

Abstract
Alzheimer’s disease (AD) and vascular dementia (VAD) are the major forms of dementia affecting elderly people, in which the levels of many metabolites are altered in cerebrospinal fluid (CSF) and serum. These metabolites could be risk factors or potential biomarkers, but the significance of some of these are not clearly understood in the context of the disease pathogenesis. In the present study serum levels of homocysteine, dehydroepiandrosterone sulphate (DHEA-S) and lipoprotein (a) or Lp(a) have been measured by ELISA using commercial kits in AD (n = 40), VAD (n = 40) and age matched control subjects (n = 40). The data are compared by ANOVA and post-hoc analysis. The serum homocysteine is markedly elevated compared to control both in AD and VAD subjects, but to a significantly higher extent in the latter. Lp(a) is increased in the serum of VAD subjects only compared to control. Likewise, serum DHEA-S level is lowered in AD but not in VAD compared to control. The analysis of the present data and those published by others suggest that alterations in homocysteine and Lp(a) in serum are indicators of vascular pathology in AD or VAD, while the lowering of serum DHEA-S is a consequence of AD pathology.
Keywords: Alzheimer’s disease, dehydroepiandrosterone sulphate, differential diagnosis, homocysteine, lipoprotein (a), vascular dementia
Alzheimer’s disease (AD) and vascular dementia (VAD) together account for the majority cases of dementia in the elderly population of various countries with nearly 70 – 75% of dementia patients belonging to AD and 15% to VAD and the societal burden for these disorders is becoming enormous with the progressive increase in the life span of general population [1]. In developed countries the prevalence of AD is around 4.4% but the prevalence is somewhat lower in the developing countries [2, 3]. On the other hand, the prevalence of VAD has been reported to vary from less than 1% to approximately 2% in different countries above the age of 65 years [2, 3]. Although studies in transgenic animals and cell based models of AD and the analysis of post-mortem AD brains have provided insights into the pathological mechanisms of this disease which comprises of abnormal amyloid beta accumulation, mitochondrial dysfunction, oxidative stress and inflammatory response in the brain, the etiopathogenesis of sporadic AD which accounts for 90 –95% of all AD cases is far from understood [4]. Thus, the role of metabolic and environmental risk factors in the disease pathogenesis should be carefully evaluated for AD. On the other hand, VAD is caused by the hemorrhagic, ischemic or hypoperfusive injury to the brain as a result of the diseases of large or small blood vessels or arterioles and various vascular risk factors may play critical roles in the disease pathogenesis [5, 6].
Epidemiological studies have established several risk factors and biomarkers in blood and CSF of both AD and VAD patients which assist in the diagnosis of the disease and also may provide useful clues to understand the pathogenesis or to develop therapeutic and preventive measures. The altered levels in CSF of amyloid beta peptides (Aβ42, Aβ40), tau and phosphorylated tau in both AD and VAD have been studied most widely [7, 8, 9, 10]. On the other hand elevated homocysteine in blood and altered levels of DHEA metabolites in brain, CSF and blood have been shown to be associated with AD and VAD in several studies [11, 12, 13, 14, 15]. The size and serum level of Lp(a) which is a well established risk factor for cardio-vascular diseases has also been linked with increased risk of both and AD and VAD in a few studies [16, 17]. However, it is not clear whether these alterations of homocysteine, DHEA-S and Lp(a) are linked to pathogenesis of the diseases or simply reflect the consequences of the biochemical changes taking place in the diseased brain. Further, no study has attempted to assess how the extent of changes in one or more of these parameters could be related to either AD or VAD. The present case-control study has attempted to address some of these issues while comparing the serum levels of homocysteine, DHEA-S and Lp(a) in AD and VAD subjects with age-matched controls.
