

Abstract
Systemic administration of sulfated cholecystokinin-8 (CCK) activates neurons within the hindbrain nucleus of the solitary tract (NTS) that project directly to the paraventricular nucleus of the hypothalamus (PVN), and these projections underlie the ability of exogenous CCK to activate the hypothalamic-pituitary-adrenal (HPA) stress axis. CCK inhibits food intake, increases NTS neuronal cFos expression, and activates the HPA axis in a dose-dependent manner. While the hypophagic effects of exogenous CCK are attenuated in food-deprived rats, CCK dose-response relationships for NTS and hypothalamic activation in fed and fasted rats are unknown. Within the NTS, noradrenergic A2 and glucagon-like peptide-1 (GLP-1) neurons express cFos after high doses of CCK, and both neuronal populations project directly to the medial parvocellular (mp) PVN. We hypothesized that increasing and correlated proportions of A2, GLP-1, and mpPVN neurons would express cFos in rats after increasing doses of CCK, and that food deprivation would attenuate both hindbrain and hypothalamic neural activation. To test these hypotheses, ad libitum-fed (ad lib) and overnight food-deprived (DEP) rats were anesthetized and perfused with fixative 90 minutes after i.p. injection of 1.0 ml saline vehicle containing CCK at doses of 0, 3, or 10 μg/kg BW. Additional ad lib and DEP rats served as non-handled (NH) controls. Brain tissue sections were processed for dual immunocytochemical localization of cFos and dopamine-β-hydroxylase to identify A2 neurons, or cFos and GLP-1. Compared to negligible A2 cFos activation in NH control rats, i.p. vehicle and CCK dose-dependently increased A2 activation, and this was significantly attenuated by DEP. DEP also attenuated mpPVN cFos expression across all treatment groups, and A2 activation was strongly correlated with mpPVN activation in both ad lib and DEP rats. In ad lib rats, large and similar numbers of GLP-1 neurons expressed cFos across all i.p. treatment groups, regardless of CCK dose. Surprisingly, DEP nearly abolished baseline GLP-1 cFos expression in NH controls, and also in rats after i.p. injection of vehicle or CCK. We conclude that CCK-induced hypothalamic cFos activation is strongly associated with A2 activation, whereas the relationship between mpPVN and GLP-1 activation is less clear. Furthermore, activation of A2, GLP-1, and mpPVN neurons is significantly modulated by feeding status, suggesting a mechanism through which food intake and metabolic state might impact hypothalamic neuroendocrine responses to homeostatic challenge.
Keywords: CCK, Nucleus of the solitary tract, Paraventricular nucleus of the hypothalamus, Hypothalamic-pituitary-adrenal axis, cFos
1. Introduction
It is well established that moderate to high systemic doses (i.e., > 3 μg/kg BW) of cholecystokinin-8 (CCK) suppress food intake [1, 2] and activate hypothalamic endocrine neurons [3–6]. These behavioral and endocrine responses are critically dependent on CCK-A receptor-mediated activation of vagal sensory neurons that provide glutamatergic axonal input to the caudal nucleus of the solitary tract (NTS) [7–19]. While neural circuits contained within the brainstem are sufficient for the hypophagic effects of CCK [20], the ability of CCK to recruit hypothalamic endocrine neurons requires ascending projections from the NTS to the hypothalamus [21].
A large variety of neurotransmitter molecules are expressed by neurons within the caudal NTS [22]. Subsets of these NTS neurons directly modulate the neuroendocrine hypothalamic-pituitary-adrenal (HPA) axis via axonal projections to the medial parvocellular paraventricular nucleus of the hypothalamus (mpPVN) [22, 23]. Most of these projections arise from two phenotypically distinct populations of NTS neurons: noradrenergic (NA) neurons of the A2 cell group, and glucagon-like peptide-1 (GLP-1) neurons [24–29]. A2 and GLP-1 neurons densely innervate the mpPVN [29–32], synapsing directly onto corticotropin-releasing hormone (CRH)-positive neurons at the apex of the HPA axis [33, 34]. Furthermore, intraventricular and local parenchymal injections of either GLP-1 or norepinephrine (NE) induce cFos expression within the mpPVN and increase plasma levels of corticosterone (CORT) [26, 28, 35]. Accumulated evidence suggests that CCK-induced activation of A2 and GLP-1 neurons underlies the ability of CCK to activate the HPA axis and generate other neuroendocrine responses [5, 30, 36–39]. Several published studies have reported dose-response relationships for the ability of systemic CCK to inhibit food intake [1, 40], increase NTS cFos expression [8, 17, 36], and activate the HPA axis [5, 7]. However, none of these reports included phenotypic identification of the NTS neurons activated by CCK, and none has assessed the degree to which NTS cFos expression is associated with cFos activation within the mpPVN.
Interestingly, food deprivation attenuates the ability of systemic CCK to suppress food intake [41, 42], perhaps due to reduced circulating levels of leptin in food-deprived rats [43–45]. Leptin potentiates the excitatory effects of exogenous CCK on vagal sensory afferents [46, 47] and NTS neurons [48–50], including A2 neurons [51], and the ability of food deprivation to attenuate CCK-induced hypophagia is abolished by systemic leptin administration [41]. While it is clear that food deprivation reduces the hypophagic potency of CCK, the ability of CCK to activate phenotypically-identified A2 and GLP-1 neurons in rats after food deprivation has not been assessed, and there are no published reports regarding the effect of food deprivation on CCK-induced activation of the HPA axis. Considering this, the present study was designed to test three hypotheses: first, that increasing doses of CCK activate increasing numbers of A2, GLP-1, and mpPVN neurons to express cFos; second, that A2, GLP-1, and mpPVN neuronal activation occurs in a correlated manner; and third, that food deprivation significantly attenuates the ability of CCK to increase cFos expression within these populations of hindbrain and hypothalamic neurons.
2. Materials and methods
2.1. Animals
Adult male Sprague-Dawley rats (Harlan, IN; 225–275g BW; n = 51) were housed singly in hanging stainless steel wire mesh cages in a temperature-controlled room (20–22°C) with a 12/12 hr light/dark cycle (lights on at 0700 hr). Rats had ad libitum access to pelleted chow (Purina 5001) and water, except as noted. All experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
2.2. Injections and perfusion
Rats were weighed one day before CCK administration to determine proper dosage. Rats were returned to their home cage with ad libitum chow access (ad lib; n = 28), or were deprived of food (but not water) for 16–18 hours overnight in their home cage (DEP; n = 23) before CCK or vehicle treatment. On the day of the experiment rats from each group were removed from their home cage between 0830 and 1030 hr and injected intraperitoneally (i.p.) with 1.0 ml of sterile 0.15 M NaCl containing sulfated CCK-8 (Bachem; H-2080) as follows: 0 μg/kg BW (n = 9 ad lib; n = 9 DEP); 3 μg/kg BW (n = 7 ad lib; n = 4 DEP); 10 μg/kg BW (n = 7 ad lib; n = 4 DEP). These CCK doses significantly inhibit food intake in rats [21, 41, 52], stimulate pituitary hormone secretion [7, 52], and activate cFos expression by CRH-positive and other PVN neurons [5, 53, 54]. CCK was dissolved in vehicle just before injection, and rats were returned to their home cage immediately after injection. Additional nonhandled (NH) control rats (n = 5 ad lib; n = 6 DEP) received no i.p. injection and remained undisturbed in their home cages until perfusion within the same time period. Ninety minutes after i.p. injection, rats were deeply anesthetized with pentobarbital sodium (39 mg/1.0 ml i.p., Fatal Plus Solution; Butler Schein) and perfused transcardially with a brief saline rinse followed by fixative (100 ml of 2% paraformaldehyde and 1.5% acrolein in 0.1M phosphate buffer, followed by 100 ml of 2% paraformaldehyde alone) [55]. Brains were post-fixed in situ overnight at 4°C, then removed from the skull and cryoprotected for 24–48 hr in 20% sucrose. Brains were blocked and sectioned coronally (35 μm) on a freezing stage microtome. Sections were collected in six serial sets, and stored at −20°C in cryopreservant solution [56] until immunocytochemical processing.
