurement due to its stability to outliers or noise.
Results
We propose a maximum correntropy criterion (MCC)-based NMF method (NMF-MCC) for gene expression data-based cancer clustering. Instead of minimizing the l2 norm or the Kullback-Leibler distance, NMF-MCC maximizes the correntropy between the product of the two matrices and the original matrix. The optimization problem can be solved by an expectation conditional maximization algorithm.
Conclusions
Extensive experiments on six cancer benchmark sets demonstrate that the proposed method is significantly more accurate than the state-of-the-art methods in cancer clustering.
Background
Because cancer has been a leading cause of death in the world for several decades, the classification of cancers is becoming more and more important to cancer treatment and prognosis [1,2]. With advances in DNA microarray technology, it is now possible to monitor the expression levels of a large number of genes at the same time. There have been a variety of studies on analyzing DNA microarray data for cancer class discovery [3-5]. Such methods are demonstrated to outperform the traditional, morphological appearance-based cancer classification methods. In such studies, different cancer classes are discriminated by their corresponding gene expression profiles [1].
Several clustering algorithms have been used to identify groups of similar expressed genes. Non-negative matrix factorization (NMF) was recently introduced to analyze gene expression data and this method demonstrated superior performance in terms of both accuracy and stability [6-8]. Gao and Church [3] reported an effective unsupervised method for cancer clustering with gene expression profiles via sparse NMF (SNMF). Carmona et al. [9] presented a methodology that was able to cluster closely related genes and conditions in sub-portions of the data based on non-smooth non-negative matrix factorization (nsNMF), which was able to identify localized patterns in large datasets. Zheng et al. [5,7] applied penalized matrix decomposition (PMD) to extract meta-samples from gene expression data, which could captured the inherent structures of samples that belonged to the same class.
NMF approximates a given gene data matrix, X, as a product of two low-rank nonnegative matrices, H and W, as X≈HW. This is usually formulated as an optimization problem, where the objective function is to minimize either the l2 norm or the Kullback-Leibler (KL) distance [10] between X and HW. Most of the improved NMF algorithms are also based on the minimization of these two distances while adding the sparseness term [3], the graph regularization term [11], etc. Sandler and Lindenbaum [12] argued that measuring the dissimilarity of W and HW by either the l2 norm or the KL distance, even with additional bias terms, was inappropriate in computer vision applications due to the nature of errors in images. Sandler and Lindenbaum [12] proposed a novel NMF with earth mover’s distance (EMD) metric by minimizing the EMD error between X and HW. The proposed NMF-EMD algorithm demonstrated significantly improved performance in two challenging computer vision tasks, i.e., texture classification and face recognition. Liu et al. [4] tested a family of NMF algorithms using α-divergence with different α values as dissimilarities between X and HW for clustering cancer gene expression data.
It is widely acknowledged that DNA microarry data contain many types of noise, especially experimental noise. Recently, correntropy was shown to be an effective similarity measurement in information theory due to its stability to outliers or noise [13]. However, it has not been used in the analysis of microarray data. In this paper, we propose a novel form of NMF that maximizes the correntropy. We introduce a new NMF algorithm with a maximum correntropy criterion (MCC) [13] for the gene expression data-based cancer clustering problem. We call it NMF-MCC. The goal of NMF-MCC is to find a meta-sample matrix, H, and a coding matrix, W, such that the gene expression data matrix, X, is as correlative to the product of H and W as possible under MCC.