MATERIALS AND METHODS
Samples
The study was carried out in the Department of Biochemistry of IPGMER, Kolkata and the patients were selected from the Dementia Clinic of Department of Neurology, BIN, Kolkata, which is a sister institute. The control subjects selected were part of an ongoing collaborative population based study on aging, dietary habits and cognitive impairment undertaken by the Department of Biochemistry, IPGMER and Indian Statistical Institute, Kolkata, India. The study was approved by the institutional ethics committee of IPGMER which follows the guidelines set by the Helsinki declaration. The study consisted of 40 cases of AD and VAD each and 40 control subjects recruited over a period of 18 months from August 2009 to February 2011. After fully explaining the study, a written informed consent was obtained from every control subject and patient or a close relative of the latter in case the patient was not in a position to give the consent.
The age, sex distribution and Mini Mental State Examination (MMSE) scores of the control subjects and the patients are presented in Fig. 1. The diagnosis of VAD was established according to the NINDS-AIREN (National Institute of Neurological Disorders and Stroke and the Association Internationale pour la Recherche et l’Enseignement en Neurosciences) criteria for probable vascular dementia. All patients were subjected to Magnetic Resonance Imaging (MRI), which showed subcortical lacunae and/or multiple involvements in the white matter. The diagnosis of probable AD was established following the criteria of DSM-IV (Diagnostic and Statistical Manual of Mental Disorders). The diagnosis was corroborated by cerebral MRI studies which showed atrophy in cortical and hippocampal regions, and normal aspects of the white matter. All healthy controls used in this study were examined to exclude any cognitive impairment. All controls and patients under the study were examined clinically and routine biochemical tests were performed for each subject. However, MRI of brain was routinely carried out in this study for AD and VAD patients but not for control subjects. The exclusion criteria for all the groups were overt ischemic heart disease, renal failure, diabetes, chronic liver disease, cancer, and any other neurological disorders. At the time of sample collections all AD subjects were under treatment with donepezil and memantine, while all the VAD subjects were taking anti-platelet agents, statins, amlodipine and also donepezil.
Fig. 1.
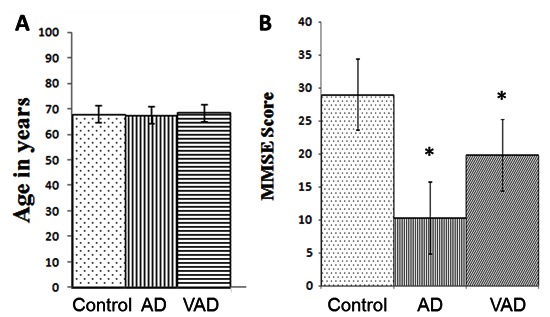
Demographic profile of AD and VAD subjects. Values are the means ± SD for the number of cases (n) for (A) Age in years and (B) MMSE scores. Control, n= 40, M/F = 26/14; AD, n= 40, M/F = 28/12; VAD, n= 40, M/F = 25/15. *p<0.001 vs. Control.
Biochemical assays
Fasting venous blood samples were obtained from both patients and controls. The sera were stored at −20°C and analysed within 15 days. Serum Hcy assay was done using a commercial kit (Axis, UK) in which both free homocysteine and protein bound homocysteine were enzymatically converted to S-adenosylhomocysteine which was measured by a sandwich ELISA. Serum DHEA-S and Lp(a) were estimated by ELISA (Adaltis, Italia) and immunoturbidimetry (Daichi, Japan) respectively following manufacturer’s protocol.
Routine biochemical parameters were measured for all the subjects under study using automated clinical analyzer (model Daytona, Randox).
Statistical analysis
Statistical analysis of different parameters in 3 groups was performed by means of one way analysis of variance (ANOVA). These comparisons were followed by post-hoc comparisons between groups by means of the Tukey’s test.
RESULTS
The demographic profiles of all the subjects under study are shown in Fig. 1. Both controls and cases were in the similar age group (Fig. 1A). When the age groups were analyzed according to sex, no significant difference was observed between the males and the females either in controls or in cases (data not shown). The MMSE scores of AD and VAD subjects were considerably lower than the controls (Fig. 1B), but the scores were not significantly different between the males and the females within each group (data not presented). The representative MRI scans of AD and VAD subjects are shown in Fig. 2.