2.3. Immunohistochemistry
Primary and secondary antisera were diluted in 0.1 M phosphate buffer containing 0.3% Triton X-100 and 1% normal donkey serum. One set of tissue sections from each rat was incubated in a rabbit polyclonal antiserum against cFos (1:50,000; kindly provided by Dr. Philip Larsen, Denmark), followed by biotinylated donkey anti-rabbit IgG (1:500; Jackson ImmunoResearch). Sections were treated with Elite Vectastain ABC reagents (Vector) and reacted with diaminobenzidine (DAB) intensified with nickel sulfate to produce a blue-black nuclear cFos reaction product. To identify NA neurons of the A2 cell group, cFos-labeled sections were subsequently processed to localize dopamine-β-hydroxylase (DβH) using mouse anti-DβH (1:50,000; Millipore, MAB308) followed by biotinylated donkey anti-mouse IgG (1:500; Jackson ImmunoResearch). Sections were reacted using Elite Vectastain ABC reagents and plain DAB to produce a brown cytoplasmic reaction product.
A second set of sections from each rat was incubated in a rabbit polyclonal antiserum against GLP-1 (1:10,000; Bachem, T-4363), followed by biotinylated donkey anti-rabbit IgG (1:500; Jackson ImmunoResearch), Elite Vectastain ABC reagents, and plain DAB to produce a brown cytoplasmic reaction product. These sections then were processed for immunofluorescent localization of cFos using a higher primary antibody concentration (1:5,000) followed by incubation in Cy3-conjugated donkey anti-rabbit IgG (1:300, Jackson ImmunoResearch) to produce a red fluorescent nuclear signal.
2.4. Quantification of cFos expression by NTS A2 neurons
Hindbrain tissue sections were analyzed using a light microscope and a 40X objective to determine the number of double-labeled, DβH/cFos-positive neurons within the NTS. Double-labeled neurons were counted bilaterally through the rostrocaudal extent of the A2 cell group (i.e., from the cervical spinal cord through the NTS just rostral to the area postrema; ~15.46 mm to 13.15 mm caudal to bregma), and then averaged as counts per section. Criteria for counting a neuron as double-labeled included brown DβH cytoplasmic labeling and a nucleus that contained visible blue-black cFos immunolabeling, regardless of intensity.
2.5. Quantification of cFos expression by GLP-1 neurons
As there is no current evidence that GLP-1 neurons residing in the lateral reticular formation are anatomically or functionally distinct from their counterparts residing in the NTS, all visible GLP-1 neurons were imaged in the NTS and adjacent reticular formation using a 20X objective on an Olympus microscope equipped for brightfield and epifluorescence, and photographed using a digital camera (Hamamatsu Photonics, Hamamatsu, Japan). Neurons were counted in photographic images using Adobe Photoshop CS4 image software. Criteria for counting a neuron as GLP-1-positive included brown cytoplasmic labeling and a visible nucleus. Neurons were considered cFos positive if their nucleus contained red fluorescent cFos immunolabeling, regardless of intensity. Counts of GLP-1 neurons were performed throughout the rostrocaudal extent of the GLP-1 cell group (i.e., from the cervical spinal cord through the NTS just rostral to the area postrema; ~15.46 mm to 13.15 mm caudal to bregma), and then averaged as counts per section.
2.6. Quantification of cFos expression in the mpPVN
cFos/GLP-1-labeled sections were viewed on the Olympus photomicroscope described above. Using a 10X objective, photographic images were captured from a single selected rostrocaudal level of the mpPVN (approximately 1.78 mm caudal to bregma). This selected level was characterized by dense GLP-1 terminal labeling that clearly defined the boundaries of the mpPVN (see Figure 1). cFos-positive neurons within this defined region of the mpPVN were counted bilaterally on images using Adobe Photoshop CS4 image software. The criterion for counting a neuron as cFos-positive was the presence of visible red fluorescent nuclear immunolabeling, regardless of intensity.
Figure 1.
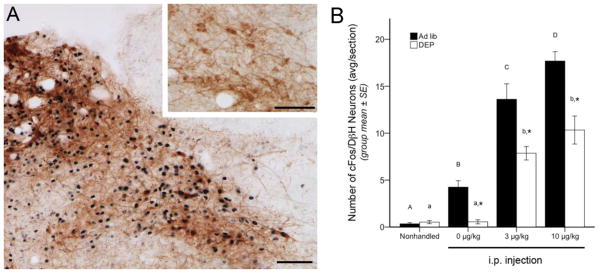
Representative color images and summary data for neuronal cFos expression (black nuclear label) within the A2 region of the caudal NTS. A, NTS cFos activation at the rostrocaudal level of the area postrema, approximately 14.16 mm caudal to bregma. In the larger image, robust cFos expression is present in a rat after CCK (10 μg/kg BW), including activation of DβH-positive (brown) neurons comprising the A2 cell group. Inset, little or no cFos activation is present in an ad lib-fed rat after i.p. vehicle (0 μg/kg BW CCK). B, bar graph illustrating the number of double labeled (i.e., both cFos- and DβH-positive) NTS neurons in ad lib-fed rats (solid bars) or food-deprived rats (DEP; open bars) after no i.p. injection (nonhandled) or after injection of CCK at doses of 0 (vehicle), 3, or 10 μg/kg BW. See Table 1 for two-way ANOVA results. In both feeding status groups, CCK dose-dependently increased cFos expression in DβH-positive A2 neurons, but cFos activation was significantly attenuated in food-deprived rats after vehicle or CCK injections. Asterisks over DEP bars indicate significant differences (p < .05) in the number of double-labeled NTS neurons compared to ad lib-fed rats in the same i.p. treatment group. Within the same feeding status group (i.e., ad lib or DEP), bars with different letters are significantly different (p < .05). Scale bars in A = 100 μm.
2.7. Statistics
Two-way multivariate ANOVA was used to reveal main effects of, or interactions between, CCK injection group (NH, 0, 3, & 10 μg/kg BW) and feeding status (ad lib vs. DEP) on cFos counts within the hindbrain and hypothalamus. When F-values indicated significant interactions or main effects, the ANOVA was followed by post hoc tests with Bonferroni correction for multiple comparisons. Differences were considered significant when p < 0.05. Pearson’s R correlation coefficient was used to determine whether significant correlations existed between treatment-induced activation of A2 neurons, GLP-1 neurons, and mpPVN neurons. Outlier tests identified 4 animals with data that lay more than 2 standard deviations from the group mean for a given dependent variable. Data from these animals were excluded from all analyses and do not contribute to the sample sizes reported in section 2.2., or to the results.
3. Results
Two-way ANOVA revealed significant main effects of CCK injection group and feeding status on cFos expression in DβH- and GLP-1-positive hindbrain neurons, and on cFos expression within the mpPVN. There also were significant interactions between injection group and feeding status on cFos activation in A2 and GLP-1 neurons, but not on mpPVN cFos activation (Table 1).
Table 1.