Related works
He et al. [13] recently developed a face recognition algorithm, correntropy-based sparse representation (CESR), based on MCC. CESR tries to find a group of sparse combination coefficients to maximize the correntropy between the facial image vector and the linear combination of faces in the database. He et al. [13] demonstrated that CESR was much more effective in dealing with the occlusion and corruption problems of face recognition than the state-of-the-art methods. However, CESR learns only the combination coefficients while the basis faces (the faces in the database) are fixed. Comparing to CESR, NMF-MCC can learn both the combination coefficients and the basis vectors jointly, which allows the algorithm to obtain more basis vectors for better representation of the data points. Zafeiriou and Petrou [14] addressed the problem of NMF with kernel functions instead of inner products and proposed the projected gradient kernel nonnegative matrix factorization (PGK-NMF) algorithm. Both NMF-MCC and PGK-NMF employ kernel functions to map the linear data space to a non-linear space. However, as we show later, NMF-MCC computes different kernels for different features, while PGK-NMF computes a single kernel for the whole feature vector. Thus, NMF-MCC allows the algorithm to assign different weights to different features and emphasizes the discriminant features with high weights, thus achieving feature selection. In contrast, like most kernel based methods, PGK-NMF simply replaces the inner product by the kernel-function and treats the features equally, thus there is no feature selection function.
Methods
In this section, we first briefly introduce the traditional NMF method. We then propose our novel NMF-MCC algorithm by maximizing the correntropy in NMF. We further propose a expectation conditional maximization-based approach to solve the optimization problem.
Nonnegative matrix factorization
NMF is a matrix factorization algorithm that focuses on the analysis of data matrices whose elements are nonnegative. Consider a gene expression dataset that consists of D genes in N samples. We denote it by a matrix of size D×N, and each column of X is a sample vector containing D genes. NMF aims to find two non-negative matrices, and , whose product closely approximates the original matrix X:
| (1) |
Matrix H is of size D×K, with each of the K columns defining a meta-sample and each entry, hdk, in H representing the expression level of gene d over meta-sample k. Matrix W is of size K×N, with each of the n columns representing the meta-sample expression pattern of the corresponding sample, and each entry, wkn, representing the coefficient of meta-sample k over sample n. Figure 1 shows an example of the factorization of a gene expression matrix X with D=2308 genes and N=83 samples as the product of the meta-sample matrix H with K=4 meta-samples and the coding matrix W.
Figure 1.
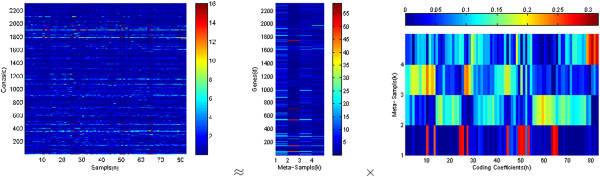
The l2 norm distance-based non-negative matrix factorization on the SRBCT dataset [29]. The gene expression data matrix, X, is factorized as the product of the meta-sample matrix, H, and the coding matrix, W.
The factorization is quantified by an objective function that minimizes some distance measure, such as:
•l2norm distance: One simple measure is the square of the l2 norm distance (also known as the Frobenius norm or the Euclidean distance) between two matrices, which is defined as:
| (2) |
•Kullback - Leibler (KL) divergence: The second one is the divergence between two matrices [10], which is defined as:
| (3) |
Maximum correntropy criterion for NMF
Another thing that has to be changed is that the definition of correntropy is not subject to the kernel being Gaussian as they seem to imply through the text, so for instance when they define they can say E(k(x-y)) and one of the common choices of k is the Gaussian kernel giving....
Correntropy is a nonlinear similarity measure between two random variables, x and y[13,15,16], defined as
| (4) |
where kσ is a kernel that satisfies the Mercer theory and E[·] is the expectation. One of the common choices of kσ is the Gaussian kernel given as .
In practice, the joint probability density function of x and y is unknown and only a finite amount of data {(xi,yi)},i=1,⋯,I is available. Therefore, the sample correntropy is estimated by
| (5) |
Based on Eq. (5), a general similarity measurement between any two discrete gene expression vectors was proposed [17]. They introduced the correntropy induced metric (CIM) for any two gene sample vectors x=[x1,⋯,xD]⊤ and y=[y1,⋯,yD]⊤, as:
| (6) |
where ed=xd−yd is defined as the error. For adaptive systems, we can define the maximum correntropy criterion (MCC) [18] as
| (7) |
where Θ is a parameter to be specified later. We must notice the difference between MCC and common kernel criterion used in [14]. The Gaussian kernel function of vectors x and y is defined as
| (8) |
We can see that the kernel is applied to the entire feature vector, x, and each feature xd,d=1⋯,D is treated equally with the same kernel parameter. However, in (7), kernel functions are applied to different functions. This can allow the algorithm to learn different kernel parameters as we will introduce later. In this way, we can assign different weights to different features and thus implement feature selection.