Fig. 2.
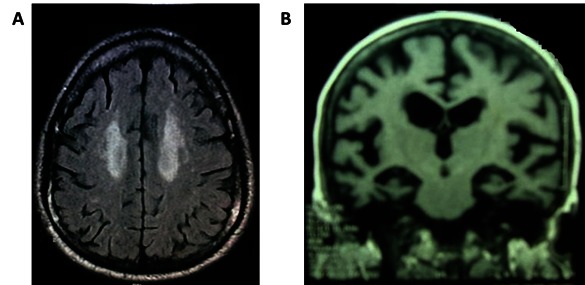
Typical MRI Scans of AD and VAD subjects. A. Vascular dementia; Axial flair image showing confluent hyper intensity in bilateral subcortical white matter suggesting ischemic changes. B. Alzheimer’s disease; coronal T1 MRI picture showing medial temporal lobe atrophy.
The results presented in Fig. 3A show higher levels of Hcy both in VAD and AD compared to that in the control subjects which were statistically significant (p<0.001). In case of serum DHEA-S (Fig. 3B), a lower level compared to control (statistically significant, p<0.001) was observed in AD subjects, but not in VAD. On the other hand, the serum level of Lp(a) (Fig. 3C) was found to be significantly higher compared to control (p<0.001) in the VAD cases, but not in AD patients. In each group the levels of serum Lp(a) among different individuals showed a large variation indicated by a high standard deviation (SD). Further, the serum levels of Hcy, DHEA-S and Lp(a) differed significantly between AD and VAD subjects (Fig. 3). When the serum levels of Hcy, DHEA-S and Lp(a) were compared between males and females within each group, no significant sex difference could be observed for any of the studied parameters (data not shown).
Fig. 3.
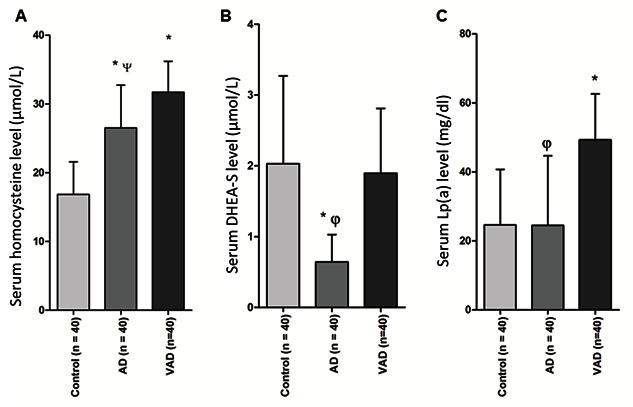
Serum levels of (A) homocysteine, (B) DHEA-S and (C) Lp(a) in AD and VAD subjects. The values are expressed as the means ± SD for the number of cases (n) in each group of subjects. Statistical comparisons were made by one way Anova followed by post-hoc analysis as described in the methods. *p< 0.001 vs. Control; ψ p< 0.005 vs. VAD; φ p< 0.001 vs. VAD.