Multivariate ANOVA main effects, interactions, and p-values for cFos counts in ad lib fed or DEP rats following NH, 0 μg/kg, 3 μg/kg, and 10 μg/kg CCK.
| Effect Source | Double-Label Count | F | p value |
|---|---|---|---|
| CCK Injection [F(3,41)] | Number of cFos/DβH Neurons – NTS Total | 87.18 | p < .00* |
| NH vs. 0 μg/kg vs. 3 μg/kg vs. 10 μg/kg | Number of cFos/GLP-1 Neurons | 5.52 | p < .00* |
| Number of cFos Neurons in mpPVN | 108.92 | p < .00* | |
| Feeding Status [F(1,41)] | Number of cFos/DβH Neurons – NTS Total | 35.77 | p < .00* |
| Ad lib vs. DEP | Number of cFos/GLP-1 Neurons | 59.16 | p < .00* |
| Number of cFos Neurons in mpPVN | 16.08 | p < .00* | |
| Interaction [F(3,41)] | Number of cFos/DβH Neurons – NTS Total | 5.15 | p < .00* |
| CCK Injection X Feeding Status | Number of cFos/GLP-1 Neurons | 3.26 | p = .03* |
| Number of cFos Neurons in mpPVN | 0.58 | p = .63 |
statistically significant
3.1. A2 neuronal activation
CCK increased cFos expression within the caudal NTS (Fig. 1A), consistent with previous reports [8, 17, 36]. Our new findings indicate that this includes a dose-dependent recruitment of DβH-positive A2 neurons (Fig. 1B). Very few A2 neurons expressed cFos in NH control rats, regardless of feeding status. A2 activation was significantly increased in ad-lib fed rats, but not in DEP rats, after i.p. injection of saline vehicle (0 μg/kg CCK). CCK delivered at doses of 3 or 10 μg/kg further increased cFos expression by A2 neurons in both feeding status groups, but these effects were significantly attenuated in DEP rats (Fig. 1B).
3.2. GLP-1 neuronal activation
In contrast to the cFos activation profile for DβH-positive A2 neurons (3.1), GLP-1 neurons did not respond to CCK in a dose-dependent manner (Fig. 2). In ad lib-fed rats, GLP-1 cFos activation was robustly increased by every i.p. treatment, with no additional effect of CCK (Fig. 2C). In DEP rats, cFos expression by GLP-1 neurons was virtually abolished in NH controls and after i.p. injection of vehicle or 3 μg/kg CCK. DEP rats displayed a trend towards increased GLP-1 activation after the 10 μg/kg dose of CCK (Fig. 2C), but this did not reach significance.
Figure 2.
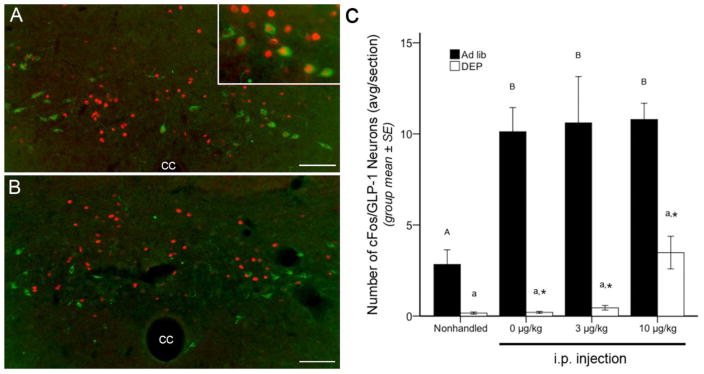
Representative color images and summary data for neuronal cFos expression (red fluorescence) within GLP-1-positive neurons (green fluorescence) of the caudal NTS. A, in an ad lib-fed rat, many GLP-1 neurons express cFos after i.p. injection of CCK (3 μg/kg BW). Inset, higher magnification view of several double-labeled neurons. B, in a food-deprived rat, GLP-1 neurons are not activated to express cFos after CCK (3 μg/kg BW), despite activation of other caudal NTS neurons. C, bar graph illustrating the number of double labeled (i.e., both cFos- and GLP-1-positive) neurons within the NTS and adjacent reticular formation in ad lib-fed rats (solid bars) or food-deprived rats (DEP; open bars) after no i.p. injection (nonhandled) or after injection of CCK at doses of 0 (vehicle), 3, or 10 μg/kg BW. See Table 1 for two-way ANOVA results. In ad lib-fed rats, a moderate number of GLP-1 neurons express cFos in the nonhandled control condition, and this number more than doubled after i.p. injection of vehicle or CCK, regardless of dose. Food deprivation markedly suppressed cFos expression by GLP-1 neurons, with a non-significant trend towards increased activation seen after the highest CCK dose (10 μg/kg BW). Asterisks indicate significantly reduced (p < .05) GLP-1 cFos expression in DEP rats compared to GLP-1 cFos expression in ad lib-fed rats in the same i.p. treatment group. Within the same feeding status group (i.e., ad lib or DEP), bar values with different letters are significantly different (p < .05). cc, central canal. Scale bars in A and B = 100 μm.
3.3. mpPVN neuronal activation
The mpPVN displayed relatively low levels of cFos labeling in both ad lib-fed and DEP rats in the NH and vehicle-injected control groups (Fig. 3A,C), and cFos was markedly increased in rats from both feeding status groups after 3 or 10 μg/kg of CCK (Fig. 3B,C). DEP rats, however, displayed less mpPVN cFos labeling overall compared to labeling in ad lib-fed rats (see Table 1).
Figure 3.
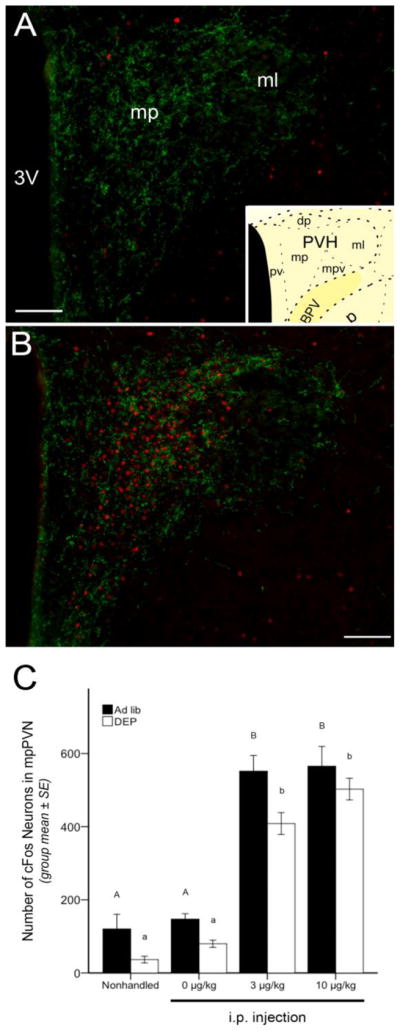
Representative color images and summary data for neuronal cFos expression (red fluorescence) within the GLP-1 (green fluorescence) terminal-rich region of the mpPVN. A, little cFos is present in the mpPVN of an ad lib-fed, nonhandled control rat. B, robust mpPVN cFos activation is present in an ad lib-fed rat injected with CCK (10 μg/kg BW). mpPVN images were captured and cFos quantified in sections approximately 1.78 mm caudal to bregma, as schematized in the inset in panel A [77]. C, bar graph illustrating the number of cFos-positive mpPVN neurons in ad lib-fed rats (solid bars) or food-deprived rats (DEP; open bars) after no i.p. injection (nonhandled) or after injection of CCK at doses of 0 (vehicle), 3, or 10 μg/kg BW. See Table 1 for two-way ANOVA results. In both feeding status groups, low levels of mpPVN cFos activation were present in nonhandled or vehicle-injected rats, and CCK delivered at doses of 3 and 10 μg/kg produced marked activation of mpPVN cFos. Food-deprived rats displayed less mpPVN cFos labeling overall compared to labeling in ad lib-fed rats. Within the same feeding status group (i.e., ad lib or DEP), bars with different letters indicate significant i.p. treatment-induced differences (p < .05) in mpPVN cFos activation. mp = medial parvocellular subdivision of the PVN; ml = lateral magnocellular subdivision of the PVN; 3V = third ventricle. Scale bars in A and B = 100 μm.