Our goal is to find a meta-sample matrix, H, and a coding matrix, W, such that HW is as correlative to X as possible under MCC as described in Eq. (7). To extend MCC from vector space RD to matrix space RD×N, we replace ed=(xd−yd) with the l2 norm distance between the samples of X and Y=HW as , where ydn is the (d,n)-th item of Y, and . Moreover, the factorization system parameter should be set to Θ=(H,W) under the framework of NMF-MCC. By substituting newly defined ed and Θ to (7), we can formulate the problem of NMF-MCC as the following optimization problem:
| (9) |
We should notice the significant difference between NMF-MCC and CESR. As a supervised learning algorithm, the CESR represents a test data point, xt, as a linear combination of all the the training data points as and wt=[w1t,⋯,wNt]⊤ is the combination coefficient vector. CESR aims to find the optimal wt to maximize the correntropy between xt and Xwt. Similarly, NMF-MCC also tries to represent a data point xn as a linear combination of some basis vectors as and wn=[w1n,⋯,wKn]⊤ is the combination coefficient vector. Differently from CESR, NMF-MCC aims to find not only the optimal wn but also the basis vectors in H to maximize the correntropy between xn and Hwn, n=1,⋯,N. The internal difference between NMF-MCC and CESR lies in whether to learn basis vectors or not.
In order to solve the optimization problem, we recognize that the expectation conditional maximization (ECM) method [19] can be applied. Based on the theory of convex conjugate functions [20], we can derive the following proposition that forms the basis to solve the optimization problem in (9):
Proposition 1
There exists a convex conjugate function of g(z,σ) such that
| (10) |
and for a fixed z, the supremum is reached at ϱ=−g(z,σ).
By substituting Eq. (10) into (9), we have the augmented objective function in an enlarged parameter space
| (11) |
where superscript φ is the convex conjugate function φ of g(z) defined in Proposition 1, and ρ=[ρ1,⋯,ρD]⊤ are the auxiliary variables.
According to Proposition 1, for fixed H and W, the following equation holds:
| (12) |
It follows that
| (13) |
That is, maximizing F(H,W) is equivalent to maximizing the augmented function .
The NMF-MCC Algorithm
The traditional NMF can be solved by the expectation-maximization (EM) algorithm [21]. However, in the case of MCC-based NMF, EM must be replaced by ECM because there is more than one parameter. Figure 2 shows the outline of ECM, which is described in more detail below.
Figure 2.
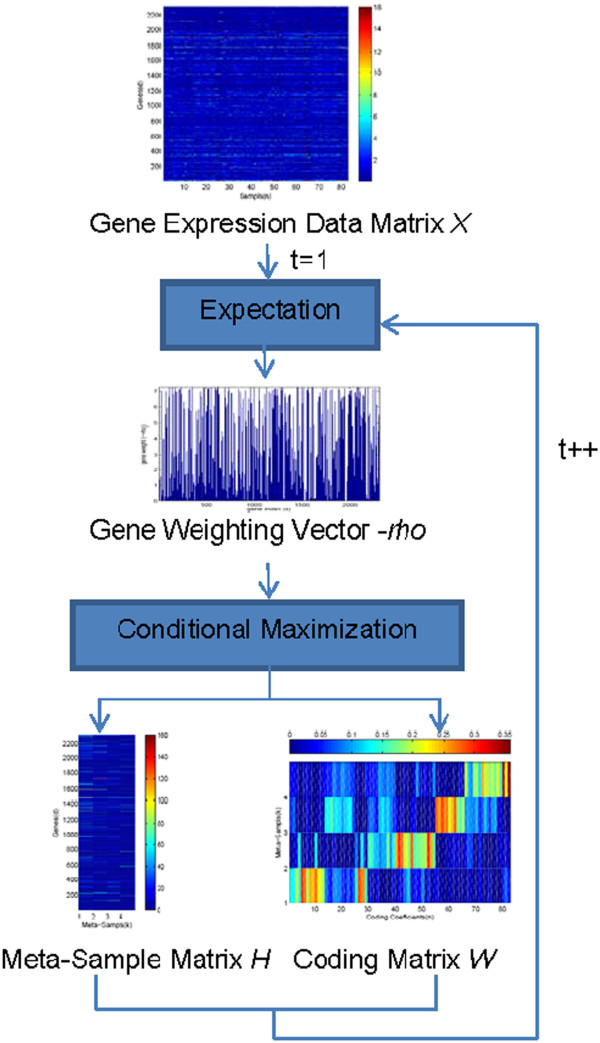
Outline of the ECM-based NMF-MCC algorithm.