DISCUSSION
The higher levels of serum homocysteine in AD and VAD compared to that in age-matched controls as observed in this study confirm several earlier reports which have shown strong association of this serum parameter with both these disease conditions [11, 12, 13, 18]. Hyperhomocysteinemia is a well known vascular risk factor and a higher level of serum homocysteine in VAD subjects is probably causal to the vascular lesions underlying this disorder [19, 20]. In case of AD a primary neurodegenerative process in which amyloid beta accumulation plays a critical role co-exists with varying degrees of cerebrovascular dysfunctions [21, 22]. The elevated level of serum homocysteine in AD cases compared to age-matched controls as seen in this and other studies probably contributes to the genesis of vascular pathology of AD. This view of hyperhomocysteinemia accounting for the vascular component of AD pathology has also been proposed by others [19, 23]. It is interesting to notice in our present results (Fig. 3A) that the VAD subjects who presumably have greater degrees of cerebrovascular pathologies than AD patients have correspondingly higher serum homocysteine level than the latter. Similar higher values of serum homocysteine in VAD compared to AD have also been reported earlier [20]. Further, the degree of cognitive decline as evident from MMSE scores is much greater in AD than in VAD in this study population (Fig. 1), but homocysteine is elevated to a greater degree in VAD cases (Fig. 3A). This suggests that the level of homocysteine does not directly reflect the extent or the severity of the brain damage. However, the direct neurotoxic effects of homocysteine have been shown in a large number of studies using cultured SHSY5Y cells, primary culture of hippocampal neurons and rat brain cortical slices [24, 25, 26]. Methionine-rich diet induces hyperhomocysteinemia in transgenic AD mice and aggravates behavioural deficits, but when such animals are switched to normal diet, hyperhomocysteinemia is corrected along with improvement of brain amyloidosis as well as cognitive impairment [27]. Thus an elevated serum homocysteine level in AD patients may also imply a primary contribution of homocysteine to the neurodegenerative process of AD. Several recent MRI based studies have shown that elevated serum homocysteine is associated with greater hippocampal and cortical atrophy in MCI subjects [28]. On the other hand, while the deficiency of folate, or vitamin B 12 or pyridoxine is responsible for hyperhomocysteinemia in many cases, the supplementation of these vitamins in high doses in AD patients has failed to produce clinical improvement of dementia, although the hyperhomocysteinemia is corrected [29]. Thus the role of hyperhomocysteinemiavis-a vis AD pathogenesis remains controversial.
Lp(a) has not been studied much in the context of AD or VAD, but the elevated level of this lipoprotein which contains both apoB of LDL and a special apoproteinapo(a) is considered as an independent genetic risk factor for cardiovascular diseases [30]. There are complex size variations of apo(a) isoforms ranging from 300 – 800 kDa and large differences in blood levels of Lp(a) among individuals in a population with an inverse relation between apo(a) size and blood levels of Lp(a) [30, 31]. In agreement with these studies, we observed that serum Lp(a) levels varied over a wide range among individuals within each group (Fig. 3C). Some studies have shown that smaller isoforms and higher blood levels of Lp(a) are associated with increased risk of VAD and AD [ 32 ]. The present study demonstrates a significantly higher level of Lp(a) in VAD but not in AD which agrees well with the fact that in VAD the pathogenesis is predominantly vascular in nature and Lp(a) potentiates the atherogenic processes in blood vessels through multiple mechanisms [ 30,31 ].
The neurosteroid DHEA and its metabolite DHEA-S are produced in the brain in significant amounts, although adrenal cortex and gonads are the major sources of these compounds in the body [33, 34]. There are consistent reports of higher brain and CSF levels of DHEA in AD, which have been correlated to the neuropathological stages of the disease, but there are variable reports of altered DHEA in blood in AD [14, 15, 35, 36]. On the other hand, the level of DHEA-S derived from DHEA has been reported to be significantly less in CSF and blood in AD compared to control, although the studies are limited in number [15, 36, 37, 38]. Both DHEA and DHEA-S have been shown to have neuroprotective effects in different experimental models and are also known to improve cognitive functions in animals, but it is not clear whether the altered levels of these neurosteroids in brain or CSF in AD represent physiological adaptive responses or are causally related to the pathogenesis [14, 15, 39, 40]. Recent studies on the other hand have demonstrated that in brain two possible pathways of DHEA biosynthesis exist -- one pathway requiring the enzyme cytochrome P450 dependent 17 α- hydroxylase (P450c17) and the other one mediated by iron and oxygen free radicals [33, 34, 35]. The ROS dependent pathway is active in AD brain producing DHEA from an unidentified precursor [41]. Thus, the higher content of DHEA in brain and CSF in AD patients is a consequence rather than a causal factor. It is further interesting to note that reduced glutathione is required for the function of hydroxysteroid sulphotransferase which converts DHEA to DHEA-S [42]. It is plausible that the decreased availability of reduced glutathione because of the accompanying oxidative stress in AD brain will lead to a reduced formation of DHEA-S in the latter which could be reflected as a lower level of this neurosteroid in CSF and blood. A lower level of serum DHEA-S as observed here and in other studies is, therefore, probably a consequence of pathologic process of AD rather than a risk factor for the disease. It is, therefore, not surprising that DHEA-S supplementation has failed to produce any significant improvement in clinical cases of AD. On the other hand it will be interesting to follow in longitudinal studies the decrease in CSF or serum DHEA-S levels during the progress of AD. The serum level of DHEA-S is not altered compared to control in VAD subjects in this study, which is in contrast to some earlier reports[36].