3.4. Correlational analyses
Activation of NTS DβH-positive A2 neurons correlated strongly with activation of mpPVN neurons (Fig. 4A). This positive correlation existed in both ad lib-fed and DEP rats when data from these groups were analyzed independently (see Table 2 for a breakdown of Pearson correlation values and significance by injection and feeding status group). The number of cFos-positive GLP-1 neurons also significantly correlated with cFos expression in the mpPVN (Fig. 4B), although the GLP-1/mpPVN cFos correlation was less robust than the DβH/mpPVN cFos correlation (Fig. 4A). The GLP-1/mpPVN correlation also was significant in both ad lib-fed and DEP animals when these groups were analyzed independently (Table 2). In addition, the number of cFos-positive GLP-1 neurons correlated positively with cFos expression in DβH neurons (Fig. 4C), and this GLP-1/DβH cFos correlation also remained significant when the ad lib-fed and DEP groups were analyzed independently (Table 2).
Figure 4.
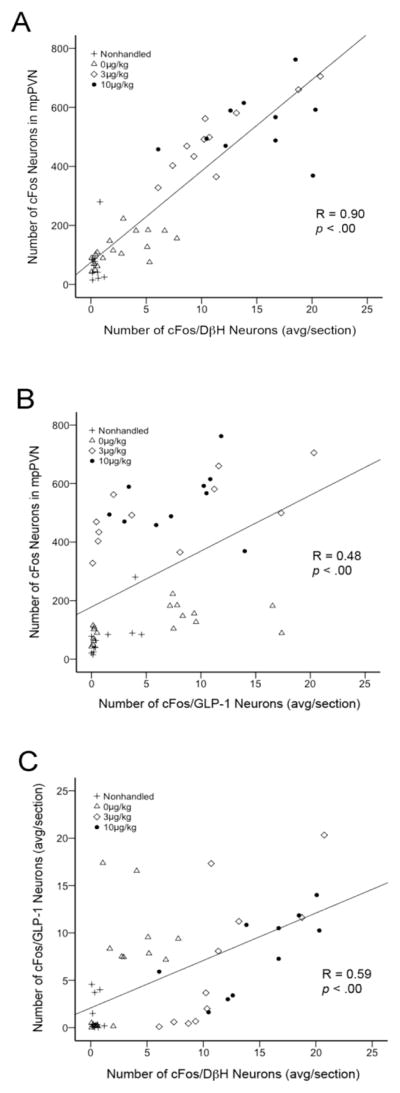
Scatter plots depicting significant positive correlations between treatment-induced activation of (A) mpPVN and NTS DβH neurons, (B) mpPVN and GLP-1 neurons, and (C) GLP-1 and DβH neurons. In each plot, individual symbols represent data from a single animal. Different symbol styles represent different i.p. treatment groups, as indicated in the key. See Table 2 for a breakdown of correlation R values and significance by i.p. treatment and feeding status groups.
Table 2.
Correlations in cFos expression between NTS DβH neurons, brainstem GLP-1 neurons, and mpPVN neurons.
| n | DβH/mpPVN | GLP-1/mpPVN | DβH/GLP-1 | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| R value | p value | R value | p value | R value | p value | |||
| Ad Lib | Nonhandled | 5 | 0.93 | .02* | 0.44 | .46 | 0.29 | .64 |
| 0 μg/kg CCK | 9 | 0.22 | .55 | −0.35 | .36 | −0.34 | .37 | |
| 3 μg/kg CCK | 7 | 0.81 | .03* | 0.45 | .31 | 0.64 | .12 | |
| 10 μg/kg CCK | 6 | −0.22 | .67 | −0.11 | .83 | 0.40 | .44 | |
| Total | 27 | 0.88 | < .00* | 0.38 | .05* | 0.48 | .01* | |
| DEP | Nonhandled | 6 | −0.22 | .68 | −0.15 | .77 | −0.09 | .87 |
| 0 μg/kg CCK | 8 | 0.57 | .14 | 0.44 | .27 | −0.20 | .64 | |
| 3 μg/kg CCK | 4 | 0.90 | .10 | 0.71 | .29 | 0.79 | .21 | |
| 10 μg/kg CCK | 4 | 0.61 | .39 | −0.26 | .74 | −0.73 | .27 | |
| Total | 22 | 0.97 | < .00* | 0.67 | < .00* | 0.60 | < .00* | |
statistically significant
Pearson correlation values and significance for relationships between cFos expression in NTS DβH neurons and mpPVN neurons (DβH/mpPVN), between brainstem GLP-1 neurons and mpPVN neurons (GLP-1/mpPVN), and between NTS DβH neurons and brainstem GLP-1 neurons (DβH/GLP-1).
4. Discussion
Results from previous studies indicate that CCK elicits neuroendocrine responses via vagally-mediated recruitment of NTS neurons, including NA neurons of the A2 cell group, that project to the hypothalamus [8, 21, 37]. Hindbrain GLP-1 neurons are also known to be activated in rats receiving a very high (i.e., 100 μg/kg BW) dose of CCK [30], and GLP-1 axonal projections target the mpPVN [29, 31, 57], suggesting an additional role for GLP-1 signaling in CCK-induced stimulation of the HPA axis. Although we did not measure stress hormone release in the present study, CCK at doses lower, higher, and similar to those used here stimulates pituitary hormone secretion [7, 52] and activates cFos expression by CRH-positive and other PVN neurons in rats [5, 53, 54]. However, the degree to which lower doses of CCK recruit phenotypically identified A2 and GLP-1 neurons, and the specific relationship between activation of these brainstem neurons and those in the mpPVN, has not been examined. Results from the present study reveal a CCK dose-dependent increase in A2 neuronal activation that strongly correlates with activation of mpPVN neurons. Conversely, i.p. injection by itself robustly increased the number of GLP-1 neurons expressing cFos, with no additional effect of CCK. Overnight food deprivation nearly abolished cFos expression in GLP-1 neurons, significantly attenuated CCK-induced cFos expression in A2 neurons, and had an overall effect to reduce cFos expression within the mpPVN.
Our results point to heterogeneity between A2 and GLP-1 neurons in their sensitivity to i.p. injection stress and CCK, although both hindbrain neuronal populations are clearly sensitive to changes in feeding status, as are neurons within the mpPVN. We propose that the apparent metabolic sensitivity of A2 and GLP-1 neurons contributes to the overall suppressive effect of food deprivation on mpPVN cFos expression. Since i.p. injection and exogenous CCK are capable of recruiting stress-sensitive hindbrain and hypothalamic neurons, the robust effect of an overnight fast to dampen treatment-induced cFos expression suggests that central neuronal responses to other stressful stimuli may also be modulated by feeding status. Indeed, fasting has been reported to interfere with the ability of animals to mount an HPA axis response to psychological stress [58, 59].
4.1. A2 neurons
A2 neurons rarely expressed cFos in non-manipulated, ad lib-fed rats, but i.p. vehicle and CCK dose-dependently activated this neuronal population. These new findings extend previous reports that CCK dose-dependently increases global NTS cFos expression [8, 17]. Interestingly, while food deprivation attenuated A2 cFos expression across all i.p. injection groups, deprivation did not abolish CCK dose-dependent increases in A2 cFos activation. Thus, while A2 neurons are sensitive to feeding status, this neuronal population retains CCK sensitivity in food-deprived rats. Since rats in the present study were not acclimated to handling or i.p. injection before experimental treatment, it is possible that fasting attenuated A2 neural sensitivity to the stress of the injection procedure without altering sensitivity to CCK, or vice versa. Additionally, injection stress may interact non-additively with CCK to elicit A2 cFos activation, obfuscating the precise contribution of each stimulus to A2 cFos recruitment in non-acclimated rats. It will be important to empirically assess these possibilities in future studies.