1. E-Step: Compute ρ given the current estimations of the meta-sample matrix H and the coding matrix W as:
| (14) |
where t means the t-th iteration. In this study, the kernel size (bandwidth) σ2t is computed by
| (15) |
where Θ is a parameter to control the sparseness of .
2. CM-steps: In the CM-step, given , we try to optimize the following function respect to H and W:
| (16) |
where diag(·) is an operator that converts the vector ρ to a diagonal matrix.
By introducing a dual objective function,
| (17) |
the optimal problem in (16) can be reformulated as the following dual problem:
| (18) |
Let ϕdk and ψkn be the Lagrange multiplier for constraints hdk≥0 and wkn≥0, respectively, and Φ=[ϕdk] and Ψ=[ψkn]. The Lagrange is
| (19) |
The partial derivatives of with respect to H and W are
| (20) |
and
| (21) |
Using the Karush-Kuhn-Tucker optimal conditions, i.e., ϕdkhdk=0 and ψknwkn=0, we get the following equations for hdk and wkn:
| (22) |
and
| (23) |
These equations lead to the following updating rules to maximize the expectation in (13).
••The meta-sample matrix H, conditioned on the coding matrix W:
| (24) |
••The coding matrix W conditioned on the newly estimated meta-sample matrix Ht+1:
| (25) |
We should note that if we exchange the numerator and denominator in (24) and (25), new update formulas will be yield. The new update rules are dual for (24) and (25), and our experimental results show that the dual update rules achieve similar clustering performances as (24) and (25).
Algorithm 1 summarizes the optimization procedure.
Algorithm 1 NMF-MCC Algorithm
Proof of convergence
In this section, we will prove that the objective function in (16) is nonincreasing under the updating rules in (24) and (25).
Theorem 1
The objective function in (16) is nonincreasing under the update rules (24) and (25).
To prove the above theorem, we first define an auxiliary function.
Definition 1
G(w,w′) is an auxiliary function for F(w) if the conditions
| (26) |
are satisfied.
The auxiliary function is quite useful because of the following lemma:
Lemma 1
If G is an auxiliary function of F, then F is nonincreasing under the update
| (27) |
We refer the readers to [22] for the proof of this lemma. Now, we show that the updating rule of (25) is exactly the update in (27) with a proper auxiliary function. We denote the objective function in (16) as O:
| (28) |
Considering any element, wkn, in W, we use Fkn to denote the part of the objective function in (16) that is relevant only to wkn. It is easy to check that
| (29) |
Since the updating rule is essentially based on elements, it is sufficient to show that each Fkn is nonincreasing under the update step of (25).
Lemma 2
Function
| (30) |
is an auxiliary function for Fkn, which is relevant only to wkn.
Proof
Since G(w,w)=Fkn(w) is obvious, we only need to show that . To do this, we compare the Taylor series expansion of Fkn(w),
| (31) |
with (30) to find that is equivalent to
| (32) |
We have
| (33) |
Thus, (32) holds and . □
We can now demonstrate the convergence of Theorem 1.
Proof of Theorem 1
Replacing G(w,wt) in (27) by (30) results in the update rule
| (34) |
Since (30) is an auxiliary function, Fkn is nonincreasing under this update rule as in (25).
Similarly, we can also show that O is nonincreasing under the updating steps in (24).