In summary, this work has attempted to understand the biological implications of the differences in blood levels of homocysteine, DHEA-S and Lp(a) in AD and VAD cases in the context of disease pathogenesis and consequences. However, more inputs are necessary from studies in post-mortem brains or experimental disease models to understand the pathological significance of the alterations of serum parameters in case-control studies. One important limitation of this case-control study is the non-availability of MRI findings for control subjects which precludes any discussion regarding any direct link between altered levels of these serum parameters and changes in brain morphology. Another shortcoming of the present study is the absence of data on serum levels of folic acid, vitamin B12 or pyridoxine in controls and cases which might have influenced the serum levels of homocysteine to different degrees among the groups. Another important confounding factor in this study is the drug history of the patients. At the time of blood collection, the subjects of AD and VAD were on different drugs and within each group the patients were on different dosage schedule. However, it is important to realize that even with such limitations the present study has shown that serum levels of Hcy, DHEA-S and Lp(a) can effectively separate AD and VAD from each other as well as from the controls implying that these parameters have causal links with these disorders. Moreover, it will be interesting to examine if these serum parameters which are differentially altered in AD and VAD can be used as biomarkers to distinguish clinical cases of AD and VAD. Because of the overlapping nature of clinical presentations and some commonalties in pathogenic processes of AD and VAD, it is often difficult to differentiate these two disorders clinically or even by MRI findings. The identifications of serum parameters like homocysteine, Lp(a) and DHEA-S as reported here may be useful for the differential diagnosis, but a larger sample size needs to be studied and methods to determine sensitivity and specificity of these serum parameters with respect to each disease have to be determined for this purpose.
Acknowledgments
The work was supported in part by a grant to Prof. Sasanka Chakrabarti from Department of Science and Technology (DST), Govt. of India, New Delhi (Sanction No. SR/SO/HS-78/2008).
We acknowledge the help of Dr. Avijit Hazra, Department of Pharmacology, IPGMER in the statistical analysis of the data. We also thank Prof. Barun Mukhopadhyay (Indian Statistical Institute, Kolkata) who is a collaborator of Dr. Keya Pal in an ongoing population based study on aging, dietary habits and cognitive impairment for help and suggestions.