The strong correlation between CCK-induced activation of A2 and mpPVN neurons suggests, but does not demonstrate, a causal relationship between these variables. Additional evidence supporting this suggestion comes from studies in which loss or interruption of NA projections from the NTS to the PVN markedly reduces the ability of visceral stimuli – including CCK – to recruit the HPA stress axis [21, 27, 60, 61]. A2 neurons are also necessary for exogenous CCK to reduce food intake in rats [21]. Thus, our finding that fasting attenuates treatment-induced A2 neuronal activation may reveal a mechanism through which feeding status modulates both behavioral (e.g., hypophagic) and neuroendocrine responses to CCK and other visceral stimuli. Importantly, however, NTS inputs to the hypothalamus – and thus HPA axis activation – are unnecessary for CCK-induced hypophagia, as chronic decerebrate and neonatal rats (both of which lack functional connectivity between the NTS and PVN) display hypophagia after CCK treatment [20, 62, 63]. Moreover, it is likely that the A2 neurons responsible for brainstem-mediated hypophagic responses are distinct from those that mediate HPA axis responses to exogenous CCK, as toxin-induced loss of PVN-projecting A2 neurons does not alter CCK-induced hypophagia [64]. We interpret these collective results as evidence that exogenous CCK recruits A2 neurons that inhibit food intake via local brainstem projections, while also recruiting A2 neurons that project to the hypothalamus to elicit neuroendocrine activation.
4.2. GLP-1 neurons
Hindbrain GLP-1 neurons displayed moderate baseline levels of cFos expression (i.e., ~2–3 activated neurons/section, corresponding to ~20% of all GLP-1 neurons) in non-manipulated, ad lib-fed rats. In contrast to the A2 population, GLP-1 neurons were markedly and similarly activated to express cFos in ad lib-fed rats subjected to any i.p. injection, regardless of CCK content. Approximately 10 GLP-1 neurons per section (i.e., ~60% of all GLP-1 neurons) were activated in each i.p. treatment group. Thus, GLP-1 neurons appear to be uniquely sensitive to the mild stress of handling and/or i.p. injection, which might have masked any additional activation by CCK.
Surprisingly, GLP-1 neural cFos expression was nearly abolished in rats after overnight food deprivation, evidence that the baseline cFos expression and robust response to i.p. injection displayed by GLP-1 neurons is strongly modulated by feeding status. Despite an apparent trend towards increased GLP-1 cFos activation in food-deprived rats after the 10 μg/kg dose of CCK, this increase did not reach significance. A higher CCK dose (e.g., 50–100 μg/kg) might be sufficient to overcome the dampening effect of food deprivation on GLP-1 cFos activation, but this remains to be determined. Although GLP-1 and mpPVN cFos expression was positively correlated, the correlation was much stronger for A2 and mpPVN cFos expression, suggesting that GLP-1 neurons have less direct effects on mpPVN responses to CCK. Consistent with this, previous results from our laboratory indicate that in the absence of normal A2 neuronal signaling, the ability of exogenous CCK to activate hindbrain GLP-1 neurons is preserved, but is insufficient to support either CCK-induced hypophagia or PVN cFos expression [21]. Nevertheless, central GLP-1 receptor signaling has been implicated in HPA stress axis and anxiogenic responses to visceral stress [26], perhaps through gating the post-synaptic effects of another transmitter molecule [65]. Thus, the observed attenuation of GLP-1 cFos activation following deprivation suggests that reduced GLP-1 signaling might contribute to the blunted neuroendocrine stress responses that have been previously reported in food-deprived rats [58, 59].
4.3. mpPVN neurons
Considering the well-documented excitatory effects of NA and GLP-1 signaling on mpPVN neural activity [23, 35, 66, 67] and the markedly blunted A2 and GLP-1 cFos activation in food-deprived rats, it is surprising that food deprivation generated only a mild overall attenuation of mpPVN cFos expression under all treatment conditions. In both ad lib-fed and food-deprived rats, mpPVN cFos activation occurred in a step-wise manner, with comparable levels of cFos observed in NH and i.p. vehicle-injected control groups, and substantially increased cFos after CCK administered at either the 3 or 10 μg/kg dose. It is possible that the comparable magnitude of mpPVN cFos activation after both CCK doses is due to a ceiling effect, as a previous study reported that doses of 5 and 10 μg/kg CCK elicit large but similar increases in plasma adrenocorticotropic hormone (ACTH) [7]. The similar number of cFos-positive mpPVN neurons after both CCK doses may also reflect an inherent limitation in interpreting cFos data, as the visible presence or absence of cFos protein in a given neuron is binary, poorly reflecting the degree to which the neuron has been activated, and providing no information about that neuron’s spike frequency or amplitude. Furthermore, we do not know the extent to which hypophysiotropic CRH-positive mpPVN neurons were activated after each CCK dose. Nevertheless, the overall blunted mpPVN cFos expression observed in food-deprived rats in the present report is consistent with prior evidence that fasting blunts ACTH responses to stress [58, 59], which may result from reduced excitatory drive to the mpPVN from hindbrain A2 and GLP-1 neurons.
4.4. Potential signals of feeding status
Attenuation of A2, GLP-1, and mpPVN neural cFos expression in food-deprived rats could result from loss of an excitatory signal that is present in ad lib-fed rats, generation of an inhibitory signal associated with food deprivation, or both. Fasting produces a multitude of metabolic and physical changes [44], with reductions in circulating leptin, increases in circulating ghrelin, and reduced gastric distention standing out as potential candidate effectors of deprivation-induced changes in central neural activity. Leptin levels fall in rats after overnight food deprivation [43, 44], and leptin enhances CCK signaling at the level of the vagus nerve [68, 69], the NTS [48, 50] (particularly A2 neurons [51]), and the PVN [48, 50]. Reduced gastric distention signals in food deprived rats might also attenuate central neural cFos responses to CCK, since CCK and gastric distention act synergistically to activate vagal afferent inputs [70, 71] that modulate NTS neuronal activity [14]. It is less likely that increased ghrelin signaling after fasting [72] is responsible for attenuated central neural responses to CCK, since i.p. ghrelin does not suppress the ability of CCK to activate cFos expression in the NTS or PVN [73].
5. Conclusions
Our results show that CCK- and i.p. injection-induced cFos activation of A2, GLP-1, and mpPVN neurons is strongly modulated by short-term fasting. As these neural populations are activated following a diverse array of stressors [30, 74], including “cognitive-type” restraint stress [75, 76], we expect that food deprivation would also alter hindbrain and hypothalamic neural responses to restraint and other stressors. Considering that A2, GLP-1, and mpPVN neurons play important roles in generating behavioral and physiological responses to stress, feeding status may impact an animal’s ability to mount these responses by altering the sensitivity of hindbrain and hypothalamic neurons to their afferent inputs. It will be critical for future studies to identify the mechanisms by which food deprivation alters neural activation in these brainstem and hypothalamic populations, and to assess the functional consequences of altered neuronal signaling.
Highlights.
CCK dose-dependently increases cFos expression in A2 neurons.
I.p. injection alone robustly activates GLP-1 neurons to express cFos.
Food deprivation attenuates A2 activation and nearly abolishes GLP-1 activation.
Food deprivation reduces mpPVN cFos expression.
A2 and mpPVN neuronal activation is strongly correlated.