Experiments
Datasets
To test the proposed algorithm, we carry out extensive experiments on six cancer-related gene expression datasets. The six datasets consist of five multi-class sets as used in [4,23] and one binary class set [24]. The descriptions of the six datasets are summarized in Table 1. In these datasets, besides the gene expression data samples, the labels are also given. They were obtained from the diagnosis results and reported in different studies [23].
Table 1.
Summary of the six cancer gene expression datasets used to test the NMF-MCC algorithm
| Dataset name | Diagnostic task | Samples ( N ) | Genes ( D ) | Cancer Classes ( K ) | Ref |
|---|---|---|---|---|---|
| Leukemia |
Acute myelogenous leukemia |
72 |
5327 |
3 |
[25] |
| Brain Tumor |
5 human brain tumor types |
90 |
5920 |
5 |
[26] |
| Lung Cancer |
4 lung cancer types and normal tissues |
203 |
12600 |
5 |
[27] |
| 9 Tumors |
9 various human tumor types |
60 |
5726 |
9 |
[28] |
| SRBCT |
Small, round blue cell tumors |
83 |
2308 |
4 |
[29] |
| DLBCL | Diffuse large B-cell lymphomas | 77 | 5469 | 2 | [24] |
Performance metric
The proposed NMF-MCC algorithm will be used to represent gene expression data for k-means clustering. The clustering results are evaluated by comparing the obtained label of each sample with the label provided by the dataset. The clustering accuracy is used to measure the clustering performance. Given a micro-array dataset containing N samples that belong to K classes, we assume that K is given in all the algorithms tested here. For each sample, xn, let cn be the cluster label predicted by an algorithm and rn be the cancer type label provided by the dataset. The accuracy of the algorithm is defined as:
| (35) |
where I(A,B) returns 1 if A=B and 0 otherwise.
Tested methods
We first compared the MCC with other loss functions between X and HW for the NMF algorithm on the cancer clustering problem, including l2 norm distance, KL distance [10], α-divergence [4], and earth mover’s distance (EMC) [12]. We further compared the proposed NMF-MCC algorithm with other NMF-based algorithms, including the penalized matrix decomposition (PMD) algorithm [7], the original NMF algorithm [22], the sparse non-negative matrix factorization (SNMF) algorithm [3], the non-smooth non-negative matrix factorization (nsNMF) algorithm [9] and the projected gradient kernel nonnegative matrix factorization (PGK-NMF).
Results
Since the initial H and W are selected randomly, we performed 100 independent trials and computed the average and the standard deviations of the accuracy for each loss function. The results from the comparison of MCC with other loss functions are presented in Figure 3. As shown in Figure 3, MCC consistently performed the best on all the six datasets. The other loss functions performed well on some datasets, but poorly on the others. It seems that the improvement of MCC increased when the number of genes increased. The standard deviation on the accuracy of MCC was much smaller than the standard deviation on the other loss functions, indicating that MCC is the most stable. On the other hand, EMD, although worked quite well in computer vision tasks [12], it did not perform well on gene expression data due to the significant difference between the image data and the gene expression data.
Figure 3.

The boxplots of the clustering accuracies for NMF with different loss functions over 100 runs on the six gene expression datasets: (a) Leukemia, (b) Brain Tumor, (c) Lung Cancer, (d) 9 Tumors, (e) SRBCT, (f) DLBCL.