References
- [1].Groves WC, Brandt J, Steinberg M, Warren A, Rosenblatt A, Baker A, Lyketsos CG. Vascular dementia and Alzheimer’s disease: is there a difference? A comparison of symptoms by disease duration. J Neuro Psychiatry Clin Neurosci. 2000;12:305–315. doi: 10.1176/jnp.12.3.305. [DOI] [PubMed] [Google Scholar]
- [2].Kalaria RN, Maestre GE, Arizaga R, Friedland RP, Galasko D, Hall K, et al. Alzheimer’s disease and vascular dementia in developing countries: prevalence, management, and risk factors. Lancet Neurol. 2008;7:812–826. doi: 10.1016/S1474-4422(08)70169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lobo A, Launer LJ, Fratiglioni L, Andersen K, Di Carlo A, Breteler MM, et al. Prealence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54:S4–S9. [PubMed] [Google Scholar]
- [4].Swerdlow RH. Pathogenesis of Alzheimer’s disease. Clin Interv Aging. 2007;2:347–359. [PMC free article] [PubMed] [Google Scholar]
- [5].Román GC. Vascular dementia: distinguishing characteristics, treatment, and prevention. J Am Geriatr Soc. 2003;51:S296–S304. doi: 10.1046/j.1532-5415.5155.x. [DOI] [PubMed] [Google Scholar]
- [6].Francis P. Targeting cell death in dementia. Alzheimer Dis Assoc Disord. 2006;20:S3–S7. doi: 10.1097/01.wad.0000213803.82058.46. [DOI] [PubMed] [Google Scholar]
- [7].Andreasen N, Minthon L, Davidsson P, Vanmechelen E, Vanderstichele H, Winblad B, Blennow K. Evaluation of CSF-tau and CSF-Abeta42 as diagnostic markers for Alzheimer disease in clinical practice. Arch Neurol. 2001;58:373–379. doi: 10.1001/archneur.58.3.373. [DOI] [PubMed] [Google Scholar]
- [8].Spies PE, Slats D, Sjögren JM, Kremer BP, Verhey FR, Rikkert MG, Verbeek MM. The cerebrospinal fluid amyloid beta42/40 ratio in the differentiation of Alzheimer’s disease from non-Alzheimer’s dementia. Curr Alzheimer Res. 2010;7:470–476. doi: 10.2174/156720510791383796. [DOI] [PubMed] [Google Scholar]
- [9].de Jong D, Jansen RW, Kremer BP, Verbeek MM. Cerebrospinal fluid amyloid beta42/ phosphorylated tau ratio discriminates between Alzheimer’s disease and vascular dementia. J Gerontol A Biol Sci Med Sci. 2006;61:755–758. doi: 10.1093/gerona/61.7.755. [DOI] [PubMed] [Google Scholar]
- [10].Nägga K, Gottfries J, Blennow K, Marcusson J. Cerebrospinal fluid phospho-tau, total tau and beta-amyloid (1–42) in the differentiation between Alzheimer’s disease and vascular dementia. Dement Geriatr Cogn Disord. 2002;14:183–190. doi: 10.1159/000066023. [DOI] [PubMed] [Google Scholar]
- [11].Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D’Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- [12].Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, Brunetti N, Porcellini E, Licastro F. Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr. 2005;82:636–643. doi: 10.1093/ajcn.82.3.636. [DOI] [PubMed] [Google Scholar]
- [13].Leblhuber F, Walli J, Artner-Dworzak E, Vrecko K, Widner B, Reibnegger G, Fuchs D. Hyperhomocysteinemia in dementia. J Neural Transm. 2000;107:1469–1474. doi: 10.1007/s007020070010. [DOI] [PubMed] [Google Scholar]
- [14].Naylor JC, Hulette CM, Steffens DC, Shampine LJ, Ervin JF, Payne VM, et al. Cerebrospinal fluid dehydroepiandrosterone levels are correlated with brain dehydroepiandrosterone levels, elevated in Alzheimer’s disease, and related to neuropathological disease stage. J Clin Endocrinol Metab. 2008;93:3173–3178. doi: 10.1210/jc.2007-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Aldred S, Mecocci P. Decreased dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS) concentrations in plasma of Alzheimer’s disease (AD) patients. Arch Gerontol Geriatr. 2010;51:e16–e18. doi: 10.1016/j.archger.2009.07.001. [DOI] [PubMed] [Google Scholar]