Acknowledgments
The authors would like to thank Victoria Maldovan for excellent technical assistance. This research was supported by NIH grant MH59911 and is based on work initially presented during the 2012 Annual Meeting of the Society for the Study of Ingestive Behavior, July 10–14, 2012, made possible in part by generous donations from Novo Nordisk A/S, Research Diets, Inc., Sanofi, Inc., and TSE, Inc..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gibbs J, Young RC, Smith GP. Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol. 1973;84:488–95. doi: 10.1037/h0034870. [DOI] [PubMed] [Google Scholar]
- 2.Gibbs J, Smith GP. Cholecystokinin and satiety in rats and rhesus monkeys. Am J Clin Nutr. 1977;30:758–61. doi: 10.1093/ajcn/30.5.758. [DOI] [PubMed] [Google Scholar]
- 3.Parrott RF, Ebenezer IS, Baldwin BA, Forsling ML. Central and peripheral doses of cholecystokinin that inhibit feeding in pigs also stimulate vasopressin and cortisol release. Exp Physiol. 1991;76:525–31. doi: 10.1113/expphysiol.1991.sp003518. [DOI] [PubMed] [Google Scholar]
- 4.Katsuura G, Ibii N, Matsushita A. Activation of CCK-A receptors induces elevation of plasma corticosterone in rats. Peptides. 1992;13:203–5. doi: 10.1016/0196-9781(92)90163-w. [DOI] [PubMed] [Google Scholar]
- 5.Verbalis JG, Stricker EM, Robinson AG, Hoffman GE. Cholecystokinin activates cFos expression in hypothalamic oxytocin and corticotropin-releasing hormone neurons. Journal of Neuroendocrinology. 1991;3:205–13. doi: 10.1111/j.1365-2826.1991.tb00264.x. [DOI] [PubMed] [Google Scholar]
- 6.Chen DY, Deutsch JA, Gonzalez MF, Gu Y. The induction and suppression of c-fos expression in the rat brain by cholecystokinin and its antagonist L364,718. Neurosci Lett. 1993;149:91–4. doi: 10.1016/0304-3940(93)90355-o. [DOI] [PubMed] [Google Scholar]
- 7.Kamilaris TC, Johnson EO, Calogero AE, Kalogeras KT, Bernardini R, Chrousos GP, et al. Cholecystokinin-octapeptide stimulates hypothalamic-pituitary-adrenal function in rats: role of corticotropin-releasing hormone. Endocrinology. 1992;130:1764–74. doi: 10.1210/endo.130.4.1312423. [DOI] [PubMed] [Google Scholar]
- 8.Monnikes H, Lauer G, Arnold R. Peripheral administration of cholecystokinin activates c-fos expression in the locus coeruleus/subcoeruleus nucleus, dorsal vagal complex and paraventricular nucleus via capsaicin-sensitive vagal afferents and CCK-A receptors in the rat. Brain Res. 1997;770:277–88. doi: 10.1016/s0006-8993(97)00865-2. [DOI] [PubMed] [Google Scholar]
- 9.Smith GP, Jerome C, Norgren R. Afferent axons in abdominal vagus mediate satiety effect of cholecystokinin in rats. Am J Physiol. 1985;249:R638–41. doi: 10.1152/ajpregu.1985.249.5.R638. [DOI] [PubMed] [Google Scholar]
- 10.Smith GP, Jerome C, Cushin BJ, Eterno R, Simansky KJ. Abdominal vagotomy blocks the satiety effect of cholecystokinin in the rat. Science. 1981;213:1036–7. doi: 10.1126/science.7268408. [DOI] [PubMed] [Google Scholar]
- 11.Moran TH, Norgren R, Crosby RJ, McHugh PR. Central and peripheral vagal transport of cholecystokinin binding sites occurs in afferent fibers. Brain Res. 1990;526:95–102. doi: 10.1016/0006-8993(90)90253-8. [DOI] [PubMed] [Google Scholar]
- 12.Corp ES, McQuade J, Moran TH, Smith GP. Characterization of type A and type B CCK receptor binding sites in rat vagus nerve. Brain Res. 1993;623:161–6. doi: 10.1016/0006-8993(93)90024-h. [DOI] [PubMed] [Google Scholar]
- 13.Raybould HE, Gayton RJ, Dockray GJ. CNS effects of circulating CCK8: involvement of brainstem neurones responding to gastric distension. Brain Res. 1985;342:187–90. doi: 10.1016/0006-8993(85)91373-3. [DOI] [PubMed] [Google Scholar]
- 14.Raybould HE, Gayton RJ, Dockray GJ. Mechanisms of action of peripherally administered cholecystokinin octapeptide on brain stem neurons in the rat. J Neurosci. 1988;8:3018–24. doi: 10.1523/JNEUROSCI.08-08-03018.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz GJ, McHugh PR, Moran TH. Pharmacological dissociation of responses to CCK and gastric loads in rat mechanosensitive vagal afferents. American Journal of Physiology. 1994;267:R303–R8. doi: 10.1152/ajpregu.1994.267.1.R303. [DOI] [PubMed] [Google Scholar]
- 16.Sutton GM, Patterson LM, Berthoud HR. Extracellular signal-regulated kinase 1/2 signaling pathway in solitary nucleus mediates cholecystokinin-induced suppression of food intake in rats. J Neurosci. 2004;24:10240–7. doi: 10.1523/JNEUROSCI.2764-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zittel TT, Glatzle J, Kreis ME, Starlinger M, Eichner M, Raybould HE, et al. C-fos protein expression in the nucleus of the solitary tract correlates with cholecystokinin dose injected and food intake in rats. Brain Res. 1999;846:1–11. doi: 10.1016/s0006-8993(99)01842-9. [DOI] [PubMed] [Google Scholar]
- 18.Appleyard SM, Marks D, Kobayashi K, Okano H, Low MJ, Andresen MC. Visceral afferents directly activate catecholamine neurons in the solitary tract nucleus. The Journal of Neuroscience. 2007;27:13292–302. doi: 10.1523/JNEUROSCI.3502-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ritter RC. A tale of two endings: modulation of satiation by NMDA receptors on or near central and peripheral vagal afferent terminals. Physiol Behav. 2011;105:94–9. doi: 10.1016/j.physbeh.2011.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grill HJ, Smith GP. Cholecystokinin decreases sucrose intake in chronic decerebrate rats. American Journal of Physiology. 1988;253:R853–R6. doi: 10.1152/ajpregu.1988.254.6.R853. [DOI] [PubMed] [Google Scholar]
- 21.Rinaman L. Hindbrain noradrenergic lesions attenuate anorexia and alter central cFos expression in rats after gastric viscerosensory stimulation. The Journal of Neuroscience. 2003;23:10084–92. doi: 10.1523/JNEUROSCI.23-31-10084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rinaman L. Hindbrain noradrenergic A2 neurons: diverse roles in autonomic, endocrine, cognitive, and behavioral functions. Am J Physiol Regul Integr Comp Physiol. 2011;300:R222–35. doi: 10.1152/ajpregu.00556.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rinaman L. Visceral sensory inputs to the endocrine hypothalamus. Frontiers in Neuroendocrinology. 2007;28:50–60. doi: 10.1016/j.yfrne.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plotsky P. Facilitation of immunoreactive corticotropin-releasing factor secretion into the hypophysial-portal circulation after activation of catecholaminergic pathways or central norepinephrine injection. Endocrinology. 1987;121:924–30. doi: 10.1210/endo-121-3-924. [DOI] [PubMed] [Google Scholar]
- 25.Plotsky PM, Cunningham ET, Jr, Widmaier EP. Catecholaminergic modulation of corticotropin-releasing factor and adrenocorticotropin secretion. Endocrine Reviews. 1989;10:437–58. doi: 10.1210/edrv-10-4-437. [DOI] [PubMed] [Google Scholar]