The results of the comparison of NMF-MCC with other related NMF methods are presented in Figure 4. Figure 4 shows the performance of different algorithms on the six datasets. The NMF-MCC algorithm outperformed the other algorithms on five out of the six datasets. The NMF-MCC algorithm could correctly cluster more than 88% and 78% of the samples in the Leukemia and DLBCL datasets, respectively, in a completely unsupervised manner. In contrast, the l2 norm distance-based NMF algorithm performed even worse than the baseline PMD algorithm on the Leukemia and DLBCL datasets, i.e., an average accuracy of 73% and 67%, respectively. This verifies that correntropy is a much better measure of cancer clustering data. Note that NMF-MCC significantly outperformed the other algorithms on the Lung Cancer dataset, which contains a large number of genes. This implies that among the large number of genes, only a small fraction is likely to be relevant to cancerous tumor growth or spread. In NMF-MCC, the auxiliary variables −ρ acts as the feature selectors, we was able to select the relevant genes. Although the SNMF and nsNMF algorithms also improved on the performance of the baseline NMF algorithm, the improvement was much less than that of the NMF-MCC algorithm. A possible reason is that many genes exhibit similar patterns across all of the samples with only a few genes differentiating different cancer classes. They are likely to be sampled from a nonlinear manifold. Hence, the loss function defined by a linear kernel with either the l2 norm or the KL distance could not capture them. In contrast, the NMF-MCC algorithm had a loss function that was defined by the correntropy and a Gaussian kernel, which could capture the nonlinear manifold structure much more effectively. By mapping the gene expression data into the nonlinear dataspace by a Gaussian kernel, the PGK-NMF outperformed the original NMF. However, our NMF-MCC could even further improve the PGK-NMF by applying different kernels to different features.
Figure 4.

The boxplots of the clustering accuracies for different versions of NMF algorithms over 100 runs on the six gene expression datasets: (a) Leukemia, (b) Brain Tumor, (c) Lung Cancer, (d) 9 Tumors, (e) SRBCT, (f) DLBCL.
To understand what genes were selected by the NMF-MCC algorithm, we drew the gene weight figure on the SRBCT dataset (Figure 5). It can be seen that the −ρ vector is sparse, which shows the significance of certain genes. The resulting meta-sample matrix weighted by −ρ with the corresponding coding matrix is shown in Figure 6. By comparing to the coding matrix learned by the original NMF with the l2 norm distance in Figure 1, we determine that the coding matrix learned by the NMF-MCC algorithm is much more discriminative among different cancer classes. On this dataset, the NMR-MCC algorithm achieved an average clustering accuracy of 63%.
Figure 5.

The gene weight vector learned by NMF-MCC with −ρ on the SRBCT dataset.
Figure 6.
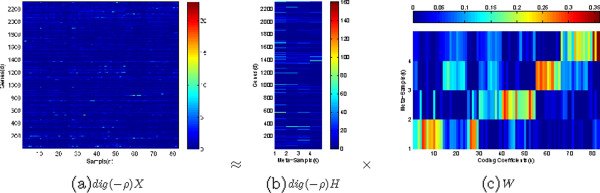
The meta-sample matrix, H, weighted by dig(−ρ) and the corresponding coding matrix,W, obtained from the NMF-MCC algorithm for the SRBCT dataset.
Discussion
Traditional unsupervised learning techniques select features with features selection algorithms and then do clustering using the selected features. The NMF-MCC algorithm proposed here achieves both goals simultaneously. The learned gene weight vector reflects the importance of the genes in the gene clustering task, and the coding matrix encodes the clustering results for the samples.
Our experimental results demonstrate that the improvement of NMR-MCC over the other methods increases when the number of genes increases. This shows the ability of the proposed algorithm to effectively select the important genes and cluster samples. This is an important property because high-dimensional data analysis has become increasingly frequent and important in diverse fields of sciences and engineering, and social sciences, ranging from genomics and health sciences to economics, finance and machine learning. For instance, in genome-wide association studies, hundreds of thousands of SNPs are potential covariates for phenotypes such as cholesterol level or height. The large number of features presents an intrinsic challenge to many classical problems, where usual low-dimensional methods no longer apply. The NMF-MCC algorithm has been demonstrated to work well on the datasets with small numbers of samples but large numbers of features. It can therefor provide a powerful tool to study high-dimensional problems, such as genome-wide association studies.
Conclusion
We have proposed a novel NMF-MCC algorithm for gene expression data-based cancer clustering. Experiments demonstrate that correntropy is a better measure than the traditional l2 norm and KL distances for this task, and the proposed algorithm significantly outperforms the existing methods.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JW designed and implemented the algorithm, conducted the experiments, performed data analysis and drafted the manuscript. XW revised the manuscript. XG supervised the study and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Jim Jing-Yan Wang, Email: jimjywang@gmail.com.