- [16].Panza F, D’Introno A, Colacicco AM, Capurso C, Pichichero G, Capurso SA, Capurso A, Solfrizzi V. Lipid metabolism in cognitive decline and dementia. Brain Res Rev. 2006;51:275–292. doi: 10.1016/j.brainresrev.2005.11.007. [DOI] [PubMed] [Google Scholar]
- [17].Emanuele E, Peros E, Tomaino C, Feudatari E, Bernardi L, Binetti G, Maletta R, Micieli G, Bruni AC, Geroldi D. Relation of apolipoprotein(a) size to Alzheimer’s disease and vascular dementia. Dement Geriatr Cogn Disord. 2004;18:189–196. doi: 10.1159/000079200. [DOI] [PubMed] [Google Scholar]
- [18].Zhuo JM, Wang H, Praticò D. Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol Sci. 2011;32:562–571. doi: 10.1016/j.tips.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chacón IJ, Molero AE, Pino-Ramírez G, Luchsinger JA, Lee JH, Maestre GE. Risk of dementia associated with elevated plasma homocysteine in a latin american population. Int J Alzheimers Dis. 20092009 doi: 10.4061/2009/632489. pii 632489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Malaguarnera M, Ferri R, Bella R, Alagona G, Carnemolla A, Pennisi G. Homocysteine, vitamin B12 and folate in vascular dementia and in Alzheimer disease. Clin Chem Lab Med. 2004;42:1032–1035. doi: 10.1515/CCLM.2004.208. [DOI] [PubMed] [Google Scholar]
- [21].de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke. 2002;33:1152–1162. doi: 10.1161/01.str.0000014421.15948.67. [DOI] [PubMed] [Google Scholar]
- [22].Kalaria RN, Ballard C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord. 1999;13:S115–123. doi: 10.1097/00002093-199912003-00017. [DOI] [PubMed] [Google Scholar]
- [23].Miller JW, Green R, Mungas DM, Reed BR, Jagust WJ. Homocysteine, vitamin B6, and vascular disease in AD patients. Neurology. 2002;58:1471–1475. doi: 10.1212/wnl.58.10.1471. [DOI] [PubMed] [Google Scholar]
- [24].Ho PI, Ortiz D, Rogers E, Shea TB. Multiple aspects of homocysteine neurotoxicity: glutamate excitotoxicity, kinase hyperactivation and DNA damage. J Neurosci Res. 2002;70:694–702. doi: 10.1002/jnr.10416. [DOI] [PubMed] [Google Scholar]
- [25].Jara-Prado A, Ortega-Vazquez A, Martinez-Ruano L, Rios C, Santamaria A. Homocysteine-induced brain lipid peroxidation: effects of NMDA receptor blockade, antioxidant treatment, and nitric oxide synthase inhibition. Neurotox Res. 2003;5:237–243. doi: 10.1007/BF03033381. [DOI] [PubMed] [Google Scholar]
- [26].Oldreive CE, Doherty GH. Effects of nitric oxide on the survival and neuritogenesis of cerebellar Purkinje neurons. Neurosci Lett. 2007;413:52–57. doi: 10.1007/s12031-011-9590-7. [DOI] [PubMed] [Google Scholar]
- [27].Zhuo JM, Praticò D. Normalization of hyperhomocysteinemia improves cognitive deficits and ameliorates brain amyloidosis of a transgenic mouse model of Alzheimer’s disease. FASEB J. 2010;24:3895–3902. doi: 10.1096/fj.10-161828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].den Heijer T, Vermeer SE, Clarke R, Oudkerk M, Koudstaal PJ, Hofman A, Breteler MM. Homocysteine and brain atrophy on MRI of non-demented elderly. Brain. 2003;126:170–175. doi: 10.1093/brain/awg006. [DOI] [PubMed] [Google Scholar]