- 26.Kinzig KP, D’Alessio DA, Herman JP, Sakai RR, Vahl TP, Figueredo HF, et al. CNS Glucagon-like peptide-1 receptors mediate endocrine and anxiety responses to interoceptive and psychogenic stressors. The Journal of Neuroscience. 2003;23:6163–70. doi: 10.1523/JNEUROSCI.23-15-06163.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bienkowski MS, Rinaman L. Noradrenergic inputs to the paraventricular hypothalamus contribute to hypothalamic-pituitary-adrenal axis and central Fos activation in rats after acute systemic endotoxin exposure. Neuroscience. 2008;156:1093–102. doi: 10.1016/j.neuroscience.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larsen PJ, Tang-Christensen M, Jessop DS. Central administration of glucagon-like peptide-1 activates hypothalamic neuroendocrine neurons in the rat. Endocrinology. 1997;138:4445–55. doi: 10.1210/endo.138.10.5270. [DOI] [PubMed] [Google Scholar]
- 29.Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in rat hypothalamus and brainstem. Neuroscience. 1997;77:257–70. doi: 10.1016/s0306-4522(96)00434-4. [DOI] [PubMed] [Google Scholar]
- 30.Rinaman L. Interoceptive stress activates glucagon-like peptide-1 neurons that project to the hypothalamus. American Journal of Physiology. 1999;277:R582–R90. doi: 10.1152/ajpregu.1999.277.2.R582. [DOI] [PubMed] [Google Scholar]
- 31.Tauchi M, Zhang R, D’Alessio DA, Stern JE, Herman JP. Distribution of glucagon-like peptide-1 immunoreactivity in the hypothalamic paraventricular and supraoptic nuclei. J Chem Neuroanat. 2008;36:144–9. doi: 10.1016/j.jchemneu.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cunningham ET, Jr, Sawchenko PE. Anatomical specificity of noradrenergic inputs to the paraventricular and supraoptic nuclei of the rat hypothalamus. Journal of Comparative Neurology. 1988;274:60–76. doi: 10.1002/cne.902740107. [DOI] [PubMed] [Google Scholar]
- 33.Sarkar S, Fekete C, Legradi G, Lechan RM. Glucagon like peptide-1 (7-36) amide (GLP-1) nerve terminals densely innervate corticotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Brain Research. 2003;985:163–8. doi: 10.1016/s0006-8993(03)03117-2. [DOI] [PubMed] [Google Scholar]
- 34.Liposits Z, Sherman D, Phelix C, Paull WK. A combined light and electron microscopic immunocytochemical method for the simultaneous localization of multiple tissue antigens. Tyrosine hydroxylase immunoreactive innervation of corticotropin releasing factor synthesizing neurons in the paraventricular nucleus of the rat. Histochemistry. 1986;85:95–106. doi: 10.1007/BF00491754. [DOI] [PubMed] [Google Scholar]
- 35.Cole RL, Sawchenko PE. Neurotransmitter regulation of cellular activation and neuropeptide gene expression in the paraventricular nucleus of the hypothalamus. The Journal of Neuroscience. 2002;22:959–69. doi: 10.1523/JNEUROSCI.22-03-00959.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Babic T, Townsend RL, Patterson LM, Sutton GM, Zheng H, Berthoud HR. Phenotype of neurons in the nucleus of the solitary tract that express CCK-induced activation of the ERK signaling pathway. American Journal of Physiology Regulatory Integrative and Comparative Physiology. 2009;296:R845–R54. doi: 10.1152/ajpregu.90531.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rinaman L, Hoffman GE, Dohanics J, Le WW, Stricker EM, Verbalis JG. Cholecystokinin activates catecholaminergic neurons in the caudal medulla that innervate the paraventricular nucleus of the hypothalamus in rats. The Journal of Comparative Neurology. 1995;360:246–56. doi: 10.1002/cne.903600204. [DOI] [PubMed] [Google Scholar]
- 38.Verbalis JG, Baker EA, Rinaman L, Stricker EM, Hoffman GE. c-Fos activation of catecholaminergic neurons in the dorsal vagal complex after gastric distension or CCK administration in rats. Society for Neuroscience Abstracts. 1995;21:7. [Google Scholar]
- 39.Kendrick K, Leng G, Higuchi T. Noradrenaline, dopamine and serotonin release in the paraventricular and supraoptic nuclei of the rat in response to intravenous cholecystokinin injections. Journal of Neuroendocrinology. 1991;3:139–44. doi: 10.1111/j.1365-2826.1991.tb00255.x. [DOI] [PubMed] [Google Scholar]
- 40.Moran TH, Baldessarini AR, Salorio CF, Lowery T, Schwartz GJ. Vagal afferent and efferent contributions to the inhibition of food intake by cholecystokinin. American Journal of Physiology. 1997;272:R1245–R51. doi: 10.1152/ajpregu.1997.272.4.R1245. [DOI] [PubMed] [Google Scholar]
- 41.McMinn JE, Sindelar DK, Havel PJ, Schwartz MW. Leptin deficiency induced by fasting impairs the satiety response to cholecystokinin. Endocrinology. 2000;141:4442–8. doi: 10.1210/endo.141.12.7815. [DOI] [PubMed] [Google Scholar]
- 42.Stein LJ, Porte D, Jr, Figlewicz DP, Woods SC. Effect of fasting interval on CCK-8 suppression of food intake in the baboon. Am J Physiol. 1986;250:R851–5. doi: 10.1152/ajpregu.1986.250.5.R851. [DOI] [PubMed] [Google Scholar]
- 43.Pico C, Sanchez J, Oliver P, Palou A. Leptin production by the stomach is up-regulated in obese (fa/fa) Zucker rats. Obes Res. 2002;10:932–8. doi: 10.1038/oby.2002.127. [DOI] [PubMed] [Google Scholar]
- 44.Dallman MF, Akana SF, Bhatnagar S, Bell ME, Choi S, Chu A, et al. Starvation: early signals, sensors, and sequelae. Endocrinology. 1999;140:4015–23. doi: 10.1210/endo.140.9.7001. [DOI] [PubMed] [Google Scholar]
- 45.Mastronardi CA, Walczewska A, Yu WH, Karanth S, Parlow AF, McCann SM. The possible role of prolactin in the circadian rhythm of leptin secretion in male rats. Proc Soc Exp Biol Med. 2000;224:152–8. doi: 10.1046/j.1525-1373.2000.22414.x. [DOI] [PubMed] [Google Scholar]
- 46.Peters JH, Ritter RC, Simasko SM. Leptin and CCK selectively activate vagal afferent neurons innervating the stomach and duodenum. American Journal of Physiology Regulatory Integrative and Comparative Physiology. 2006;290:R1544–R9. doi: 10.1152/ajpregu.00811.2005. [DOI] [PubMed] [Google Scholar]
- 47.Peters JH, Simasko SM, Ritter RC. Modulation of vagal afferent excitation and reduction of food intake by leptin and cholecystokinin. Physiol Behav. 2006;89:477–85. doi: 10.1016/j.physbeh.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 48.Wang L, Martinez V, Barrachina MD, Tache Y. Fos expression in the brain induced by peripheral injection of CCK or leptin plus CCK in fasted lean mice. Brain Res. 1998;791:157–66. doi: 10.1016/s0006-8993(98)00091-2. [DOI] [PubMed] [Google Scholar]
- 49.Wang L, Barachina MD, Martinez V, Wei JY, Tache Y. Synergistic interaction between CCK and leptin to regulate food intake. Regul Pept. 2000;92:79–85. doi: 10.1016/s0167-0115(00)00153-1. [DOI] [PubMed] [Google Scholar]
- 50.Emond M, Schwartz GJ, Ladenheim EE, Moran TH. Central leptin modulates behavioral and neuronal responsivity to CCK. American Journal of Physiology Regulatory Int Comp Physiol. 1999;276:R1545–R9. doi: 10.1152/ajpregu.1999.276.5.R1545. [DOI] [PubMed] [Google Scholar]
- 51.Williams DL, Schwartz MW, Bastian LS, Blevins JE, Baskin DG. Immunocytochemistry and laser capture microdissection for real-time quantitative PCR identify hindbrain neurons activated by interaction between leptin and cholecystokinin. J Histochem Cytochem. 2008;56:285–93. doi: 10.1369/jhc.7A7331.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCann MJ, Verbalis JG, Stricker EM. LiCl and CCK inhibit gastric emptying and feeding and stimulate OT secretion in rats. American Journal of Physiology. 1989;256:R463–R8. doi: 10.1152/ajpregu.1989.256.2.R463. [DOI] [PubMed] [Google Scholar]