Xiaolei Wang, Email: xiaolei.wang@kaust.edu.sa.
Xin Gao, Email: xin.gao@kaust.edu.sa.
Acknowledgements
The study was supported by a grant from King Abdullah University of Science and Technology, Saudi Arabia. We would like to thank Dr. Ran He for the discussion about the maximum correntropy criterion at ICPR 2012 conference.
References
- Shi F, Leckie C, MacIntyre G, Haviv I, Boussioutas A, Kowalczyk A. A bi-ordering approach to linking gene expression with clinical annotations in gastric cancer. BMC Bioinformatics. 2010;11:477. doi: 10.1186/1471-2105-11-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souto MCP, Costa IG, de Araujo DSA, Ludermir TB, Schliep A. Clustering cancer gene expression data: a comparative study. BMC Bioinformatics. 2008;9:497. doi: 10.1186/1471-2105-9-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Church G. Improving molecular cancer class discovery through sparse non-negative matrix factorization. Bioinformatics. 2005;21(21):3970—3975. doi: 10.1093/bioinformatics/bti653. [DOI] [PubMed] [Google Scholar]
- Liu W, Yuan K, Ye D. On alpha-divergence based nonnegative matrix factorization for clustering cancer gene expression data. Artif Intell Med. 2008;44(1):1–5. doi: 10.1016/j.artmed.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Zheng CH, Ng TY, Zhang L, Shiu CK, Wang HQ. Tumor classification based on non-negative matrix factorization using gene expression data. IEEE Trans Nanobioscience. 2011;10(2):86–93. doi: 10.1109/TNB.2011.2144998. [DOI] [PubMed] [Google Scholar]
- Kim MH, Seo HJ, Joung JG, Kim JH. Comprehensive evaluation of matrix factorization methods for the analysis of DNA microarray gene expression data. BMC Bioinformatics. 2011;12(Suppl 13):S8. doi: 10.1186/1471-2105-12-S13-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng CH, Zhang L, Ng VTY, Shiu SCK, Huang DS. Molecular pattern discovery based on penalized matrix decomposition. IEEE/ACM Trans Comput Biol Bioinformcs. 2011;8(6):1592–1603. doi: 10.1109/TCBB.2011.79. [DOI] [PubMed] [Google Scholar]
- Tjioe E Berry M Homayouni R Heinrich K Using a literature-based NMF model for discovering gene functional relationships BMC Bioinformatics 200897P119732469 [Google Scholar]
- Carmona-Saez P, Pascual-Marqui R, Tirado F, Carazo J, Pascual-Montano A. Biclustering of gene expression data by non-smooth non-negative matrix factorization. BMC Bioinformatics. 2006;7:78. doi: 10.1186/1471-2105-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan R, Plastino A. Deformed statistics Kullback-Leibler divergence minimization within a scaled Bregman framework. Phys Lett A. 2011;375(48):4237–4243. doi: 10.1016/j.physleta.2011.09.021. [DOI] [Google Scholar]
- Cai D, He X, Han J, Huang TS. Graph regularized nonnegative matrix factorization for data representation. IEEE Trans Pattern Anal Mach Intell. 2011;33(8):1548–1560. doi: 10.1109/TPAMI.2010.231. [DOI] [PubMed] [Google Scholar]
- Sandler R, Lindenbaum M. Nonnegative matrix factorization with earth mover’s distance metric for image analysis. IEEE Trans Pattern Anal Mach Intell. 2011;33(8):1590–1602. doi: 10.1109/TPAMI.2011.18. [DOI] [PubMed] [Google Scholar]
- He R, Zheng WS, Hu BG. Maximum correntropy criterion for robust face recognition. IEEE Trans Pattern Anal Mach Intell. 2011;33(8):1561–1576. doi: 10.1109/TPAMI.2010.220. [DOI] [PubMed] [Google Scholar]
- Zafeiriou S, Petrou M. CVPR: 2009 IEEE Conference on Computer Vision and Pattern Recognition, Vols 1-4. Miami: IEEE Conference on Computer Vision and Pattern Recognition; 2010. Nonlinear nonnegative component analysis; pp. 2852–2857. [Google Scholar]