- [29].Aisen PS, Schneider LS, Sano M, Diaz-Arrastia R, van Dyck CH, Weiner MF, Bottiglieri T, Jin S, Stokes KT, Thomas RG, Thal LJ. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: a randomized controlled trial. JAMA. 2008;300:1774–1783. doi: 10.1001/jama.300.15.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tsimikas S, Hall JL. Lipoprotein (a) as a potential causal genetic risk factor of cardiovascular disease: a rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol. 2012;60:716–721. doi: 10.1016/j.jacc.2012.04.038. [DOI] [PubMed] [Google Scholar]
- [31].Berglund L, Ramakrishnan R. Lipoprotein(a): an elusive cardiovascular risk factor. Arterioscler Thromb Vasc Biol. 2004;24:2219–2226. doi: 10.1161/01.ATV.0000144010.55563.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Panza F, D’Introno A, Colacicco AM, Capurso C, Pichichero G, Capurso SA, Capurso A, Solfrizzi V. Lipid metabolism in cognitive decline and dementia. Brain Res Rev. 2006;51:275–292. doi: 10.1016/j.brainresrev.2005.11.007. [DOI] [PubMed] [Google Scholar]
- [33].Maninger N, Wolkowitz OM, Reus VI, Epel ES, Mellon SH. Neurobiological and neuropsychiatric effects of dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEAS) Front Neuroendocrinol. 2009;30:65–91. doi: 10.1016/j.yfrne.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rammouz G, Lecanu L, Papadopoulos V. Oxidative Stress-Mediated Brain Dehydroepiandrosterone (DHEA) Formation in Alzheimer’s Disease Diagnosis. Front Endocrinol. 2011;2:69. doi: 10.3389/fendo.2011.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Brown RC, Han Z, Cascio C, Papadopoulos V. Oxidative stress-mediated DHEA formation in Alzheimer’s disease pathology. Neurobiol Aging. 2003;24:57–65. doi: 10.1016/s0197-4580(02)00048-9. [DOI] [PubMed] [Google Scholar]
- [36].Yanase T, Fukahori M, Taniguchi S, Nishi Y, Sakai Y, Takayanagi R, Haji M, Nawata H. Serum dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEA-S) in Alzheimer’s disease and in cerebrovascular dementia. Endocr J. 1996;43:119–123. doi: 10.1507/endocrj.43.119. [DOI] [PubMed] [Google Scholar]
- [37].Kim SB, Hill M, Kwak YT, Hampl R, Jo DH, Morfin R. Neurosteroids: Cerebrospinal fluid levels for Alzheimer’s disease and vascular dementia diagnostics. J Clin Endocrinol Metab. 2003;88:5199–206. doi: 10.1210/jc.2003-030646. [DOI] [PubMed] [Google Scholar]
- [38].Hillen T, Lun A, Reischies FM, Borchelt M, Steinhagen-Thiessen E, Schaub RT. DHEA-S plasma levels and incidence of Alzheimer’s disease. Biol Psychiatry. 2000;47:161–163. doi: 10.1016/s0006-3223(99)00217-6. [DOI] [PubMed] [Google Scholar]
- [39].Kimonides VG, Khatibi NH, Svendsen CN, Sofroniew MV, Herbert J. Dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEAS) protect hippocampal neurons against excitatory amino acid-induced neurotoxicity. Proc Natl Acad Sci U S A. 1998;95:1852–1857. doi: 10.1073/pnas.95.4.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kurata K, Takebayashi M, Morinobu S, Yamawaki S. β-estradiol, dehydroepiandrosterone, and dehydroepiandrosterone sulfate protect against N-methyl-D-aspartate-induced neurotoxicity in rat hippocampal neurons by different mechanisms. J Pharmacol Exp Ther. 2004;311:237–245. doi: 10.1124/jpet.104.067629. [DOI] [PubMed] [Google Scholar]
- [41].Rammouz G, Lecanu L, Aisen P, Papadopoulos V. A lead study on oxidative stress-mediated dehydroepiandrosterone formation in serum: the biochemical basis for a diagnosis of Alzheimer’s disease. J Alzheimers Dis. 2011;24:5–16. doi: 10.3233/JAD-2011-101941. [DOI] [PubMed] [Google Scholar]
- [42].Ryan RA, Carrol J. Studies on a 3-beta-hydroxysteroid sulphotransferase from rat liver. Biochim Biophys Acta. 1976;429:391–401. doi: 10.1016/0005-2744(76)90287-4. [DOI] [PubMed] [Google Scholar]