- 53.Noetzel S, Stengel A, Inhoff T, Goebel M, Wisser AS, Bannert N, et al. CCK-8S activates c-Fos in a dose-dependent manner in nesfatin-1 immunoreactive neurons in the paraventricular nucleus of the hypothalamus and in the nucleus of the solitary tract of the brainstem. Regul Pept. 2009;157:84–91. doi: 10.1016/j.regpep.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 54.Day HEW, McKnight AT, Poat JA, Hughes J. Evidence that cholecystokinin induces immediate early gene expression in the brainstem, hypothalamus and amygdala of the rat by a CCKA receptor mechanism. Neuropharmacology. 1994;33:719–27. doi: 10.1016/0028-3908(94)90111-2. [DOI] [PubMed] [Google Scholar]
- 55.McLean IW, Nakane PK. Periodate-lysine-paraformldehyde fixative. A new fixative for immunoelectron microscopy. Journal of Histochemistry and Cytochemistry. 1974;22:1077–83. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- 56.Watson RE, Wiegand ST, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–9. doi: 10.1016/0196-9781(86)90076-8. [DOI] [PubMed] [Google Scholar]
- 57.Rinaman L. Ascending projections from the caudal visceral nucleus of the solitary tract to brain regions involved in food intake and energy expenditure. Brain Research. 2010;1350:18–34. doi: 10.1016/j.brainres.2010.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rohleder N, Kirschbaum C. Effects of nutrition on neuro-endocrine stress responses. Curr Opin Clin Nutr Metab Care. 2007;10:504–10. doi: 10.1097/MCO.0b013e3281e38808. [DOI] [PubMed] [Google Scholar]
- 59.De Boer SF, Koopmans SJ, Slangen JL, Van der Gugten J. Effects of fasting on plasma catecholamine, corticosterone and glucose concentrations under basal and stress conditions in individual rats. Physiol Behav. 1989;45:989–94. doi: 10.1016/0031-9384(89)90226-6. [DOI] [PubMed] [Google Scholar]
- 60.Rinaman L, Dzmura V. Experimental dissociation of neural circuits underlying conditioned avoidance and hypophagic responses to lithium chloride. American Journal of Physiology Regulatory Integrative and Comparative Physiology. 2007;293:R1495–R503. doi: 10.1152/ajpregu.00393.2007. [DOI] [PubMed] [Google Scholar]
- 61.Ritter S, Watts AG, Dinh TT, Sanchez-Watts G, Pedrow C. Immunotoxin lesion of hypothalamically projecting norepinephrine and epinephrine neurons differentially affects circadian and stressor-stimulated corticosterone secretion. Endocrinology. 2003;144:1357–67. doi: 10.1210/en.2002-221076. [DOI] [PubMed] [Google Scholar]
- 62.Rinaman L, Hoffman GE, Stricker EM, Verbalis JG. Exogenous cholecystokinin activates cFos expression in medullary but not hypothalamic neurons in neonatal rats. Developmental Brain Research. 1994;77:140–5. doi: 10.1016/0165-3806(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 63.Robinson PH, Moran TH, McHugh PR. Cholecystokinin inhibits independent ingestion in neonatal rats. American Journal of Physiology. 1988;255:R14–R20. doi: 10.1152/ajpregu.1988.255.1.R14. [DOI] [PubMed] [Google Scholar]
- 64.Ritter S, Bugarith K, Dinh TT. Immunotoxic destruction of distinct catecholamine subgroups produces selective impairment of glucoregulatory responses and neuronal activation. Journal of Comparative Neurology. 2001;432:197–216. doi: 10.1002/cne.1097. [DOI] [PubMed] [Google Scholar]
- 65.Acuna-Goycolea C, Pol Avd. Glucagon-like peptide 1 excites hypocretin/orexin neurons by direct and indirect mechanisms: implications for viscera-mediated arousal. The Journal of Neuroscience. 2004;24:8141–52. doi: 10.1523/JNEUROSCI.1607-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Daftary SS, Boudaba C, Tasker JG. Noradrenergic regulation of parvocellular neurons in the rat hypothalamic paraventricular nucleus. Neuroscience. 2000;96:743–51. doi: 10.1016/s0306-4522(00)00003-8. [DOI] [PubMed] [Google Scholar]
- 67.Day T, Ferguson AV, Renaud L. Noradrenergic afferents facilitate the activity of tuberoinfundibular neurons of the hypothalamic paraventricular nucleus. Neuroendocrinology. 1985;41:17. doi: 10.1159/000124148. [DOI] [PubMed] [Google Scholar]
- 68.Peters JH, Ritter RC, Simasko SM. Leptin and CCK modulate complementary background conductances to depolarize cultured nodose neurons. Am J Physiol Cell Physiol. 2006;290:C427–32. doi: 10.1152/ajpcell.00439.2005. [DOI] [PubMed] [Google Scholar]
- 69.Peters JH, Karpiel AB, Ritter RC, Simasko SM. Cooperative activation of cultured vagal afferent neurons by leptin and cholecystokinin. Endocrinology. 2004;145:3652–7. doi: 10.1210/en.2004-0221. [DOI] [PubMed] [Google Scholar]
- 70.Schwartz GJ, Netterville LA, McHugh PR, Moran TH. Gastric loads potentiate inhibition of food intake produced by a cholecystokinin analogue. American Journal of Physiology. 1991;261:R1141–R6. doi: 10.1152/ajpregu.1991.261.5.R1141. [DOI] [PubMed] [Google Scholar]
- 71.Schwartz GJ, McHugh PR, Moran TH. Gastric loads and cholecystokinin synergistically stimulate rat gastric vagal afferents. American Journal of Physiology. 1993;265:R872–R6. doi: 10.1152/ajpregu.1993.265.4.R872. [DOI] [PubMed] [Google Scholar]
- 72.Toshinai K, Mondal MS, Nakazato M, Date Y, Murakami N, Kojima M, et al. Upregulation of Ghrelin expression in the stomach upon fasting, insulin-induced hypoglycemia, and leptin administration. Biochem Biophys Res Commun. 2001;281:1220–5. doi: 10.1006/bbrc.2001.4518. [DOI] [PubMed] [Google Scholar]
- 73.Kobelt P, Tebbe JJ, Tjandra I, Stengel A, Bae HG, Andresen V, et al. CCK inhibits the orexigenic effect of periphral ghrelin. American Journal of Physiology - Regulatory, Integrative, and Comparative Physiology. 2005;288:R751–R8. doi: 10.1152/ajpregu.00094.2004. [DOI] [PubMed] [Google Scholar]
- 74.Li HY, Ericsson A, Sawchenko PE. Distinct mechanisms underlie activation of hypothalamic neurosecretory neurons and their medullary catecholaminergic afferents in categorically different stress paradigms. Proceedings of the National Academy of Sciences, USA. 1996;93:2359–64. doi: 10.1073/pnas.93.6.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dayas CV, Buller KM, Crane JW, Xu Y, Day TA. Stressor categorization: acute physical and psychological stressors elicit distinctive recruitment patterns in the amygdala and in medullary noradrenergic cell groups. European Journal of Neuroscience. 2001;14:1143–52. doi: 10.1046/j.0953-816x.2001.01733.x. [DOI] [PubMed] [Google Scholar]
- 76.Maniscalco JW, Gordon PJ, Rinaman L. Activation of hindbrain glucagon-like peptide-1 (GLP-1) neurons in rats after restraint stress correlates with activation in the GLP-1 terminal-rich region of the parastrial nucleus (PS) of the hypothalamic preoptic area. Society for Neuroscience; New Orleans, LA: 2012. [Google Scholar]
- 77.Swanson L. Structure of the Rat Brain. 3. Amsterdam: Elsevier; 2004. Brain Maps III. [Google Scholar]