- Yan H, Yuan X, Yan S, Yang J. Correntropy based feature selection using binary projection. Pattern Recognit. 2011;44(12):2834–2842. doi: 10.1016/j.patcog.2011.04.014. [DOI] [Google Scholar]
- He R, Hu BG, Zheng WS, Kong XW. Robust principal component analysis based on maximum correntropy criterion. IEEE Trans Image Process. 2011;20(6):1485–1494. doi: 10.1109/TIP.2010.2103949. [DOI] [PubMed] [Google Scholar]
- Chalasani R, Principe JC. Proceedings of the 2010 International Joint Conference on Neural Networks (IJCNN2010) Barcelona, Spain: ; 2010. Self organizing maps with the correntropy induced metric; pp. 1–6. [Google Scholar]
- Liu W, Pokharel PP, Principe JC. Correntropy: properties and applications in non-gaussian signal processing. IEEE Trans Signal Process. 2007;55(11):5286–5298. [Google Scholar]
- Horaud R, Forbes F, Yguel M, Dewaele G, Zhang J. Rigid and articulated point registration with expectation conditional maximization. IEEE Trans Pattern Anal Mach Intell. 2011;33(3):587–602. doi: 10.1109/TPAMI.2010.94. [DOI] [PubMed] [Google Scholar]
- BEER G. Conjugate convex-functions and the epi-distance topology. Proc Am Math Soc. 1990;108(1):117–126. doi: 10.1090/S0002-9939-1990-0982400-8. [DOI] [Google Scholar]
- Qi Y, Ye P, Bader J. Genetic interaction motif finding by expectation maximization - a novel statistical model for inferring gene modules from synthetic lethality. BMC Bioinformatics. 2005;6:288. doi: 10.1186/1471-2105-6-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DD, Seung HS. Algorithms for non-negative matrix factorization. Adv Neural Inf Process Syst. 2001;13:556–562. [Google Scholar]
- Statnikov A, Aliferis C, Tsamardinos I, Hardin D, Levy S. A comprehensive evaluation of multicategory classification methods for microarray gene expression cancer diagnosis. Bioinformatics. 2005;21(5):631–643. doi: 10.1093/bioinformatics/bti033. [DOI] [PubMed] [Google Scholar]
- Shipp M, Ross K, Tamayo P, Weng A, Kutok J, Aguiar R, Gaasenbeek M, Angelo M, Reich M, Pinkus G, Ray T, Koval M, Last K, Norton A, Lister T, Mesirov J, Neuberg D, Lander E, Aster J, Golub T. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8(1):68–74. doi: 10.1038/nm0102-68. [DOI] [PubMed] [Google Scholar]
- Golub T, Slonim D, Tamayo P, Huard C, Gaasenbeek M, Mesirov J, Coller H, Loh M, Downing J, Caligiuri M, Bloomfield C, Lander E. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Pomeroy S, Tamayo P, Gaasenbeek M, Sturla L, Angelo M, McLaughlin M, Kim J, Goumnerova L, Black P, Lau C, Allen J, Zagzag D, Olson J, Curran T, Wetmore C, Biegel J, Poggio T, Mukherjee S, Rifkin R, Califano A, Stolovitzky G, Louis D, Mesirov J, Lander E, Golub T. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415(6870):436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Richards W, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark E, Lander E, Wong W, Johnson B, Golub T, Sugarbaker D, Meyerson M. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci. 2001;98(24):13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staunton J, Slonim D, Coller H, Tamayo P, Angelo M, Park J, Scherf U, Lee J, Reinhold W, Weinstein J, Mesirov J, Lander E, Golub T. Chemosensitivity prediction by transcriptional profiling. Proc Natl Acad Sci. 2001;98(19):10787–10792. doi: 10.1073/pnas.191368598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Wei J, Ringner M, Saal L, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu C, Peterson C, Meltzer P. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7(6):673–679. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]